Introduction
Hepatocellular carcinoma (HCC) is the second most
common cause of cancer-associated mortality worldwide and the
leading cause of mortality among cirrhotic patients (1). Early-stage tumors may be curatively
treated using surgical approaches; however, they are often
undiagnosed and treatment options for advanced HCC, including
transarterial chemoembolization (TACE) and radiofrequency ablation
(RFA), are unsatisfactory. The multikinase inhibitor sorafenib has
been identified to improve the survival benefit for patients with
locally advanced or metastatic HCC; however, adverse side effects
and moderate efficacy limit its use in patients with advanced liver
disease (2). Therefore, it is
important to develop a therapy for advanced HCC that is
well-tolerated, cost-effective and poses an acceptable
risk-to-benefit ratio.
Cytokine-induced killer (CIK) cells, generated by
ex vivo expansion of peripheral blood mononuclear cells
(PBMCs), have been described as exerting a highly potent, non-major
histocompatibility complex (MHC)-restricted,
CD3+/CD56+, natural killer-like T
cell-mediated cytotoxic activity in solid tumors (3–6),
while exhibiting a low capacity to induce graft-versus-host disease
(GvHD) (7,8). Autologous immunotherapy with CIK
cells has been evaluated toward different solid tumors, including
HCC (9,10), and was demonstrated, in association
with TACE and RFA or as adjuvant therapy, to increase median time
to progression, progression-free survival and overall survival for
post-operative patients (11–14).
Randomized studies comparing TACE, RFA and/or surgery with or
without autologous immunotherapy using patient-derived CIK cells
indicated that immunotherapy significantly reduces the recurrence
rate of HCC and increases the overall and progression-free survival
of patients with HCC (15–18). A previous study performed a
meta-analysis and confirmed the benefits of combining CIK
immunotherapy with conventional treatments (19). However, a large-scale development
of this immunotherapy remains difficult to consider in an
autologous setting, due to the logistical hurdles associated with
the production of this cell therapy product (CTP). A previous study
provided the in vitro and in vivo proof-of-concept
that another CTP, termed allogenic suicide gene-modified killer
cells (aSGMKCs), may provide a potent anti-tumor effect toward HCC
(20). This CTP has been
previously demonstrated to provide an anti-leukemic effect when
infused to patients receiving bone marrow transplantation (21), with no major acute toxicity upon
infusion. Long-term monitoring of patients demonstrated the safety
of this approach (22,23). In the event of severe side effects,
including the induction of GvHD, the prior retroviral
vector-mediated transfer of a suicide gene in these cells allows
them to be efficiently killed by infusion of a prodrug, leading to
an efficient control of possible side effects (24–27).
Therefore, the production of a bank of 'ready-for-use' aSGMKCs from
healthy blood donors was proposed as an innovative approach, to
allow for the development of immunotherapies on a more convenient
and broader scale than for that of autologous therapies. This
approach is more attractive, due to several reasons. Firstly, CTP
production is easier to perform when using lymphocytes from normal
donors, as they typically expand more efficiently than those from
heavily-treated patients (28,29).
Additionally, a 'ready-for-use' allogeneic CTP may be rapidly
infused into the patient, without the time required for the
production and qualification of a patient-specific CTP (an
autologous or patient-directed CTP) (30). Furthermore, there is no risk of
losing a clinical-grade CTP batch; if a cell batch planned to be
infused to a given patient is not administered due to medical
contra-indication or non-compliance, it may still be used later for
another patient (30). Although
the gene transfer adds technical complexity during the production
of aSGMKCs, as compared with the process of CIK cell production,
previous studies have indicated the feasibility of producing large
quantities of clinical-grade aSGMKCs (31–33).
Considering their functional similarities [including
potent and non-MHC class I-restricted, NK and NK-like
T-cell-mediated cytotoxic activity and a low potential of GvHD
induction (20,34,35)], the present study aimed to compare
aSGMKCs and CIK cells.
Materials and methods
Production of effector cells
Allogeneic SGMKCs were produced as previously
described (34). Buffy coats of 35
healthy anonymous volunteer blood donors (purchased from the French
National Blood Service following receipt of their written informed
consent for research use of their blood donation) were used as a
source of PBMCs. Buffy coats were obtained by centrifugation over a
Ficoll layer (Invitrogen; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) at 360 × g for 20 min at 20°C. PMBCs were activated with
10 ng/ml CD3 monoclonal antibody (mAb) Orthoclone OKT3 (OKT3;
Janssen-Cilag, Levallois-Perret, France) and 500 IU/ml human
interleukin-2 (IL-2; Proleukin; Novartis Pharmaceuticals Canada,
Inc., Dorval, QC, Canada), and transduced on day 3 with the
MP71-T34FT retroviral vector (Eufets GmbH, Idar-Oberstein, Germany)
encoding the truncated form of the human CD34 (selection marker)
fused to the Herpes simplex virus thymidine kinase (HSV-tk) suicide
gene (36) or the
SFG.iCasp9.2A.ΔCD19 retroviral vector (provided by Professor M.K.
Brenner, Center for Cell and Gene Therapy, Baylor College of
Medicine, Houston, TX, USA) encoding the human CD19 (selection
marker) and the inducible caspase-9 (iCasp9) suicide gene (26). Cells were then immunomagnetically
selected on day 5 with an autoMACS Pro Separator (Miltenyi Biotec
SAS, Paris, France), based on the expression of the CD34 or CD19
membrane markers, and expanded until day 14. Transduced and CD34 or
CD19-selected cells were referred to as aSGMKCs, while
non-transduced cells that expanded in parallel for 14 days were
termed control cells. The transduction efficiency (percentage of
CD34 or CD19-positive cells following transduction) was 8.6±1.0%
(n=13) and 51.2±6.1% (n=3), and the purity of the
immunomagnetically selected fraction was 92.4±1.1% (n=13) and
94.8±0.4% (n=3) for CD34/HSV-tk and CD19/iCasp9-transduced cells,
respectively. Relative cell growth was calculated as the ratio of
the number of cells (counted using a B-500 microscope; Optika,
Ponteranica, Italy) of the indicated phenotype obtained at the end
of the expansion at 37°C (day 14) to the input number of cells of
the specific phenotype at initiation of the culture (day 0).
CIK cell production
The cells were produced according to three
previously published protocols: Protocol 1 (P1), PBMC activation
with 100 U/ml interferon-γ (IFN-γ; PeproTech, Inc.,
Neuilly-sur-Seine, France) at day 0 and 10 ng/ml OKT3 + 500 U/ml
IL-2 at day 1 (9,37). These cells were referred to as CIK
P1. Protocol 2 (P2), PBMC activation with 100 U/ml IFN-γ at day 0
and 10 ng/ml OKT3 + 500 U/ml IL-2 + 100 U/ml IL-1 (PeproTech, Inc.)
at D0 (11,15). These cells were referred to as CIK
P2. Protocol 3 (P3): PBMCs activation with 10 ng/ml OKT3 + 500 U/ml
IL-2 + 20 ng/ml IL-7 (PeproTech, Inc.) + 20 ng/ml IL-15 (PeproTech,
Inc.) at day 0 (38). These cells
were referred to as CIK P3. All cells were expanded until day 14 in
culture medium consisting in RPMI 1640 medium (Invitrogen; Thermo
Fisher Scientific, Inc.) supplemented with 10% v/v human serum and
500 U/ml IL-2.
Flow cytometry
Effector cells were analyzed for the relative
repartition of CD3−/CD56+ NK,
CD3+/CD56+ NK-like T and
CD3+/CD56− T cells following staining with
Pacific Blue-conjugated CD3 mAb (BD Biosciences, San Diego, CA,
USA) and phycoerythrincyanin (PE-Cy)5-conjugated CD56 mAb (Miltenyi
Biotec GmbH, Bergisch Gladbach, Germany). The purity of aSGMKCs was
evaluated by flow cytometry following staining with
PE-Cy7-conjugated mAb specific for CD34 or CD19 (BD Biosciences).
Multi-color samples were acquired on a LSRII special order research
product flow cytometer (LSRII; BD Biosciences) calibrated using a
Cytometer Setup & Tracking Beads kit (BD Biosciences) to ensure
consistency of fluorescence intensity measurement throughout all
experiments. Compensation was performed with a CompBeads kit (cat.
no. 51-90-9001229; BD Biosciences). Cell debris and dead cells were
excluded using forward scatter area and side scatter area and
doublet cells were excluded using side scatter width and side
scatter area. FACSDiva software (version 6.1.2; BD Biosciences) was
used for the final analysis and graphical output.
In vitro cytotoxicity assay
Target cells (1,500 HeLa, Hu h-7 cel ls/wel l, 2, 50
0 SK-Hep1 cel ls/wel l, 20,000 PLC-PRF/5 cells/well; American Type
Culture Collection, Molsheim, France) were co-cultured in RPMI 1640
medium (with 500 U/ml IL-2) in the presence or absence of graded
quantities of effector cells for 6 days in flat bottom 96-well
plates and, when indicated, in the presence of monoclonal mouse
anti-human tumor necrosis factor-α (TNF-α; clone MAb1; cat. no.
16-7348), monoclonal mouse anti-human IFN-γ (clone NIB42; cat. no.
16-7318), monoclonal mouse anti-human Fas-ligand (Fas-L; clone
NOK-1; cat. no. 16-9919) or monoclonal mouse anti-human TNF-related
apoptosis inducing ligand (TRAIL; clone RIK-2; cat. no. 16-9927)
mAbs (5 µg/ml; eBiosciences, Inc., San Diego, CA, USA;
1:100). Subsequently, non-adherent effector cells and dead target
cells were removed by washing with phosphate-buffered saline (PBS),
and the remaining viable, adherent target cells were stained with
crystal violet (Sigma-Aldrich, St. Louis, MO, USA) at room
temperature for 1 min. Absorbance was read at 560 nm on a Mithras
LB940 microplate reader (Berthold Technologies GmbH & Co.,
Thoiry, France). Data are expressed as percentage killing, at the
indicated effector:target (E:T) cell ratios as: [1 −
(Abs.E:T / Abs.Ctrle)] × 100, where Abs.E:T indicates
the absorbance at a given E:T ratio and Abs.Ctrle is the absorbance
of target cells alone (E:T=0). Percentage killing is then expressed
as lytic units 50% (LU50), calculated as the reverse of the number
of cells per 106 effector cells required to kill 50% of
target cells (Fig. 1). LU50 of the
experimental groups were normalized to the LU50 values of their
control group, in order to normalize inter-experimental variations,
and expressed as the mean ± standard error (20).
CD107a degranulation assay
Lymphokine-activated killer (LAK) cells were
generated by activating PBMCs with IL-2 (1,000 U/ml) for 4 days.
The effector cells (aSGMKCs or LAK cells) and target cells (Huh-7
cells or K562 cells, used as positive control of lysis) were
co-incubated at 37°C in culture medium at an E:T cell ratio of 4:1
in the presence of monoclonal mouse anti-human PE-CD107a (cat. no.
555801) or isotype control antibody (10 µl per
106 effector cells; BD Biosciences; 1:10) to a total
volume of 100 µl in a flat bottom 96-well plate. After 1 h,
50 µl culture medium containing brefeldin A (10
µg/ml) and GolgiStop (4 µl for 6×106
cells; BD Biosciences) were added for a further 3 h incubation. The
effector cells were then labeled in cytometry tubes with Pacific
Blue-conjugated CD3 mAb and PE-Cy5-conjugated CD56 mAb, washed
twice with 3 ml PBS (Invitrogen; Thermo Fisher Scientific, Inc.),
fixed with PBS supplemented with 2% v/v formaldehyde (Merck
Millipore, Darmstadt, Germany), and analyzed by flow cytometry
(LSRII).
Ethical approval
Animal experimentations were performed following
approval by the local ethical committee (Comité Régional d'Ethique
en Matière d'Expérimentation Animale de Strasbourg; approval no.
AL/34/41/02/13) on November 7, 2012, according to European
guidelines (directive 2010/63/UE) and relevant national rules. For
ethical reasons, mice were not monitored until tumor-associated
mortality but were sacrificed at the end of experiments by cervical
dislocation following general anesthesia with 3% isoflurane.
In vivo cytotoxicity assays
In vivo cytotoxicity assays were performed as
previously described (20).
Luciferase-expressing Huh-7 target cells (1×106
Huh-7-Luc cells) were subcutaneously co-injected into the right
flank of 86 severe combined immunodeficient-beige (SCIB-bg)
recipient mice (Taconic Biosciences, Inc., Ejby, Denmark) in the
absence (PBS, n=13) or presence of 10×106
iCasp9-expressing aSGMKCs effector cells (CIK P1, n=14; CIK P2,
n=17; CIK P3, n=13; iCasp9-expressing aSGMKCs, n=17). Daily
intraperitoneal injections of IL-2 (106 IU/kg) were
administered throughout the duration of the experiment. When
indicated, an intraperitoneal injection of the iCasp9 prodrug
[AP20187; also termed chemical inducer of death (CID); Clontech
Laboratories, Saint Germain en Laye, France] at 2.5 mg/kg was
performed together with the aSGMKCs injection (n=12).
Eight-to-twelve week-old male and female mice were used and
randomly distributed in experimental groups. The mice were housed
under specific pathogen-free conditions with food and drinking
water containing 1 mg/ml paracetamol (Doliprane 300;
Sanofi-Aventis, Paris, France) ad libitum. Luciferase
activity of Huh-7-Luc cells, determined by bioluminescence imaging
(BLI) using an IVIS Lumina Series II camera (PerkinElmer, Roissy,
France) following an intraperitoneal injection of 100 µl
luciferin (20 mg/ml; Caliper Lifesciences), was determined at the
time of target cell injection on days 0, 4, 7, 11 and 14. Tumor
cell bioluminescence, analyzed with Living Image software (version
3.1; PerkinElmer), was quantified in a round-shape region of
interest centered on the cell injection site and was expressed as
photons/sec/cm2/steradian (p/sec/cm2/sr).
Relative tumor growth was obtained by calculating the ratio of
bioluminescence at the indicated day to the bioluminescence at day
0. A relative tumor growth value of ≤1 indicated tumor cell
elimination and a value of >1 indicated an expansion of tumor
cells.
Statistical analysis
Data that was not normally distributed was compared
with Mann-Whitney U or Fisher's exact non-parametric tests; when
data distribution was normal, Student's paired t-test was used.
Data are expressed as the mean ± standard error. A χ2
test was performed to compare the number of animals with and
without relapse in the experimental groups compared with the
control group. P<0.05 indicates a statistically significant
difference.
Results
Phenotypic characterization of
aSGMKCs
As previously reported (20,35),
aSGMKCs were predominantly constituted of
CD3+/CD56− T cells, then by
CD3+/CD56+ NK-like T cells and to a lesser
extent by CD3−/CD56+ NK cells. Other
mononuclear subsets, including B cells or monocytes, were not
detectable (Fig. 2A). This
repartition was similar to the one observed in non-transduced
control cells that were activated and expanded in parallel with
aSGMKCs (Fig. 2A). This indicates
that the cell culture process was responsible for these
phenotypical modifications and that the gene transfer process did
not lead to the preferential transduction or selection of a
lymphocyte population. Furthermore, the ex vivo expansion
was associated with a significantly higher relative expansion of
NK-like T cells than that observed with NK or T cells (P<0.05;
Student's paired t-test; Fig.
2B).
Mechanism of aSGMKC-mediated killing of
hepatoma cells
As previously reported, aSGMKCs may provide a potent
cytotoxic activity toward various liver-derived cell lines, such as
Huh-7, PLC-PRF5 (HCC cell lines) and SK-Hep1, and toward the
cervical carcinoma cell line HeLa (20). The cytotoxic activity of aSGMKCs,
evaluated at different E:T ratios, indicated that 50 and 90% target
cell killing were obtained at mean E:T ratios of ~3:1 and ~13:1 to
40:1, respectively (Fig. 3A),
excluding PLC-PRF5 cells, which were more sensitive to
aSGMKC-mediated killing. It was previously reported that the
cytotoxic activity of aSGMKCs, evaluated in more detail toward
Huh-7 cells, was HLA class-I independent and was predominantly
mediated by CD56+/CD3− NK and
CD56+/CD3+ NK-like T cells (20). In order to further characterize the
mechanisms of target cell killing, the involvement of the
granzyme/perforin pathway was evaluated by CD107a staining of
aSGMKCs (and of LAK cells, used as a positive control of
non-MHC-restricted cytotoxic cells) following incubation with Huh-7
cells (or K562 cells, used as a control of LAK-sensitive target
cells). Following co-incubation with Huh-7 cells, the frequency of
CD107a-positive cells, corresponding to effector cells with
degranulated lytic granules, were higher in LAK cells than in
aSGMKCs generated from the specific donors (Fig. 3B). A similar trend was observed, to
a greater extent, when effector cells were incubated with K562
cells (Fig. 3B). Regardless of the
E:T cell combinations, the frequency of CD107a-positive cells was
higher in the NK cell subset than in the NK-like T cell subset,
while no induction of CD107a expression was observed in T cells
(Fig. 3C). Overall, considering
that the frequency of NK and NK-like T cells was lower than that of
T cells (Fig. 2A), the induction
of CD107a expression in aSGMKCs following incubation with Huh-7
cells was minimal, suggesting that other killing mechanisms may be
involved.
 | Figure 3Characterization of aSGMKC cytotoxic
activity. (A) Cytotoxic activity of aSGMKCs tested at different
effector:target ratios against HCC cell lines (Huh-7, PLC-PRF5) and
non-HCC cell lines (HeLa, SK-Hep1). Data are expressed as mean ±
standard error of 6 independent experiments. (B) Percent of
CD107a-positive cells in aSGMKCs and LAK cells after a 4 h-culture
in the absence (NS) or presence of Huh-7 or K562 target cells. Data
are expressed as mean ± standard error of 3 independent
experiments. (C) Percent of CD107a-positive cells gated in NK,
NK-like T and T cell subsets in the experiments reported in B. (D)
Cytotoxic activity of aSGMKCs incubated with Huh-7 cells in the
absence or presence of blocking monoclonal antibodies against
IFN-γ, TNF-α, Fas-L or TRAIL. Data are expressed as normalized LU50
(mean ± standard error) of 7 independent experiments. Control group
: 100%=587±286 LU50 (*P<0.05 vs. control). aSGMKCs,
allogeneic suicide gene-modified killer cells; HCC, hepatocellular
carcinoma; LAK, lymphokine-activated killer; NK, natural killer;
IFN-γ, interferon-γ; TNF-α, anti-tumor necrosis factor-α; Fas-L,
fas ligand; TRAIL, TNF-related apoptosis inducing ligand; LU50,
lytic units 50%. |
Thus, in vitro cytotoxicity assays were
performed in the presence or absence of antibodies targeting IFN-γ,
TNF-α, Fas-L or TRAIL. The cytotoxic activity of aSGMKCs was not
affected by the addition of anti-IFN-γ or anti-TNF-α antibodies
alone but a significant (P<0.05; Mann-Whitney U test; Fig. 3D) reduction of the cytotoxicity
level was observed when both antibodies were added, suggesting a
synergistic involvement of IFN-γ and TNF-α in target cell killing.
TRAIL-mediated killing was also involved in aSGMKC-induced killing,
as demonstrated by the significant (P<0.05; Mann-Whitney U test;
Fig. 3D) inhibitory effect of
blocking anti-TRAIL antibodies on the cytotoxicity level. Blocking
anti-Fas-L antibodies had no significant inhibitory effect on
sSGMKCs cytotoxicity but enhanced the reduction of cytotoxicity by
anti-TRAIL antibodies (P<0.05; Fig.
3D).
Allogeneic SGMKCs are phenotypically and
functionally similar to CIK cells
It was previously reported, that aSGMKCs exhibited
reduced in vitro and in vivo alloreactivity (20,34,35),
in terms of proliferative response and potential of GvHD induction
and that their cytotoxicity was non-MHC class I-restricted and
predominantly mediated by NK and NK-like T cells (20). Together with the present
observations indicating the involvement of TRAIL in the killing
mechanism, this prompted the evaluation of similarities between
aSGMKCs and CIK cells, another immunotherapy product identified to
provide potent non-MHC class I-restricted anti-tumor activity
toward hematologic and solid tumors. Indeed, CIK cells have also
previously exhibited NK and NK-like T cell-mediated cytotoxicity
and low potential of GvHD induction (39). Thus, the phenotypic and cytotoxic
activity of aSGMKCs and CIK cells were compared, generated in
parallel from the same healthy donors using three different cell
expansion protocols for CIK cell production. There were no
significant differences between aSGMKCs and CIK cells in terms of
repartition of T, NK-like T and NK cell subsets (Fig. 4A). Furthermore, the cytotoxic
activity of aSGMKCs, evaluated against Huh-7 cells (or HeLa cells,
used as a positive control of target cell killing) was similar, or
even greater than that of CIK cells, when evaluated following 1 or
6 days of E:T cell co-culture (Fig.
4B).
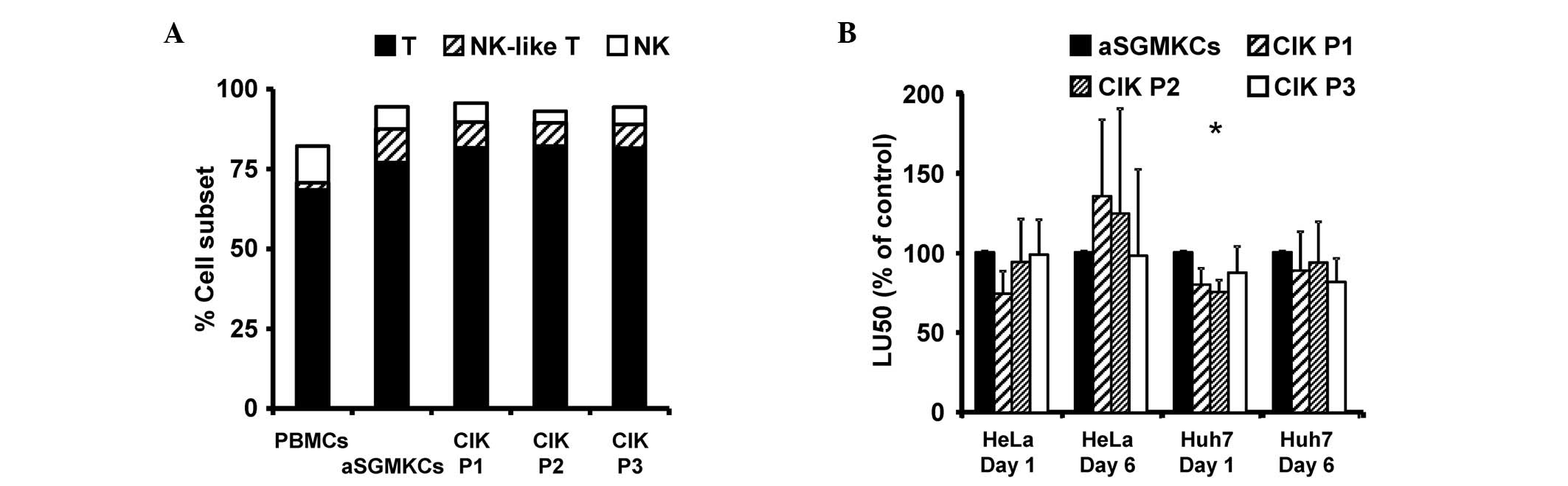 | Figure 4Comparison of aSGMKCs and CIK cells
produced according to three different protocols (P1-P3). (A)
Frequency of NK (white bars), NK-like T and T cells in aSGMKCs, CIK
cells and PBMCs. In a single experiment, aSGMKCs, CIK cells and
PBMCs were isolated and produced from the same healthy donor. Data
are expressed as the mean of four independent experiments. (B)
Cytotoxic activity of aSGMKCs and CIK cells evaluated after day 1
or 6 of co-culture with HeLa and Huh-7 target cells. Data are
expressed as normalized LU50 (mean ± standard error) of five
independent experiments. Control groups (aSGMKCs): 100%=258±137
LU50 (HeLa, day 1), 752±362 LU50 (HeLa, day 6), 90±28 LU50 (Huh-7,
day 1) and 162±53 LU50 (Huh-7, day 6) (*P<0.05 vs.
aSGMKCs). aSGMKCs, allogeneic suicide gene-modified killer cells;
CIK, cytokine-induced killer; P1/2/3, protocol 1/2/3; NK, natural
killer; PBMCs, peripheral blood mononuclear cells; LU50, lytic
units 50%. |
In vivo anti-tumor effect of aSGMKCs
toward hepatoma cells
The data presented was generated using aSGMKCs
transduced with an HSV-tk-expressing retroviral vector. However, a
new generation of suicide genes, based on the inducible activation
of caspase domains, has been recently developed, including the
iCasp9 gene (40), whose efficacy
has been demonstrated in clinical trials (26,27).
This transgene induces rapid killing of aSGMKCs in the presence of
their prodrug, CID, as >95% of cells are killed within 2 h, as
determined by flow cytometry following Annexin V/propidium iodide
staining (unpublished data). This is more rapid than the killing of
HSV-tk-expressing aSGMKCs by its prodrug, ganciclovir (20,41).
Therefore, iCasp9-expressing aSGMKCs were compared with CIK cells
using an in vivo cytotoxicity assay. Immunodeficient SCID-bg
mice were subcutaneously injected with Huh-7-Luc target cells, in
the absence or presence of aSGMKCs or CIK cells, and monitored for
tumor growth by BLI. Fig. 5
represents the data obtained for one representative mouse of each
group.
When Huh-7-Luc cells were injected alone, the median
relative tumor growth was >1 at all time points, indicating the
expansion of tumor cells (Table
I). When CIK cells were co-injected with target cells, the
median relative tumor growth values were <1 (P<0.05,
Mann-Whitney U test), indicating that CIK cells exhibited a
significant antitumor effect. When aSGMKCs were co-injected with
target cells, a similar anti-tumor effect was also observed
(P<0.05, Mann-Whitney U test). Notably, the antitumor effect of
aSGMKCs was reversed by CID administration, confirming that the
effect was aSGMKC-mediated (Table
I). However, tumor escape occurred in certain (12–38%) mice
injected with aSGMKCs or CIK cells. Therefore, the frequency of
mice with tumor regression (a relative tumor growth value <1)
was also evaluated. As indicated by Table I, this frequency was 23.1% in the
control group injected with Huh-7-Luc cells alone, indicating a
lack of engraftment in these mice. However, this frequency
significantly (P<0.05, χ2 test) increased to
61.5–88.2% of mice injected with aSGMKCs or CIK cells, indicating
tumor rejection in these groups. It is of note that the frequency
reversed to 25.0% in mice receiving aSGMKCs when the prodrug CID
was administered at the same time (Table I), indicating that aSGMKCs were
rapidly killed, before having time to exert their anti-tumor
effects.
 | Table IEffect of varied treatments on tumor
growth. |
Table I
Effect of varied treatments on tumor
growth.
| Treatment | Relative tumor
growth [median (min-max)]a
| Regressing
tumors%b |
|---|
| Day 0 | Day 4 | Day 7 | Day 11 | Day 14 |
|---|
| PBS (n=13) | 1.0 (1.0–1.0) | 2.2
(0.1–290.6) | 5.9
(0.1–440.5) | 27.8
(0.1–1231.0) | 33.3
(0.0–1739.3) | 23.1 |
| CIK P1 (n=14) | 1.0 (1.0–1.0) | 0.1c (0.0–22.9) | 0.3c (0.0–21.5) | 0.2c (0.0–46.2) | 0.3c (0.0–104.6) | 71.4d |
| CIK P2 (n=17) | 1.0 (1.0–1.0) | 0.1c (0.0–22.5) | 0.1c (0.0–193.2) | 0.1c (0.0–297.6) | 0.1c (0.0–137.2) | 88.2d |
| CIK P3 (n=13) | 1.0 (1.0–1.0) | 0.1c (0.0–1.0) | 0.1c (0.0–1.4) | 0.1c (0.0–26.4) | 0.1c (0.0–26.0) | 61.5d |
| SGMKC (n=17) | 1.0 (1.0–1.0) | 0.1c (0.0–16.0) | 0.1c (0.0–41.8) | 0.1c (0.0–109.9) | 0.1c (0.0–110.4) | 76.5d |
| SGMKC + CID
(n=12) | 1.0 (1.0–1.0) | 0.8
(0.1–114.7) | 1.4 (0.0–97.2) | 0.3
(0.0–136.2) | 0.7
(0.0–330.7) | 25.0 |
Discussion
A previous study demonstrated (20) that aSGMKCs produced following CD3
plus IL-2 activation, a combination previously described (31) and used in our clinical trial of
aSGMKC infusion at time of bone marrow transplantation (24), exhibit a potent non-MHC class
I-restricted cytotoxic activity towards HCC cell lines that is
mediated primarily by NK and NK-like T cells. Numerous studies have
indicated that ex vivo-activated autologous PBMCs,
previously expanded in similar conditions as the current aSGMKCs
[namely CIK cells (3,5)], however without gene transfer,
exhibit phenotypic and functional properties similar to those of
the current aSGMKCs, including high cytotoxic activity (6), particularly against HCC cell lines
(4,5), and low potential for GvHD induction
(7,8). The cytotoxic activity of CIK cells is
non-MHC-restricted and is primarily mediated by NK-like T cells
(3). To the best of our knowledge,
the current study is the first to indicate that, by comparing cells
expanded according to protocols used for aSGMKC and CIK cell
production, aSGMKCs are suicide-gene-modified CIK cells. The
repartition of NK, NK-like T and T cells within aSGMKCs is similar
to that observed in CIK cells, and the in vitro and in
vivo cytotoxic activity of aSGMKCs is as potent as that
obtained with CIK cells. Although further experiments are required,
the results of the present study suggest that the association
between IFN-γ, TNF-α and TRAIL, possibly in synergy with Fas-L, all
previously demonstrated to be involved in CIK-mediated cell
killing, may be involved as an effector mechanism of aSGMKC-induced
target cell killing (42–44). Overall, the current results support
the hypothesis that aSGMKCs, proposed to be used clinically in an
allogeneic setting, are similar to CIK cells, which are currently
used clinically in an autologous setting.
Various clinical studies (11–18),
several of which were randomized (15–18),
comparing TACE, RFA and/or surgery with or without CIK therapy,
have demonstrated the ability of this autologous adoptive
immunotherapy to reduce HCC recurrence and to increase median time
to relapse, relapse-free survival, and overall survival in patients
with HCC. These clinical results strengthen the hypothesis that
aSGMKCs may exert an anti-tumor effect against HCC, particularly as
they are allogeneic; the MHC-restricted and/or non-MHC-restricted
alloreactivity of aSGMKCs may provide an additional mechanism for
tumor recognition.
Another issue to consider for aSGMKC-based adoptive
allogeneic immunotherapy is alloimmunization. As the patients are
immunocompetent, an anti-aSGMKC immune response leading to aSGMKC
rejection should occur. This is an additional safety element that
would limit the risk of long-term aSGMKC toxicity, but it may also
limit short-term anti-tumor efficacy. This means that aSGMKCs must
be sufficiently cytotoxic to operate quickly and destroy tumor
cells prior to being rejected by the patient's immune system. The
observations of the current study indicate that high cytotoxic
activity in aSGMKCs may be detected within 1 day in vitro
and 4 days in vivo (prior to the development of an immune
response leading to their rejection). In phase I/II clinical trials
of infusion of partially HLA-matched EBV-specific allogeneic T
cells for the treatment of post-transplant EBV-induced lymphoma,
performed by Haque et al (45,46)
infused cells may be detected for up to 44 days following infusion
(45), with patients experiencing
withdrawal or reduction of immunosuppression for the previous 2–6
weeks. However, in vivo survival was typically limited to ~1
week. In the phase II study reported by Haque et al
(46) complete responses were
observed in 14 out of 33 patients despite alloimmunization,
indicating that alloimmunization may not represent a strong
limitation to adoptive allogeneic immunotherapy if short-term
anti-tumor effects are selected. Similarly, Slavin et al
(47) performed a phase I trial
investigating the infusion of 'intentionally HLA-mismatched IL-2
activated killer' (IMAK) cells produced from normal donors to
patients with solid tumors. Rejection of infused IMAK cells was
expected to occur within 1 week and was considered a safety
mechanism to avoid GvHD. Infusion of IMAK cells was determined to
be safe and, among the 35 patients with metastatic solid tumors,
only one grade I GvHD was observed.
Taking advantage of the resistance of aSGMKCs to
calcineurin inhibitors, such as ciclosporin A and FK506 (20,48,49),
it was previously reported in a surrogate mouse model of aSGMKCs
that alloimmunization of such effector cells may be prevented in
immunocompetent mice, leading to an anti-tumor effect toward a
syngeneic HCC cell line (20).
However, other methods for preventing alloimmunization should be
considered by eliminating or inhibiting regulatory T cells, using
cyclophosphamide or fludarabine-based lymphodepletion protocols
(50,51) [as performed by Slavin et al
(47) at the time of IMAK
infusion] or mTOR inhibitors such as rapamycin [known to also
exhibit anti-tumor activity (52,53)]. Induction of aSGMKC-specific
tolerance by co-infusion of non-depleting CD3 or CD4 monoclonal
antibodies (54,55) or by administration of CTLA4-Ig
(56) may also be considered.
In conclusion, the present study has demonstrated
that aSGMKCs are CIK cells with a high cytotoxic activity towards
solid tumors. Therefore, the production of a bank of 'ready-to-use'
aSGMKCs for the treatment of solid tumors, particularly for
indications of hepatic localization, should be considered.
Additionally, their advantages include rapid availability and low
potential for GvHD induction, as well as the fact that no clinical
batch in the bank may be lost due to last-minute contra-indication
from the recipient. To prevent secondary effects leading to
toxicity, the suicide gene will allow the specific elimination of
aSGMKCs through the administration of the prodrug. The primary
issue remaining to be determined is the clinical efficacy of
aSGMKCs in immunocompetent animal HCC models, provided that
alloimmunization may be controlled by an immunosuppressive and/or
lympho-ablative conditioning regimen.
Acknowledgments
The authors would like to thank Mr. Nicolas Brignon
and Mr. Richard Pidl for their excellent animal care. This study
was supported by Inserm, the Fondation pour la Recherche Médicale
(Comité Alsace), the Ligue Nationale Contre le Cancer (Conférence
de Coordination Inter-Régionale Grand-Est; grant no. 1FI10005LBKD),
the Association pour la recherche sur le Cancer (ARC; grant no.
SFI20111203529). Dr Eric Robinet was supported by the Agence
Nationale pour la Recherche sur le SIDA et les Hépatites Virales
(grant no 2008 059 ULP). Dr Tao Wu was a recipient of a fellowship
of the Alsace Region and Dr Céline Leboeuf was a recipient of
fellowships of the Fondation Transplantation and the ARC (grant no.
DOC20110603384).
References
|
1
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology.
142:1264–1273.e1. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Ricci S, Mazzaferro V, Hilgard
P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A,
et al SHARP Investigators Study Group: Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med. 359:378–390. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmidt-Wolf IG, Lefterova P, Mehta BA,
Fernandez LP, Huhn D, Blume KG, Weissman IL and Negrin RS:
Phenotypic characterization and identification of effector cells
involved in tumor cell recognition of cytokine-induced killer
cells. Exp Hematol. 21:1673–1679. 1993.PubMed/NCBI
|
|
4
|
Wang FS, Liu MX, Zhang B, Shi M, Lei ZY,
Sun WB, Du QY and Chen JM: Antitumor activities of human autologous
cytokine-induced killer (CIK) cells against hepatocellular
carcinoma cells in vitro and in vivo. World J Gastroenterol.
8:464–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim HM, Lim J, Yoon YD, Ahn JM, Kang JS,
Lee K, Park SK, Jeong YJ, Kim JM, Han G, et al: Anti-tumor activity
of ex vivo expanded cytokine-induced killer cells against human
hepatocellular carcinoma. Int Immunopharmacol. 7:1793–1801. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu PH and Negrin RS: A novel population of
expanded human CD3+CD56+ cells derived from T
cells with potent in vivo antitumor activity in mice with severe
combined immunodeficiency. J Immunol. 153:1687–1696.
1994.PubMed/NCBI
|
|
7
|
Nishimura R, Baker J, Beilhack A, Zeiser
R, Olson JA, Sega EI, Karimi M and Negrin RS: In vivo trafficking
and survival of cytokine-induced killer cells resulting in minimal
GVHD with retention of antitumor activity. Blood. 112:2563–2574.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baker J, Verneris MR, Ito M, Shizuru JA
and Negrin RS: Expansion of cytolytic CD8(+) natural killer T cells
with limited capacity for graft-versus-host disease induction due
to interferon gamma production. Blood. 97:2923–2931. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Olioso P, Giancola R, Di Riti M, Contento
A, Accorsi P and Iacone A: Immunotherapy with cytokine induced
killer cells in solid and hematopoietic tumours: A pilot clinical
trial. Hematol Oncol. 27:130–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi M, Zhang B, Tang ZR, Lei ZY, Wang HF,
Feng YY, Fan ZP, Xu DP and Wang FS: Autologous cytokine-induced
killer cell therapy in clinical trial phase I is safe in patients
with primary hepatocellular carcinoma. World J Gastroenterol.
10:1146–1151. 2004.PubMed/NCBI
|
|
11
|
Hao MZ, Lin HL, Chen Q, Ye YB, Chen QZ and
Chen MS: Efficacy of transcatheter arterial chemoembolization
combined with cytokine-induced killer cell therapy on
hepatocellular carcinoma: a comparative study. Chin J Cancer.
29:172–177. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang ZM, Li W, Li S, Gao F, Zhou QM, Wu
FM, He N, Pan CC, Xia JC, Wu PH and Zhao M: Cytokine-induced killer
cells in combination with transcatheter arterial chemoembolization
and radiofrequency ablation for hepatocellular carcinoma patients.
J Immunother. 36:287–293. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pan K, Li YQ, Wang W, Xu L, Zhang YJ,
Zheng HX, Zhao JJ, Qiu HJ, Weng DS, Li JJ, et al: The efficacy of
cytokine-induced killer cell infusion as an adjuvant therapy for
postoperative hepatocellular carcinoma patients. Ann Surg Oncol.
20:4305–4311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cui J, Wang N, Zhao H, Jin H, Wang G, Niu
C, Terunuma H, He H and Li W: Combination of radiofrequency
ablation and sequential cellular immunotherapy improves
progression-free survival for patients with hepatocellular
carcinoma. Int J Cancer. 134:342–351. 2014. View Article : Google Scholar
|
|
15
|
Weng DS, Zhou J, Zhou QM, Zhao M, Wang QJ,
Huang LX, Li YQ, Chen SP, Wu PH and Xia JC: Minimally invasive
treatment combined with cytokine-induced killer cells therapy lower
the short-term recurrence rates of hepatocellular carcinomas. J
Immunother. 31:63–71. 2008. View Article : Google Scholar
|
|
16
|
Yu X, Zhao H, Liu L, Cao S, Ren B, Zhang
N, An X, Yu J, Li H and Ren X: A randomized phase II study of
autologous cytokine-induced killer cells in treatment of
hepatocellular carcinoma. J Clin Immunol. 34:194–203. 2014.
View Article : Google Scholar
|
|
17
|
Lee JH, Lee JH, Lim YS, Yeon JE, Song TJ,
Yu SJ, Gwak GY, Kim KM, Lee JW and Yoon JH: Adjuvant immunotherapy
with autologous cytokine-induced killer cells for hepatocellular
carcinoma. Gastroenterology. 148:1383–1391.e6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takayama T, Sekine T, Makuuchi M, Yamasaki
S, Kosuge T, Yamamoto J, Shimada K, Sakamoto M, Hirohashi S, Ohashi
Y and Kakioze T: Adoptive immunotherapy to lower postsurgical
recurrence rates of hepatocellular carcinoma: A randomised trial.
Lancet. 356:802–807. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li X, Dai D, Song X, Liu J, Zhu L and Xu
W: A meta-analysis of cytokine-induced killer cells therapy in
combination with minimally invasive treatment for hepatocellular
carcinoma. Clin Res Hepatol Gastroenterol. 38:583–591. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Leboeuf C, Mailly L, Wu T, Bour G, Durand
S, Brignon N, Ferrand C, Borg C, Tiberghien P, Thimme R, et al: In
vivo proof of concept of adoptive immunotherapy for hepatocellular
carcinoma using allogeneic suicide gene-modified killer cells. Mol
Ther. 22:634–644. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ciceri F, Bonini C, Marktel S, Zappone E,
Servida P, Bernardi M, Pescarollo A, Bondanza A, Peccatori J,
Rossini S, et al: Antitumor effects of HSV-TK-engineered donor
lymphocytes after allogeneic stem-cell transplantation. Blood.
109:4698–4707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Deschamps M, Mercier-Lethondal P, Certoux
JM, Henry C, Lioure B, Pagneux C, Cahn JY, Deconinck E, Robinet E,
Tiberghien P and Ferrand C: Deletions within the HSV-tk transgene
in long-lasting circulating gene-modified T cells infused with a
hematopoietic graft. Blood. 110:3842–3852. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou X, Di Stasi A, Tey SK, Krance RA,
Martinez C, Leung KS, Durett AG, Wu MF, Liu H, Leen AM, et al:
Long-term outcome after haploidentical stem cell transplant and
infusion of T cells expressing the inducible caspase 9 safety
transgene. Blood. 123:3895–3905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tiberghien P, Ferrand C, Lioure B, Milpied
N, Angonin R, Deconinck E, Certoux JM, Robinet E, Saas P, Petracca
B, et al: Administration of herpes simplex-thymidine
kinase-expressing donor T cells with a T-cell-depleted allogeneic
marrow graft. Blood. 97:63–72. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bonini C, Ferrari G, Verzeletti S, Servida
P, Zappone E, Ruggieri L, Ponzoni M, Rossini S, Mavilio F,
Traversari C and Bordignon C: HSV-TK gene transfer into donor
lymphocytes for control of allogeneic graft-versus-leukemia.
Science. 276:1719–1724. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Di Stasi A, Tey SK, Dotti G, Fujita Y,
Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG,
Grilley B, et al: Inducible apoptosis as a safety switch for
adoptive cell therapy. N Engl J Med. 365:1673–1683. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou X, Dotti G, Krance RA, Martinez CA,
Naik S, Kamble RT, Durett AG, Dakhova O, Savoldo B, Di Stasi A, et
al: Inducible caspase-9 suicide gene controls adverse effects from
alloreplete T cells after haploidentical stem cell transplantation.
Blood. 125:4103–4113. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ott PA, Herzog BA, Quast S, Hofstetter HH,
Boehm BO, Tary-Lehmann M, Durinovic-Bello I, Berner BR and Lehmann
PV: Islet-cell antigen-reactive T cells show different expansion
rates and Th1/Th2 differentiation in type 1 diabetic patients and
healthy controls. Clin Immunol. 115:102–114. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Imataki O, Heike Y, Makiyama H, Iizuka A,
Ikarashi Y, Ishida T, Wakasugi H and Takaue Y: Insufficient ex vivo
expansion of Valpha24(+) natural killer T cells in malignant
lymphoma patients related to the suppressed expression of CD1d
molecules on CD14(+) cells. Cytotherapy. 10:497–506. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vickers MA, Wilkie GM, Robinson N, Rivera
N, Haque T, Crawford DH, Barry J, Fraser N, Turner DM, Robertson V,
et al: Establishment and operation of a good manufacturing
practice-compliant allogeneic Epstein-Barr virus (EBV)-specific
cytotoxic cell bank for the treatment of EBV-associated
lymphoproliferative disease. Br J Haematol. 167:402–410. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Robinet E, Certoux JM, Ferrand C, Maples
P, Hardwick A, Cahn JY, Reynolds CW, Jacob W, Hervé P and
Tiberghien P: A closed culture system for the ex vivo transduction
and expansion of human T lymphocytes. J Hematother. 7:205–215.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tey SK, Dotti G, Rooney CM, Heslop HE and
Brenner MK: Inducible caspase 9 suicide gene to improve the safety
of allodepleted T cells after haploidentical stem cell
transplantation. Biol Blood Marrow Transplant. 13:913–924. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kühlcke K, Ayuk FA, Li Z, Lindemann C,
Schilz A, Schade UM, Fauser AA, Zander AR, Eckert HG and Fehse B:
Retroviral transduction of T lymphocytes for suicide gene therapy
in allogeneic stem cell transplantation. Bone Marrow Transplant.
25(Suppl 2): S96–S98. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mercier-Letondal P, Montcuquet N, Sauce D,
Certoux JM, Jeanningros S, Ferrand C, Bonyhadi M, Tiberghien P and
Robinet E: Alloreactivity of ex vivo-expanded T cells is correlated
with expansion and CD4/CD8 ratio. Cytotherapy. 10:275–288. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sauce D, Tonnelier N, Duperrier A,
Petracca B, de Carvalho Bittencourt M, Saadi M, Saas P, Ferrand C,
Herve P, Tiberghien P and Robinet E: Influence of ex vivo expansion
and retrovirus-mediated gene transfer on primary T lymphocyte
phenotype and functions. J Hematother Stem Cell Res. 11:929–940.
2002. View Article : Google Scholar
|
|
36
|
Fehse B, Kustikova OS, Li Z, Li Z, Wahlers
A, Bohn W, Beyer WR, Chalmers D, Tiberghien P, Kühlcke K, Zander AR
and Baum C: A novel 'sort-suicide' fusion gene vector for T cell
manipulation. Gene Ther. 9:1633–1638. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chan JK, Hamilton CA, Cheung MK, Karimi M,
Baker J, Gall JM, Schulz S, Thorne SH, Teng NN, Contag CH, et al:
Enhanced killing of primary ovarian cancer by retargeting
autologous cytokine-induced killer cells with bispecific
antibodies: A preclinical study. Clin Cancer Res. 12:1859–1867.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi Y, Wu W, Wan T, Liu Y, Peng G, Chen Z
and Zhu H: Impact of polyclonal anti-CD3/CD28-coated magnetic bead
expansion methods on T cell proliferation, differentiation and
function. Int Immunopharmacol. 15:129–137. 2013. View Article : Google Scholar
|
|
39
|
Rutella S and Locatelli F: Is there a role
for cytokine-induced killer cells in cancer immunotherapy?
Immunotherapy. 4:867–869. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Straathof KC, ulè MA, Yotnda P, Dotti G,
Vanin EF, Brenner MK, Heslop HE, Spencer DM and Rooney CM: An
inducible caspase 9 safety switch for T-cell therapy. Blood.
105:4247–4254. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Marin V, Cribioli E, Philip B, Tettamanti
S, Pizzitola I, Biondi A, Biagi E and Pule M: Comparison of
different suicide-gene strategies for the safety improvement of
genetically manipulated T cells. Hum Gene Ther Methods. 23:376–386.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rajbhandary S, Zhao MF, Zhao N, Lu WY, Zhu
HB, Xiao X, Deng Q and Li YM: Multiple cytotoxic factors involved
in IL-21 enhanced antitumor function of CIK cells signaled through
STAT-3 and STAT5b pathways. Asian Pac J Cancer Prev. 14:5825–5831.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hu L, Cao D, Li Y, He Y and Guo K:
Resveratrol sensitized leukemia stem cell-like KG-1a cells to
cytokine-induced killer cells-mediated cytolysis through NKG2D
ligands and TRAIL receptors. Cancer Biol Ther. 13:516–526. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Franceschetti M, Pievani A, Borleri G,
Vago L, Fleischhauer K, Golay J and Introna M: Cytokine-induced
killer cells are terminally differentiated activated CD8 cytotoxic
T-EMRA lymphocytes. Exp Hematol. 37:616–628. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Haque T, Wilkie GM, Taylor C, Amlot PL,
Murad P, Iley A, Dombagoda D, Britton KM, Swerdlow AJ and Crawford
DH: Treatment of Epstein-Barr-virus-positive post-transplantation
lymphoproliferative disease with partly HLA-matched allogeneic
cytotoxic T cells. Lancet. 360:436–442. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Haque T, Wilkie GM, Jones MM, Higgins CD,
Urquhart G, Wingate P, Burns D, McAulay K, Turner M, Bellamy C, et
al: Allogeneic cytotoxic T-cell therapy for EBV-positive
post-transplantation lymphoproliferative disease: results of a
phase 2 multicenter clinical trial. Blood. 110:1123–1131. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Slavin S, Ackerstein A, Or R, Shapira MY,
Gesundheit B, Askenasy N and Morecki S: Immunotherapy in high-risk
chemotherapy-resistant patients with metastatic solid tumors and
hematological malignancies using intentionally mismatched donor
lymphocytes activated with rIL-2: A phase I study. Cancer Immunol
Immunother. 59:1511–1519. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Contassot E, Robinet E, Angonin R,
Laithier V, Bittencourt M, Pavy JJ, Cahn JY, Hervé P and Tiberghien
P: Differential effects of cyclosporin A on the alloreactivity of
fresh and ex vivo-expanded T lymphocytes. Bone Marrow Transplant.
22:1097–1102. 1998. View Article : Google Scholar
|
|
49
|
Maury S, Litvinova E, Boyer O, Benard L,
Bruel S, Klatzmann D and Cohen JL: Effect of combined cytostatic
cyclosporin A and cytolytic suicide gene therapy on the prevention
of experimental graft-versus-host disease. Gene Ther. 9:201–207.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dudley ME, Wunderlich JR, Yang JC, Sherry
RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE,
Mavroukakis SA, et al: Adoptive cell transfer therapy following
non-myeloablative but lymphodepleting chemotherapy for the
treatment of patients with refractory metastatic melanoma. J Clin
Oncol. 23:2346–2357. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ghiringhelli F, Menard C, Puig PE, Ladoire
S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L and Chauffert
B: Metronomic cyclophosphamide regimen selectively depletes
CD4+CD25+ regulatory T cells and restores T
and NK effector functions in end stage cancer patients. Cancer
Immunol Immunother. 56:641–648. 2007. View Article : Google Scholar
|
|
52
|
Zhang JF, Liu JJ, Lu MQ, Cai CJ, Yang Y,
Li H, Xu C and Chen GH: Rapamycin inhibits cell growth by induction
of apoptosis on hepatocellular carcinoma cells in vitro. Transpl
Immunol. 17:162–168. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Semela D, Piguet AC, Kolev M, Schmitter K,
Hlushchuk R, Djonov V, Stoupis C and Dufour JF: Vascular remodeling
and antitumoral effects of mTOR inhibition in a rat model of
hepatocellular carcinoma. J Hepatol. 46:840–848. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Martin A, Tisch RM and Getts DR:
Manipulating T cell-mediated pathology: Targets and functions of
monoclonal antibody immunotherapy. Clin Immunol. 148:136–147. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Waldmann H, Adams E and Cobbold S:
Reprogramming the immune system: Co-receptor blockade as a paradigm
for harnessing tolerance mechanisms. Immunol Rev. 223:361–370.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fife BT and Bluestone JA: Control of
peripheral T-cell tolerance and autoimmunity via the CTLA-4 and
PD-1 pathways. Immunol Rev. 224:166–182. 2008. View Article : Google Scholar : PubMed/NCBI
|

















