Introduction
Wilms' tumor (WT), or nephroblastoma, is one of the
most prevalent types of solid tumor of the urinary tract in
childhood (1). It is estimated
that this malignant kidney tumor affects ~1/10,000 children
worldwide, which arises from undifferentiated renal precursors and
presents with a triphasic histology consisting of stromal,
epithelial and blastemal elements (2,3).
Although it is treatable with long-term survival rates, the
combination of chemo/radiotherapy and surgery can result in severe
complications in adulthood (4,5). In
addition, the molecular mechanism underlying the pathogenesis of WT
remains to be fully elucidated. Therefore, the urgent investigation
of novel therapeutic targets for developing anticancer drugs in WT
treatment is required.
The histone deacetylase (HDAC) family is comprised
of 18 proteins, which are classified into stages I–IV based on
their homology and structure (6–8). The
epigenetic regulation of gene expression by the HDAC family has
been demonstrated to be involved in tumor initiation, progression
and metastasis (9,10). Several HDAC antagonists have been
observed to inhibit the growth and induce the apoptosis of
different types of cancer cell (11–13).
In addition, preclinical studies have demonstrated the therapeutic
application of HDAC inhibitors as potential anticancer agents
(11). HDAC5 belongs to the class
II HDAC/acuc/apha family, which is critical in the regulation of
cell growth, proliferation, apoptosis and survival (14). A previous study demonstrated that
HDAC5 was overexpressed in patients with liver cancer, suggesting
that the dysfunction of HDAC5 may be significant in
hepatocarcinogenesis (15).
Another previous study observed the upregulation of HDAC5 in
patients with high-risk medulloblastoma, which was associated with
poor survival rates (16). The
present study aimed to identify the expression of HDAC5 in WT
tissue specimens and investigate the proliferation-promoting
function of HDAC5 in human WT cells.
Materials and methods
Tissue samples
A total of 23 pairs of primary WT tissues (male, 12;
female, 11; age, 3–60 months) and normal adjacent tissues (male,
13; female, 10; age, 8–64 months) were collected during therapeutic
surgery at the Department of Surgery, Childrens' Hospital
Affiliated to Soochow University, Soochow University (Suzhou,
China). All of the specimens from the WT cases had a size of
1.0×1.0×0.2 cm and were pathologically diagnosed. Informed consent
was obtained from all participants, and the present study was
approved by the Institutional Review Board of the Childrens'
Hospital Affiliated to Soochow University, Soochow University
(Suzhou, China).
Cell culture and transfection
G401 cells were obtained from American Type Culture
Collection (Manassas, VA, USA) and were cultured in RPMI 1640
medium (Gibco Life Technologies, Carlsbad, CA, USA), supplemented
with 10% fetal calf serum (Invitrogen Life Technologies), 100 IU/ml
penicillin and 100 mg/ml streptomycin (both from Sigma-Aldrich, St.
Louis, MO, USA). Cells were cultured at 37°C in a humidified
atmosphere containing 5% CO2. Plasmids encoding
HDAC5-Flag, small interfering RNA (siRNA) specific for HDAC5 (sense
5′-CAUUGCCCACGAGUUCUCACCUGAU-3′ and anti-sense
5′-AUCAGGUGAGAACUCGUGGGCAAUG-3′) and siRNA specific for c-Met
(sense 5′-AGCCAAUUUAUCAGGAGGUTT-3′ and antisense
5′-ACCUCCUGAUAAAUUGGCUTT-3′) were purchased from Shanghai
GenePharma Co., Ltd. (Shanghai, China). HDAC5 and siRNA c-Met were
purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). The
cells were transfected using Lipofectamine 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA), according to the manufacturer's
instructions. In brief, cells were seeded into six-well plates and
transfected with 30 nM siRNA oligos with 4 µl Lipofectamine
2000 at 80% confluence.
Cell Counting Kit-8 (CCK-8) assay
The cells were seeded into 96-well plates at a
density of 6.0×103 cells/well. Cell viability was
assessed using a CCK-8 assay (Beyotime Institute of Biotechnology,
Haimen, China). The absorbance of each well was read using a
spectrophotometer (4015-000; Thermo Fisher Scientific, Waltham, MA,
USA) at 450 nm.
Bromodeoxyuridine (BrdU) incorporation
analysis
BrdU incorporation assay was performed using a BrdU
Cell Proliferation kit (Roche Diagnostics, Indianapolis, IN, USA)
to analyze cell growth. Briefly, the cells were incubated with BrdU
for 6 h, rinsed and then incubated with a fluorescein
isothiocyanate-labeled antibody against BrdU for 30 min. The
stained cells were then analyzed using a fluorescence micro-plate
reader (2350; EMD Millipore, Billerica, MA, USA). Fold changes in
BrdU incorporation were normalized against the mean fluorescence
intensity of the control group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Frozen tissues or cells were homogenized using
TRIzol reagent (Invitrogen Life Technologies) and total RNA was
isolated using a Qiagen RNeasy Lipid Tissue Mini kit (Qiagen,
Hilden, Germany) according to the manufacturer's instructions.
Reverse transcription was performed using a Takara RNA PCR kit
(Takara Bio, Inc., Otsu, Japan) according to the manufacturer's
instructions. RT-qPCR was performed using SYBR Green Premix Ex Taq
(Takara Bio, Inc.) on an ABI 7500 Cycling system (Applied
Biosystems, Life Technologies, Thermo Fisher Scientific) to
determine the expression levels of the genes of interest. The
cycling parameters were as follows: Initial denaturation at 95°C
for 30 sec, followed by a two-step program of 95°C for 5 sec and
60°C for 31 sec over 40 cycles. Experiments were performed in
triplicate. The primers were obtained from Invitrogen Life
Technologies and the sequences were as follows: HDAC5 forward,
5′-CTCAAGCAGCAGCAGCAGCTCCA-3′ and reverse,
5′-CCTTCTGTTTAAGCCTCGAACG-3′; c-Met forward,
5′-CCTTCGAAAGCAACCATTTTACG-3′ and reverse,
5′-TTACTGACATACGCGGCTTGCGC-3′. The expression levels were
quantified using the 2−∆∆Ct method.
Western blot analysis
Total protein was extracted with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology) and quantified using the bicinchoninic acid assay
(BAC kit; Beyotime Institute of Biotechnology). The proteins (20
µg) were fractionated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes (EMD Millipore). The membrane
was blocked in 4% dried milk at room temperature for 1 h and
incubated with primary antibodies at 4°C overnight. Rabbit
anti-HDAC5 monoclonal antibody (cat no. 3443S; 1:1,000; Cell
Signaling Technology, Inc., Beverly, MA, USA) and mouse anti-c-Met
monoclonal antibody (cat no. sc-162; 1:1,000; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) were used as primary
antibodies. Subsequently, the membrane was incubated with
horseradish peroxidase-conjugated secondary antibodies (mouse-IgG;
1:5,000; Beyotime Institute of Biotechnology, Shanghai, China) for
1 h at room temperature and detected with an enhanced
chemiluminescence system (Roche Diagnostics), according to the
manufacturer's instructions. β-actin was purchased from Santa Cruz
Biotechnology, Inc., which was used as a loading control.
Statistical analysis
Values are expressed as the mean ± standard
deviation, based on three separate experiments. One-way analysis of
variance was used for comparison between multiple groups and
Student's t-test was used for comparison between two groups.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed using SPSS,
version 13.0 (SPSS, Inc., Chicago, IL, USA).
Results
Upregulation of HDAC5 in WT tissues
The mRNA expression levels of HDAC5 were examined in
23 paired WT and adjacent non-tumor tissues using RT-qPCR. It was
observed that the expression of HDAC5 was significantly increased
in the tumor tissues (Fig. 1A). In
addition, the protein expression level of HDAC5 was determined
using western blot analysis, which revealed increased expression
levels of HDAC5 in the WT samples, compared with the adjacent
normal samples (Fig. 1B). These
data suggested that HDAC5 may be important in WT pathogenesis.
HDAC5 promotes cellular proliferation of
WT
In order to elucidate the biological role of HDAC5
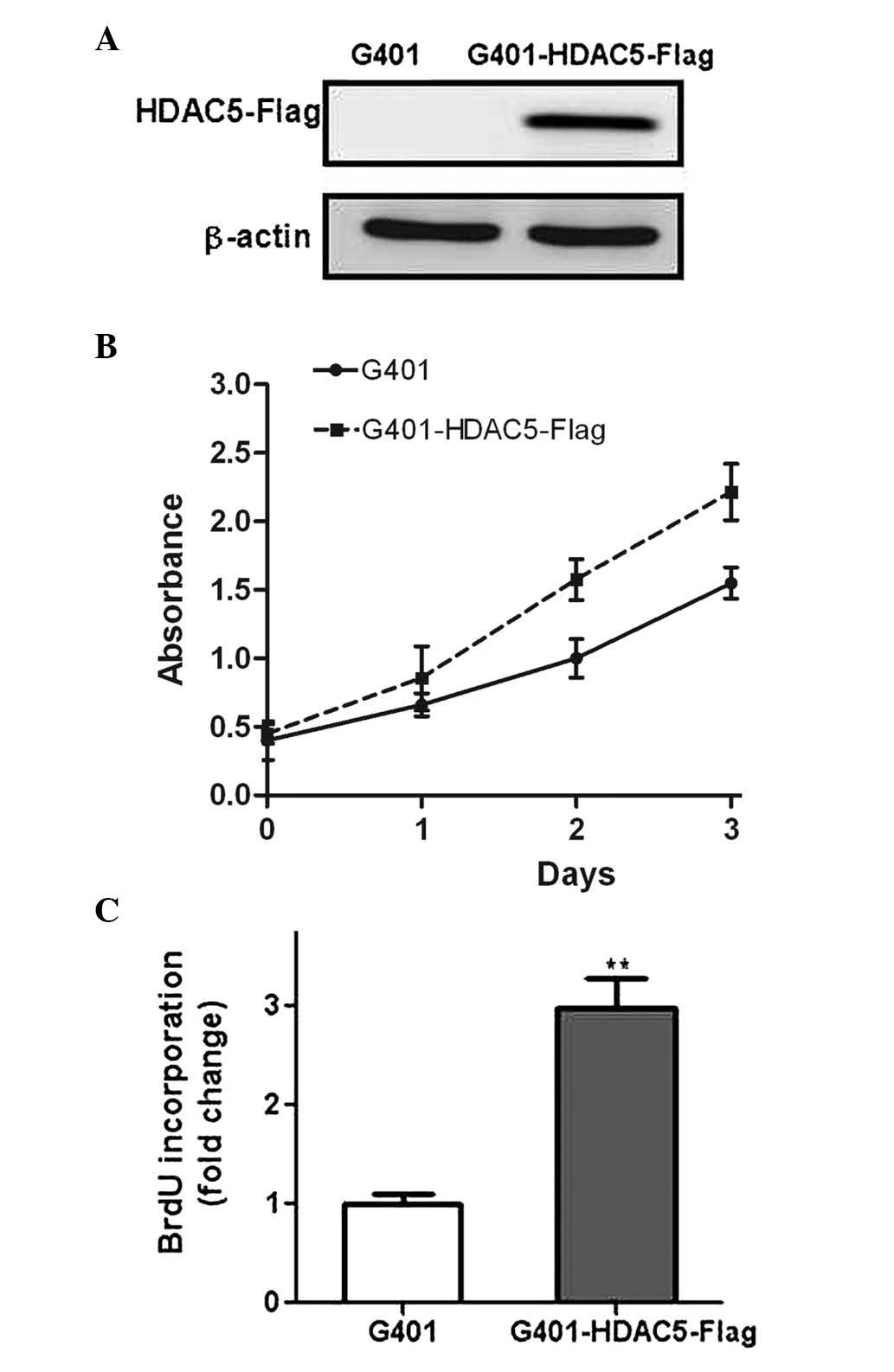
in WT, G401 cells were transfected with a recombinant plasmid
containing HDAC5 (Fig. 2A). The
subsequent CCK-8 assay demonstrated that overexpression of HDAC5
clearly promoted the proliferation of the WT cells (P<0.05)
(Fig. 2B). BrdU incorporation is a
standard method to measure DNA synthesis and is considered to be a
surrogate procedure for the evaluation of proliferation (17). In the present study, the G401 cells
overexpressing HDAC5 exhibited a faster growth rate, compared with
the control G401 cells (Fig. 2C),
consistent with the results of the CCK-8 assay. In addition, the
G401 cells were transfected with siRNA targeting HDAC5, and the
subsequent downregulation of the target gene was confirmed using
RT-qPCR and western blotting (Fig. 3A
and B). The data indicated that the downregulation of HDAC5
significantly decreased the rate of proliferation of the G401 cells
(Fig. 3C and D). Taken together,
these results indicated that HDAC5 may be a positive regulator of
cellular proliferation in WT.
HDAC5 regulates WT growth through
c-Met
The present study also investigated the molecular
mechanism underlying the proliferative effect of HDAC5, and the
results demonstrated that the mRNA expression of c-Met was
increased by >4-fold in the G401 cells overexpressing HDAC5,
compared with the control G401 cells (Fig. 4A). Western blot analysis also
confirmed the upregulation of c-Met in the HDAC5-transfected cells
(Fig. 4B and C). By contrast, the
expression of c-Met was reduced in the G401 cells following the
downregulation of HDAC5 by siRNA interference (Fig. 4D–F). Taken together, these data
suggested that HDAC5 was an upstream regulator of c-Met. In order
to ascertain whether the induction of c-Met is a prerequisite for
the proliferative function of HDAC5, the expression of c-Met was
suppressed using siRNA oligos (Fig. 5A
and B). Notably, the downregulation of c-Met inhibited the
proliferative effects of HDAC5 on the G401 cells (Fig. 5C and D), suggesting that the
proliferation-promoting function of HDAC5 was involved in the
regulation of c-Met.
Discussion
Accumulating evidence has demonstrated that several
members of the HDAC family are critical in promoting carcinogenesis
(18). However, the biological
function of HDAC5 in WT remains to be fully elucidated. In the
present study, the expression levels of HDAC5 were examined in WT
specimens and adjacent normal tissue specimens, and the
proliferative effect of HDAC5 was investigated in human WT
cells.
HDACs have been reported to remove the acetyl groups
from the N-acetyl-sites on the histone, thus modifying the
chromatin structure and modulating the expression levels of several
genes (19). The aberrant
expression levels of HDAC family members have been associated with
tumor initiation and progression (18). HDAC5 belongs to the class II HDAC
family and is a critical regulator in cellular proliferation, cell
cycle progression and apoptosis in several cancer cell lines and
animal models (20,21). A previous study demonstrated that
HDAC5 is significantly overexpressed in high-risk medulloblastoma,
compared with low-risk medulloblastoma, and its expression is
associated with poor survival rates, suggesting that HDAC5 may be
an important marker for risk stratification (18). Another study demonstrated that
HDAC5 promotes the progression of osteosarcoma through upregulation
of the twist 1 oncogene (22). In
the present study, it was demonstrated that HDAC5 was significantly
increased in human WT samples, compared with adjacent normal
tissues, suggesting that HDAC5 may be critical in the pathogenesis
of WT. In addition, in vitro investigation demonstrated that
overexpression of HDAC5 in G401 cells significantly increased the
level of cell proliferation, compared with normal G401 cells. By
contrast, HDAC5 knockdown using siRNA reduced cell growth,
suggesting that HDAC5 acts as a positive regulator in the
proliferation of WT cells.
The cell surface receptor tyrosine kinase, c-Met, is
overexpressed in a several types of malignancy, including
hepatocellular carcinoma (23),
lung cancer (24) and ovarian
carcinoma (25). c-Met is
important in cellular proliferation, migration and metastasis
(26). A number of studies have
demonstrated that increased c-Met signaling contributes to
carcinogenesis through several pathways, including the focal
adhesion kinase, phosphatidyl inositol 3-kinase and extracellular
signal-regulated kinase pathways (27–29).
It has been demonstrated that the downregulation of c-Met produces
anti-tumor effects, predominantly based on anti-proliferation and
anti-angiogenesis, in several types of cancer cell (30–32).
In the present study, the upregulation of HDAC5 was observed to
promote the mRNA and protein expression levels of c-Met in G401
cells. In addition, c-Met depletion significantly reversed the
proliferation-promoting function of HDAC5 in human WT cells.
In conclusion, the present study demonstrated that
HDAC5 promoted the proliferation of WT through upregulation of
c-Met. These findings provide further insight into the pathogenic
mechanisms of WT and suggests HDAC5 as a potential therapeutic
target.
Acknowledgments
The present study was supported by grants from the
Science Foundation of Jiangsu Province Health Department (grant
nos. Q201304 and Q201503), the Natural Science Foundation of
Jiangsu Province of China (grant nos. BK2011312 and BL2012051) and
the Natural Science Foundation of China (no. 81502496).
References
|
1
|
Turnbull C, Perdeaux ER, Pernet D, Naranjo
A, Renwick A, Seal S, Munoz-Xicola RM, Hanks S, Slade I, Zachariou
A, et al: A genome-wide association study identifies susceptibility
loci for Wilms tumor. Nat Genet. 44:681–684. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wegert J, Bausenwein S, Kneitz S, Roth S,
Graf N, Geissinger E and Gessler M: Retinoic acid pathway activity
in Wilms tumors and characterization of biological responses in
vitro. Mol Cancer. 10:1362011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tian F, Yourek G, Shi X and Yang Y: The
development of Wilms tumor: From WT1 and microRNA to animal models.
Biochim Biophys Acta. 1846:180–187. 2014.PubMed/NCBI
|
|
4
|
Sonn G and Shortliffe LM: Management of
Wilms tumor: Current standard of care. Nat Clin Pract Urol.
5:551–560. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Acipayam C, Sezgin G, Bayram İ, Yılmaz S,
Özkan A, Tuncel DA, Tanyeli A and Küpeli S: Treatment of Wilms
tumor using carboplatin compared to therapy without carboplatin.
Pediatr Blood Cancer. 61:1578–1583. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
West AC, Mattarollo SR, Shortt J, Cluse
LA, Christiansen AJ, Smyth MJ and Johnstone RW: An intact immune
system is required for the anticancer activities of histone
deacetylase inhibitors. Cancer Res. 73:7265–7276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wagner JM, Hackanson B, Lübbert M and Jung
M: Histone deacetylase (HDAC) inhibitors in recent clinical trials
for cancer therapy. Clin Epigenetics. 1:117–136. 2010. View Article : Google Scholar
|
|
8
|
Witt O, Deubzer HE, Milde T and Oehme I:
HDAC family: What are the cancer relevant targets? Cancer Lett.
277:8–21. 2009. View Article : Google Scholar
|
|
9
|
Song SH, Han SW and Bang YJ:
Epigenetic-based therapies in cancer: Progress to date. Drugs.
71:2391–2403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vigushin DM and Coombes RC: Histone
deacetylase inhibitors in cancer treatment. Anticancer Drugs.
13:1–13. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khan O and La Thangue NB: HDAC inhibitors
in cancer biology: Emerging mechanisms and clinical applications.
Immunol Cell Biol. 90:85–94. 2012. View Article : Google Scholar
|
|
12
|
Cao B, Li J, Zhu J, Shen M, Han K, Zhang
Z, Yu Y, Wang Y, Wu D, Chen S, et al: The antiparasitic clioquinol
induces apoptosis in leukemia and myeloma cells by inhibiting
histone deacetylase activity. J Biol Chem. 288:34181–34189. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lai F, Jin L, Gallagher S, Mijatov B,
Zhang XD and Hersey P: Histone deacetylases (HDACs) as mediators of
resistance to apoptosis in melanoma and as targets for combination
therapy with selective BRAF inhibitors. Adv Pharmacol. 65:27–43.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Matkovich SJ, Duan X, Diwan A,
Kang MY and Dorn GN II: Receptor-independent protein kinase C alpha
(PKCalpha) signaling by calpain-generated free catalytic domains
induces HDAC5 nuclear export and regulates cardiac transcription. J
Biol Chem. 286:26943–26951. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng GW, Dong LD, Shang WJ, Pang XL, Li
JF, Liu L and Wang Y: HDAC5 promotes cell proliferation in human
hepatocellular carcinoma by up-regulating Six1 expression. Eur Rev
Med Pharmacol Sci. 18:811–816. 2014.PubMed/NCBI
|
|
16
|
Milde T, Oehme I, Korshunov A,
Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M,
Taylor MD, von Deimling A, et al: HDAC5 and HDAC9 in
medulloblastoma: Novel markers for risk stratification and role in
tumor cell growth. Clin Cancer Res. 16:3240–3252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Actis M, Inoue A, Evison B, Perry S,
Punchihewa C and Fujii N: Small molecule inhibitors of PCNA/PIP-box
interaction suppress translesion DNA synthesis. Bioorg Med Chem.
21:1972–1977. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Z and Zhu WG: Targeting histone
deacetylases for cancer therapy: From molecular mechanisms to
clinical implications. Int J Biol Sci. 10:757–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rincon-Arano H, Halow J, Delrow JJ,
Parkhurst SM and Groudine M: UpSET recruits HDAC complexes and
restricts chromatin accessibility and acetylation at promoter
regions. Cell. 151:1214–1228. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marek L, Hamacher A, Hansen FK, Kuna K,
Gohlke H, Kassack MU and Kurz T: Histone deacetylase (HDAC)
inhibitors with a novel connecting unit linker region reveal a
selectivity profile for HDAC4 and HDAC5 with improved activity
against chemoresistant cancer cells. J Med Chem. 56:427–436. 2013.
View Article : Google Scholar
|
|
21
|
Peixoto P, Castronovo V, Matheus N, Polese
C, Peulen O, Gonzalez A, Boxus M, Verdin E, Thiry M, Dequiedt F and
Mottet D: HDAC5 is required for maintenance of pericentric
heterochromatin and controls cell-cycle progression and survival of
human cancer cells. Cell Death Differ. 19:1239–1252. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Xia J, Yu YL, Wang SQ, Wei YB,
Chen FY, Huang GY and Shi JS: HDAC5 promotes osteosarcoma
progression by upregulation of Twist 1 expression. Tumour Biol.
35:1383–1387. 2014. View Article : Google Scholar
|
|
23
|
Kondo S, Ojima H, Tsuda H, Hashimoto J,
Morizane C, Ikeda M, Ueno H, Tamura K, Shimada K, Kanai Y and
Okusaka T: Clinical impact of c-Met expression and its gene
amplification in hepatocellular carcinoma. Int J Clin Oncol.
18:207–213. 2013. View Article : Google Scholar
|
|
24
|
Lee JM, Yoo JK, Yoo H, Jung HY, Lee DR,
Jeong HC, Oh SH, Chung HM and Kim JK: The novel miR-7515 decreases
the proliferation and migration of human lung cancer cells by
targeting c-Met. Mol Cancer Res. 11:43–53. 2013. View Article : Google Scholar
|
|
25
|
Koon EC, Ma PC, Salgia R, Christensen JG,
Berkowitz RS and Mok SC: Effect of a c-Met-specific,
ATP-competitive small-molecule inhibitor SU11274 on human ovarian
carcinoma cell growth, motility and invasion. Int J Gynecol Cancer.
18:976–984. 2008. View Article : Google Scholar
|
|
26
|
Goetsch L, Caussanel V and Corvaia N:
Biological significance and targeting of c-Met tyrosine kinase
receptor in cancer. Front Biosci (Landmark Ed). 18:454–473. 2013.
View Article : Google Scholar
|
|
27
|
Lee YY, Kim HP, Kang MJ, Cho BK, Han SW,
Kim TY and Yi EC: Phosphoproteomic analysis identifies activated
MET-axis PI3K/AKT and MAPK/ERK in lapatinib-resistant cancer cell
line. Exp Mol Med. 45:e642013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nam HJ, Chae S, Jang SH, Cho H and Lee JH:
The PI3K-Akt mediates oncogenic Met-induced centrosome
amplification and chromosome instability. Carcinogenesis.
31:1531–1540. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Guessous F, Yang Y, Johnson E,
Marcinkiewicz L, Smith M, Zhang Y, Kofman A, Schiff D, Christensen
J and Abounader R: Cooperation between c-Met and focal adhesion
kinase family members in medulloblastoma and implications for
therapy. Mol Cancer Ther. 11:288–297. 2012. View Article : Google Scholar :
|
|
30
|
Hong SW, Jung KH, Park BH, Zheng HM, Lee
HS, Choi MJ, Yun JI, Kang NS, Lee J and Hong SS: KRC-408, a novel
c-Met inhibitor, suppresses cell proliferation and angiogenesis of
gastric cancer. Cancer Lett. 332:74–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang X, Yang Y, Tang S, Tang H, Yang G, Xu
Q and Wu J: Anti-tumor effect of polysaccharides from Scutellaria
barbata D. Don on the 95-D xenograft model via inhibition of the
C-met pathway. J Pharmacol Sci. 125:255–263. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang HT, Wang L, Ai J, Chen Y, He CX, Ji
YC, Huang M, Yang JY, Zhang A, Ding J and Geng MY: SOMG-833, a
novel selective c-MET inhibitor, blocks c-MET-dependent neoplastic
effects and exerts antitumor activity. J Pharmacol Exp Ther.
350:36–45. 2014. View Article : Google Scholar : PubMed/NCBI
|