Introduction
Colorectal cancer (CRC) is the third most common
type of cancer in men, and the second most common in women
(1). Following surgery for the
treatment of CRC, chemotherapy is vital, and in patients that lack
the opportunity for surgical intervention, chemotherapy is the
predominant therapy used to prolong survival. However, sensitivity
to chemotherapy varies widely between individuals; according to
statistics, ~50% of patients with CRC succumb to tumor metastasis
and recurrence (2).
Hypoxia is a common feature in various types of
solid tumor, including CRC. It has previously been reported that
hypoxic microenvironments promote the malignant progression of
solid tumors (3).
Hypoxia-inducible factor 1 (HIF-1) has an important role in the
alteration of tumor biology in response to hypoxia. The adaptive
response to hypoxia is adjusted by a family of transcription
factors; the most important member of which is HIF-1α (4). Cobalt chloride (CoCl2) has
been widely used to simulate hypoxia in in vivo and in
vitro studies (5,6). CoCl2 inhibits the
hydroxylation of HIF-1α, thus stabilizing HIF-1α by preventing it
from binding to the von Hippel-Lindau tumor suppressor protein
(7–9). In addition, CoCl2 has been
shown to activate hypoxia-dependent pathways (10), and is a chemical inducer of HIF-1
(11). In the present study, tumor
cells were treated with CoCl2 to stimulate hypoxia. It
has previously been reported that hypoxia potentiates tumor
resistance to chemotherapy and radiotherapy (12); however, how the hypoxic
microenvironment is involved in anticancer drug resistance remains
unclear.
HIF-1α induces the expression of various genes that
are associated with angiogenesis, glucose metabolism, survival and
tumor progression (13,14). Multidrug resistance is often caused
by overexpression of P-glycoprotein (P-gp) and multidrug resistance
protein (MRP). The genes multidrug resistant 1 (MDR1)/P-gp and MRP
are able to decrease the efficacy of chemotherapeutic anticancer
agents (15,16). The B-cell lymphoma 2 (Bcl-2) family
comprises anti-apoptotic and proapoptotic proteins. The Bcl-2
family members Bcl-2, Bcl-2-associated X protein (Bax) and
Bcl-2-associated agonist of cell death (Bad) are considered to be
associated with signals of cell survival and damage, and have a
critical role in apoptosis. Bcl-2 is an apoptosis suppressor gene,
whereas Bax and Bad are apoptosis-inducing genes (17,18).
However, the molecular mechanism underlying hypoxia-induced
chemoresistance in tumor cells has yet to be fully elucidated. In
the present study, the proliferation of LOVO cells and the tumor
inhibition ratio (TIR) following treatment with 5-fluorouracil
(5-FU) were determined using a
3-(4,5-dimethylthiahiazol-z-y1)-3,5-diphenyte-trazolium bromide
(MTT) assay. Furthermore, reverse transcription-polymerase chain
reaction (RT-PCR) and western blotting were used to determine mRNA
and protein expression levels, respectively. Flow cytometry (FCM)
was used to detect the accumulation and retention of Adriamycin
(ADR) under hypoxic and normoxic conditions.
Materials and methods
Cell lines and cell culture
The LOVO human colorectal cancer cell line was
obtained from Peking University Health Science Center (Beijing,
China). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc.) and antibiotics (1% penicillin and 1%
streptomycin; North China Pharmaceutical Group Corporation, Shi
Jiazhuang, China) at 37°C in a humidified incubator containing 5%
CO2.
Chemically induced hypoxia
Hypoxia was achieved by exposing cells cultured in
normoxic conditions to CoCl2 (Sigma-Aldrich, St. Louis,
MO, USA). Various CoCl2 concentrations are often used to
simulate degrees of hypoxia. In the present study, cells were
cultured in DMEM with various concentration of CoCl2, or
with the same concentration of CoCl2 for various
durations. The different concentrations of CoCl2 were
obtained by diluting the stock solution (250 µmol/1) in
culture medium.
MTT assay for the determination of LOVO
cell proliferation
The proliferation of LOVO cells was determined using
an MTT assay (BD Biosciences, Franklin Lakes, NJ, USA). The cells
were plated in a 96-well plate at a density of 1×104
cells/well, in DMEM supplemented with 10% FBS, and were cultured
for 12 h at 37°C with 5% CO2. Subsequently, the cells
were treated with various concentrations of CoCl2 (100,
150, 200 or 250 µmol/l) for 7 days. Six duplicate wells were
set up for each sample. Untreated cells were used as control cells.
At the end of the treatment, 20 µl MTT (5 mg/ml) was added
to the cells, which were incubated for a further 4 h. Dimethyl
sulfoxide (DMSO; 200 µl) was added to each well following
removal of the supernatant, and the plate was agitated for 10 min
until the crystals had dissolved. Absorbance was measured at 570 nm
using an enzyme-labeling instrument (ELx-800; BioTek, Winooski, VT,
USA). The negative control well contained no cells and was
considered the zero point of absorbance. Each assay was performed
in triplicate. Cell growth graphs were generated with time as the
abscissa and absorbance value (mean ± standard deviation) as the
ordinate.
MTT assay for determination of the TIR
following treatment with 5-FU
LOVO cells were cultured in various concentrations
of CoCl2 to simulate an hypoxic environment as
previously described. The TIR following treatment with 5-FU
(Sigma-Aldrich) was assessed using an MTT assay. LOVO cells were
plated in a 96-well plate at a density of 6×104
cells/well, in DMEM supplemented with 10% FBS, and were cultured
for 12 h at 37°C with 5% CO2. The experimental cells
were then treated with various concentrations of CoCl2
(0, 100, 150, 200 or 250 µmol/l) for 20 h. Five duplicate
wells were set up for each sample. The cells not treated with 5-FU
were used as the negative control group. The wells that did not
contain cells were measured as the blank group. Subsequently, 5-FU
was added to the experimental group and blank group cells; each
well contained a final concentration of 50 mg/l 5-FU. Following a
36 h incubation, 20 µl MTT (5 mg/ml) was added, and the
cells were incubated for a further 4 h. DMSO (200 µl) was
added to each well following removal of the supernatant, and the
plate was agitated for 10 min until the crystals had dissolved.
Absorbance was measured at 570 nm using an enzyme-labeling
instrument (EX-800). Each assay was performed in triplicate. The
average value was determined for each group: Experimental group
average value (EAV), control group average value (CAV) and blank
group average value (BAV), these values were used to determine the
TIR, as follows: TIR = [1−(EAV−BAV)/(CAV−BAV)] × 100%.
RNA isolation and RT-PCR analysis
LOVO cells were seeded in 6 cm culture capsules and
were treated with a concentration gradient of CoCl2 (0,
50, 100, 150 and 200 µmol/l) for 24 h. RNA extraction was
performed using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's protocol.
A total of 5 µg RNA was reverse transcribed using the One
Step RT-PCR kit (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. The mRNA expression
levels of HIF-1α, MDR1 and MRP were determined using a PCR kit
(Premix Taq; cat. no. RR901A; Takara Biotechnology Co. Inc.,
Dailan, China) according to the manufacturer's instructions. The
reaction system contained cDNA (2.5 µl), Premix Taq (25
µl), primers (2 µl) and sterile purified water
(≤50µ l). β-actin was used as an internal control. PCR
primers for each gene were designed based on the mRNA sequence
provided by the University of California Santa Cruz database
(https://genome.ucsc.edu/) using Oligo6 software
(www.oligo.net). All of the primers were provided
by Beijing Tsingke Bio Tech, Co., Ltd. (Beijing, China). The primer
sequences used were as follows: HIF-1α, forward
5′-GCCGCTGGAGACACAATCAT-3′, reverse 5′-GAAGTGGCTTTGGCGTTTCA-3′;
MDR1, forward 5′-GGCAAAGAAATAAAGCGACTGA-3′, reverse
5′-GGTGGACAGGCGGTGAG-3′; MRP, forward 5′-AGCCAGAAAATCCTCCACGGT-3′,
reverse 5′-CATCGCCATCACAGCATTGAC-3′; and β-actin, forward
5′-CGGGACCTGACTGACTACCTC-3′ and reverse 5′-CTAGAAGCATTTGCGGTGGA-3′.
The RT-PCR cycling conditions were as follows: Denaturation for 3
min at 94°C, followed by 25 cycles of denaturation for 30 sec at
94°C, annealing for 30 sec at 55°C, 58°C, 59°C and 59°C for HIF-1α,
MDR1, MRP and β-actin, respec tively, and extension for 10 min at
72°C (Eppendorf 5331 MasterCycler Gradient Thermal Cycler;
Eppendorf, Hamburg, Germany). The products were separated by 3%
agarose gel electrophoresis and were visualized with ethidium
bromide. The mRNA expression levels were semi-quantified with the
aid of computer software (Quantity One 4.4.0; Bio-Rad Laboratories,
Inc., Hercules, CA, USA), and the quantity of each transcript was
normalized against β-actin.
Western blot analysis
LOVO cells were treated with CoCl2 (0,
50, 100, 150 or 200 µmol/l) for 24 h. Protein extraction was
conducted as described previously (19). Briefly, the cells were washed with
cold phosphate-buffered saline (PBS) (4°C), were scraped from the
surface of the plate into PBS, and were collected by centrifugation
at 3,000 × g for 5 min at 4°C. Soluble proteins were extracted with
cell lysis buffer containing 1% NP40, 137 mM NaCl, 20 mM Tris base
(pH 7.4), 1 mM dithiothreitol, 10% glycerol, 10 mg/ml Aprotinin, 2
mM sodium vanadate and 100 µM phenylmethylsulfonyl fluoride.
Insoluble material was removed by centrifugation at 14,000 × g for
15 min at 4°C. Protein concentrations were determined using the
Pierce bicinchoninic acid protein assay kit (Thermo Fisher
Scientific, Inc.). The total protein extracts (2.5
µg/µl) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and were transferred
onto nitrocellulose membranes (Bio-Rad Laboratories, Inc.). The
membranes were blocked, and were then incubated with primary
polyclonal antibodies: Rabbit anti-human HIF-1α (1:500 dilution;
sc-10790), P-gp (1:200 dilution; 8313), MRP (1:200 dilution;
sc-13960), Bcl-2 (1:4,000 dilution; sc-783), Bax (1:4,000 dilution;
sc-7480), Bad (1:2,000 dilution; sc-783) and β-actin (1:5,000
dilution; sc-130656) (Santa Cruz Biotechnology, Inc. Dallas, TX,
USA) at 4°C overnight. After being washed in Tris-buffered saline
containing 0.05% Tween-20, the membranes were incubated with goat
anti-rabbit secondary antibody (1:400 dlution; sc-2040; Santa Cruz
Biotechnology, Inc.) for 2 h and were then visualized by
chemiluminescence using a Western Luminescent Detection kit
(Vigorous Biotechnology, Beijing, China). For quantification,
signals were densitometrically normalized to β-actin using Quantity
One image analysis software (version 4.40; Bio-Rad Laboratories,
Inc.).
FCM evaluation
Cells were prepared and treated as previously
described. ADR (Sigma-Aldrich) produces characteristic fluorescence
at an excitation wavelength of 488 nm and an emission wavelength of
575 nm. The cells were divided into two groups: The experimental
group and the negative control group. The experimental group
comprised two subgroups: The hypoxia group, cultured in 200
µmol/l CoCl2 and treated with 5 mg/l ADR; and the
normoxia group cultured in DMEM and treated with 5 mg/l ADR. The
negative control group also comprised two subgroups: The hypoxia
group, cultured in 200 µmol/l CoCl2 only; and the
normoxia group cultured in DMEM only. ADR was added to the
experimental groups following an 18 h culture at a final
concentration of 5 mg/l, and the cells were treated with ADR for
1.5 h. Subsequently, the cells were washed twice with ice-cold PBS
and were collected. FCM (FACSCalibur; BD Biosciences) was used to
analyze ADR accumulation. In addition, to measure ADR retention,
following collection the cells were resuspended in DMEM medium
containing no ADR, and were incubated for 1.5 h. Subsequently, the
cells were washed twice with ice-cold PBS, and FCM was used to
analyze ADR retention. Data were collected and analyzed using
CellQuest software (version 5.1; BD Biosciences). Overlapping
images between retention and accumulation were automatically
generated by CellQuest software.
Statistical analysis
Each experiment was performed at least three times.
All results are presented as the mean ± standard deviation.
Statistical analysis was performed using one-way analysis of
variance. All analyses were performed using SPSS software, version
19.0 (SPSS IBM, Armonk, NY, USA). P<0.05 was consid ered to
indicate a statistically significant difference.
Results
Growth curve of LOVO cells cultured with
various concentrations of CoCl2
The growth curve of LOVO cells cultured under
normoxic and hypoxic conditions exhibited an 'S' shape. Compared
with the normoxia group, the cells treated with various
concentrations of CoCl2 (100, 150 and 200 µmol/l)
exhibited slow growth during days 1–3.5, and underwent a period of
exponential growth and rapid proliferation after 4–7 days (Fig. 1). As CoCl2
concentrations increased the speed of proliferation decreased.
Proliferation was inhibited the most following treatment with 250
µmol/l CoCl2, in the growth curve the cells
exhibited significant growth inhibition. After 7 days of treatment,
a dose-dependent inhibition of cell growth was observed
(P<0.05).
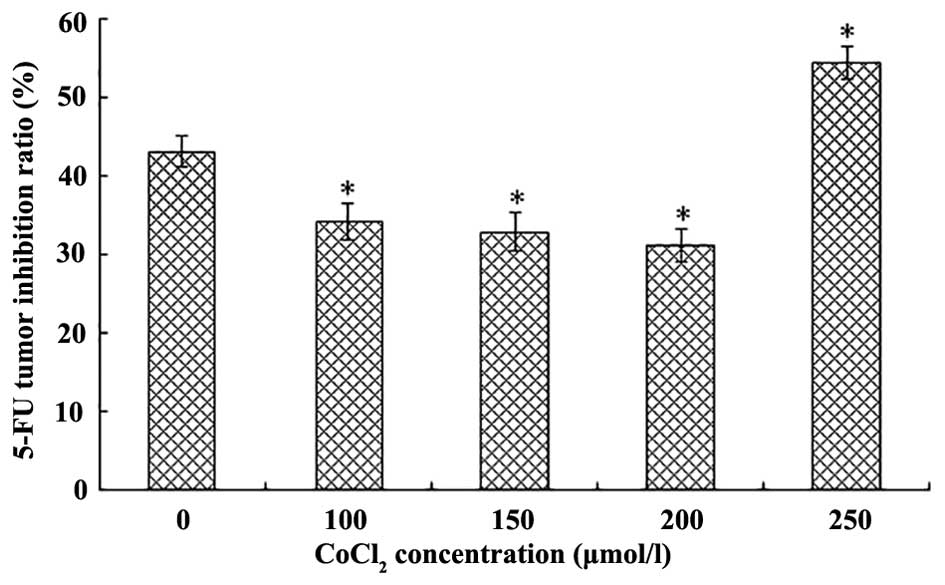
TIR following 5-FU treatment is altered
in response to various concentrations of CoCl2
Compared with the normoxia group, following
treatment with various concentrations of CoCl2 (100, 150
and 200 µmol/l), LOVO cells acquired resistance against 5-FU
as CoCl2 increased. Hypoxia was able to decrease the
sensitivity of LOVO cells to 5-FU (P<0.05), in a dose-dependent
manner. However, cells treated with 250 µmol/l
CoCl2 were more sensitive to 5-FU (P<0.05), as
compared with cells in the normoxia group (Fig. 2).
Hypoxia induces overexpression of HIF-1α,
MDR1/P-gp and MRP
Cells were treated with various concentrations of
CoCl2 (0, 50, 100, 150 and 200 µmol/l) for 24 h.
Subsequently, mRNA and protein expression levels were detected by
RT-PCR and western blot analysis (Fig.
3), with β-actin as an internal control. The expression levels
of HIF-1α, MDR1/P-gp and MRP gradually increased as
CoCl2 concentration increased. Therefore, HIF-1α,
MDR1/P-gp and MRP were increased in response to CoCl2 in
a dose-dependent manner (P<0.05).
Hypoxia downregulates Bax and Bad
expression
Cells were treated with various concentrations of
CoCl2 (0, 50, 100, 150 and 200 µmol/l) for 24 h.
Western blot analysis was then performed to detect the expression
levels of apoptosis-associated proteins (Bcl-2, Bax and Bad)
(Fig. 4). Following treatment with
CoCl2, Bax and Bad expression levels were gradually
decreased in a dose-dependent manner (P<0.05), whereas no
differences in Bcl-2 protein expression were detected (P>0.05;
Fig. 4A and B). In addition, the
ratio of Bcl-2/Bax was increased in a dose-dependent manner
(P<0.05; Fig. 4C).
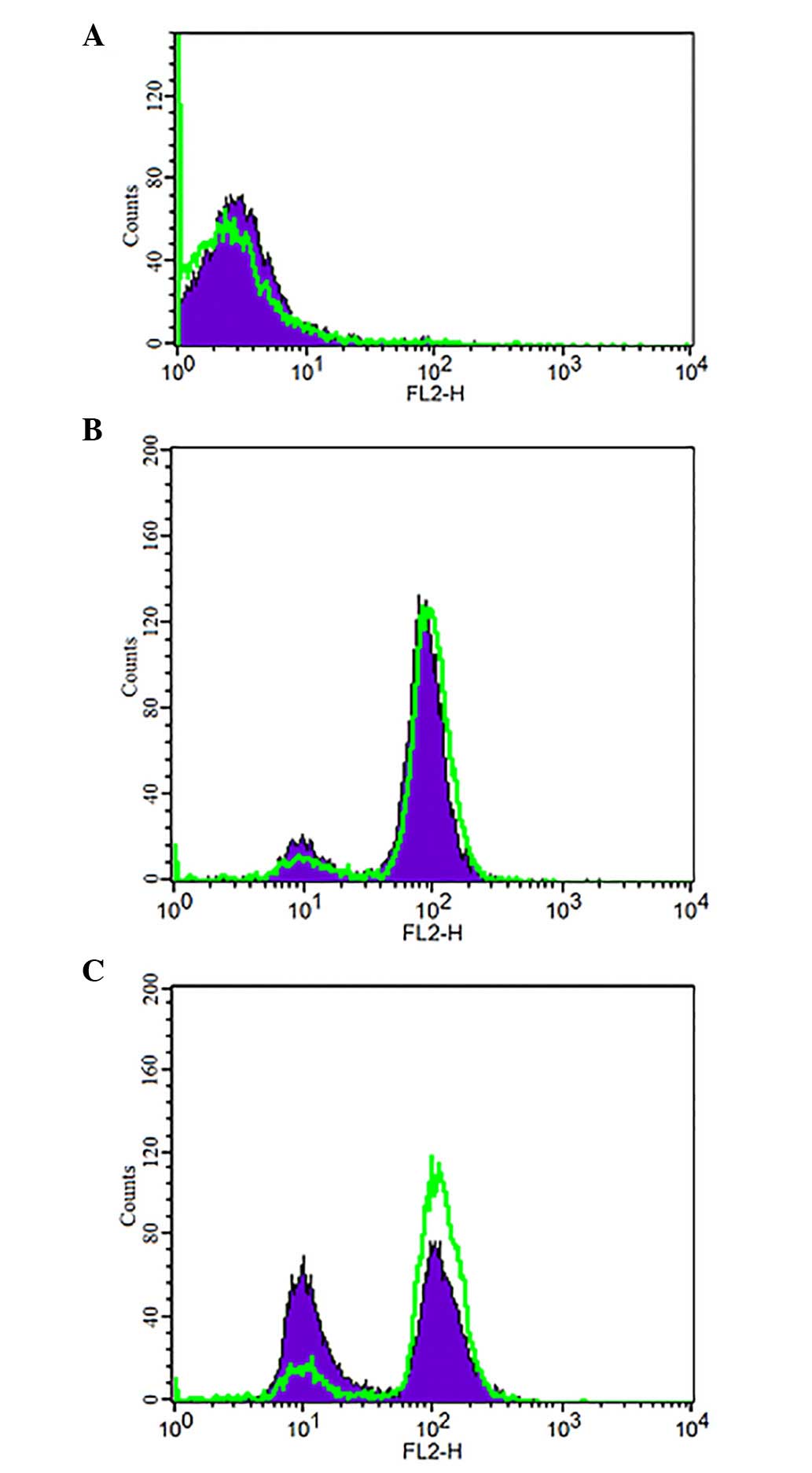
Hypoxia decreases intracellular retention
of ADR
ADR produces characteristic fluorescence at an
excitation wavelength of 488 nm and an emission wavelength of 575
nm. Under hypoxia (200 µmol/l CoCl2) FCM was
performed to detect the changes in intracellular ADR concentration
(Fig. 5). The accumulation and
retention of ADR were determined and were used to evaluate changes
to intracellular ADR. ADR intracellular accumulation exhibited no
significant difference between the hypoxia (CoCl2, 200
µmol/l; ADR, 5 mg/l) and normoxia (DMEM; ADR, 5 mg/l)
groups. Subsequently, the two groups were cultured for 1.5 h in
fresh DMEM, without ADR. The hypoxia group exhibited a marked
decrease in intracellular retention of ADR.
Discussion
The microenvironment of rapidly growing tumors is
associated with increased energy demand and diminished vascular
supply, resulting in central areas of prominent hypoxia. The
hypoxic phenomenon is considered to be a tumor microenvironment
factor that favors tumor cell survival and resistance to
chemotherapy and radiotherapy (20). Experimental and clinical studies
have demonstrated that intra-tumor hypoxia may be a key factor in
the tumor microenvironment promoting invasive growth and metastasis
(5,21). However the molecular mechanisms
underlying hypoxia-induced colorectal cancer chemoresistance remain
unclear.
CoCl2 has been used as a hypoxia mimic in
in vivo and in vitro studies. CoCl2, as a
substrate of the ferrochelatase enzyme, can combine with
hemoglobin, instead of Fe2+, and cause cells to enter a
hypoxic state (22). Hypoxia is a
common feature of several types of malignant tumor. HIF-1α is a
major transcription factor and key regulator of adaptive responses
to hypoxia, including activation of signaling pathways and
downstream genes, which promote cell survival and alter the
biological characteristics of tumors. HIF-1α mediates the response
to hypoxia by regulating the expression of genes capable of
regulating glycolysis, angiogenesis and erythropoiesis, including
erythropoietin, vascular endothelial growth factor and pyruvate
kinase (23–25). A previous study demonstrated that
HIF-1α is an important regulator of Survivin expression, which may
have a potential role as a therapeutic target in cancer (26).
In the present study, LOVO cells were cultured in a
hypoxic environment simulated using CoCl2. An MTT assay
demonstrated that as CoCl2 concentration increased,
proliferation decreased. These results suggested that in order to
adapt to the hypoxic environment, cell growth had slowed down. In
addition, at a certain extent of hypoxia (100–200 µmol/l
CoCl2), the average TIR following treatment with 5-FU
was decreased. However, severe hypoxia (250 µmol/l)
significantly inhibited growth and increased TIR following
treatment with 5-FU. These results indicated that appropriate
hypoxia may protect tumor cells; however, severe hypoxia is able to
inhibit proliferation, and may increase 5-FU-induced tumor cell
apoptosis (27,28).
Under hypoxic conditions, the key molecular player
HIF-1α was upregulated. The present study indicated that mRNA and
protein expression levels were markedly increased following
treatment with CoCl2 in a dose-dependent manner
(P<0.05), and that the level of hypoxia coincided with the
expression of HIF-1α.
Chemotherapy failure remains a major problem in many
patients with cancer. Tumor chemoresistance is usually associated
with the overexpression of drug-resistance genes. MRP and the MDR1
gene, which encodes the major transmembrane efflux transporter
P-gp, are membrane transporter proteins; their function is similar
to efflux pumps at cell membranes and they are considered to exert
a protective function against the entry of xenobiotics. The
overexpression of these genes is the classical mechanism of
multidrug resistance in several types of tumor, including lung
carcinoma, breast cancer and pancreatic carcinoma (29–31).
However, the relationship between LOVO cell hypoxia and
chemoresistance has not yet been clearly investigated. The results
of the present study demonstrated that under hypoxia HIF-1α was
overexpressed in a dose-dependent manner. In addition, under
moderate hypoxia, MDR1/P-gp and MRP were overexpressed in a
dose-dependent manner in response to a CoCl2
concentration gradient (P<0.05). P-gp and MRP are
energy-dependent efflux pumps that are responsible for decreases in
drug accumulation, which also promote multidrug resistance. In the
present study, measurement of ADR accumulation and retention was
used to confirm the chemoresistance of cells treated with
CoCl2. The FCM results demonstrated that
CoCl2-simulated hypoxia affects intracellular ADR
concentration in the presence of tumor chemoresistance. This
occurred when the intracellular ADR accumulation reached effective
concentrations; however, the duration of intracellular ADR
retention was the decisive factor in the success of chemotherapy.
In addition, the overexpression of MDR1/P-gp and MRP resulted in an
increased ability to pump anticancer drugs out of cells; this may
be considered an important method by which cancer cells survive
anticancer drug treatment under hypoxic conditions. These results
suggested that HIF-1α, MDR1/P-gp and MRP may be interactively
involved in the occurrence of tumor multidrug resistance.
Whether hypoxia promotes tumor cell apoptosis or
decreases apoptosis has yet to be elucidated. Various studies have
reached different conclusions. A previous study indicated that
overexpression of HIF-1α could promote downregulation of BH3
interacting-domain death agonist and/or Bax, thus contributing to
etoposide resistance (32).
Another study demonstrated that, under hypoxic conditions, HIF-1α
is an important regulator of Survivin, and has a great potential
capacity for cancer therapeutics (26). Conversely, HIF-1 has been shown to
induce apoptosis, via the stabilization of p53 (33) or the transactivation of
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3, which
encodes a pro-apoptotic Bcl-2 family member (34). Many contradictory reports involving
the role of HIF-1α in the regulation of apoptosis have emerged in
recent years, and the results demonstrate that the effect of tumor
cell anti-apoptosis is related to the duration and degree of
hypoxia (35,36). In the present study, LOVO cells
cultured in an hypoxic microenvironment exhibited reduced
apoptosis. This phenomenon may be due to the expression of Bcl-2
family proteins. As CoCl2 concentration increased, the
expression of proapoptotic proteins, including Bax and Bad,
decreased in the tumor cells; however, the expression of the
anti-apoptotic protein Bcl-2 was not significantly altered, thus
resulting in a decreased Bax/Bcl-2 ratio. The ratio of Bcl-2/Bax is
usually regarded as a criterion for programmed cell death (37). These results indicated that under
hypoxic conditions the downregulation of Bax and Bad provided a
significant survival advantage, since Bax and Bad are suppressors
of Bcl-2 and Bcl-extra large (38).
In conclusion, the present study reported that
hypoxia altered the phenotype of LOVO colorectal cancer cells. The
hypoxic microenvironment was simulated in the present study using
the CoCl2 method, and the expression of HIF-1α was
upregulated in a dose-dependent manner. In addition, hypoxia
exhibited an inhibitory effect on LOVO cell proliferation, and
moderate hypoxia decreased the sensitivity of LOVO cells to 5-FU.
In addition, two aspects that may have resulted in hypoxia-induced
chemotherapy resistance were identified. Firstly, MDR1/P-gp and MRP
were upregulated, which may decrease the retention of
chemotherapeutic drugs. Secondly, Bax and Bad expression was
downregulated, whereas Bcl-2 expression exhibited no significant
change, thus decreasing the Bax/Bcl-2 ratio. Therefore, LOVO cells
may escape apoptosis mediated by chemotherapeutic drugs. The
hypoxic microenvironment may promote chemoresistance and
anti-apoptosis, resulting in greater malignant behavior of tumor
cells.
Acknowledgments
The present study was supported by the Xingtai
People's Hospital Affiliated Hebei Medical University, Hebei,
China. The authors would like to thank the Molecular Biology
Laboratory, Peking University Health Science Center, for equipment
support.
References
|
1
|
Mylonas CC and Lazaris AC: Colorectal
cancer and basement membranes: Clinicopathological correlations.
Gastroenterol Res Pract. 2014:5801592014. View Article : Google Scholar
|
|
2
|
Abdalla EK, Adam R, Bilchik AJ, Jaeck D,
Vauthey JN and Mahvi D: Improving resectability of hepatic
colorectal metastases: Expert consensus statement. Ann Surg Oncol.
13:1271–1280. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ameri K, Luong R, Zhang H, Powell AA,
Montgomery KD, Espinosa I, Bouley DM, Harris AL and Jeffrey SS:
Circulating tumour cells demonstrate an altered response to hypoxia
and an aggressive phenotype. Br J Cancer. 102:561–569. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hockel M, Schlenger K, Aral B, Mitze M,
Schaffer U and Vaupel P: Association between tumor hypoxia and
malignant progression in advanced cancer of the uterine cervix.
Cancer Res. 56:4509–4515. 1996.PubMed/NCBI
|
|
5
|
Chu CY, Jin YT, Zhang W, Yu J, Yang HP,
Wang HY, Zhang ZJ, Liu XP and Zou Q: CA IX is upregulated in
CoCl2-induced hypoxia and associated with cell invasive
potential and a poor prognosis of breast cancer. Int J Oncol.
48:271–280. 2016.
|
|
6
|
Dai ZJ, Gao J, Ma XB, Yan K, Liu XX, Kang
HF, Ji ZZ, Guan HT and Wang XJ: Up-regulation of hypoxia inducible
factor-1α by cobalt chloride correlates with proliferation and
apoptosis in PC-2 cells. J Exp Clin Cancer Res. 31:282012.
View Article : Google Scholar
|
|
7
|
Semenza GL: Hypoxia-inducible factor 1
(HIF-1) pathway. Sci STKE. 2007:cm82007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yuan Y, Hilliard G, Ferguson T and
Millhorn DE: Cobalt inhibits the interaction between
hypoxia-inducible factor alpha and von Hippel-Lindau protein by
direct binding to hypoxia-inducible factor-alpha. J Biol Chem.
278:15911–15916. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Goldberg MA, Gaut CC and Bunn HF:
Erythropoietin mRNA levels are governed by both the rate of gene
transcription and posttranscriptional events. Blood. 77:271–277.
1991.PubMed/NCBI
|
|
10
|
Vengellur A and LaPres JJ: The role of
hypoxia inducible factor 1alpha in cobalt chloride induced cell
death in mouse embryonic fibroblasts. Toxicol Sci. 82:638–646.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang GL and Semenza GL: Desferrioxamine
induces erythropoietin gene expression and hypoxia-inducible factor
1 DNA-binding activity: Implications for models of hypoxia signal
transduction. Blood. 82:3610–3615. 1993.PubMed/NCBI
|
|
12
|
Ramaekers CH, van den Beucken T, Meng A,
Kassam S, Thoms J, Bristow RG and Wouters BG: Hypoxia disrupts the
Fanconi anemia pathway and sensitizes cells to chemotherapy through
regulation of UBE2T. Radiother Oncol. 101:190–197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pouysségur J, Dayan F and Mazure NM:
Hypoxia signalling in cancer and approaches to enforce tumour
regression. Nature. 441:437–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Juliano RL and Ling V: A surface
glycoprotein modulating drug permeability in Chinese hamster ovary
cell mutants. Biochim Biophys Acta. 455:152–162. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cole SP, Bhardwaj G, Gerlach JH, Mackie
JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU, Duncan AM and
Deeley RG: Overexpression of a transporter gene in a
multidrug-resistant human lung cancer cell line. Science.
258:1650–1654. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sasi N, Hwang M, Jaboin J, Csiki I and Lu
B: Regulated cell death pathways: New twists in modulation of BCL2
family function. Mol Cancer Ther. 8:1421–1429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Adams JM and Cory S: The Bcl-2 apoptotic
switch in cancer development and therapy. Oncogene. 26:1324–1337.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bhutia SK, Mallick SK, Maiti S and Maiti
TK: Antitumor and proapoptotic effect of Abrus agglutinin derived
peptide in Dalton's lymphoma tumor model. Chem Biol Interact.
174:11–18. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Toffoli S and Michiels C: Intermittent
hypoxia is a key regulator of cancer cell and endothelial cell
interplay in tumours. FEBS J. 275:2991–3002. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Semenza GL: Targeting HIF-1 for cancer
therapy. Nat Rev Cancer. 3:721–732. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goldberg MA and Schneider TJ: Similarities
between the oxygen-sensing mechanisms regulating the expression of
vascular endothelial growth factor and erythropoietin. J Biol Chem.
269:4355–4359. 1994.PubMed/NCBI
|
|
23
|
Forsythe JA, Jiang BH, Iyer NV, Agani F,
Leung SW, Koos RD and Semenza GL: Activation of vascular
endothelial growth factor gene transcription by hypoxia-inducible
factor 1. Mol Cell Biol. 16:4604–4613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gleadle JM and Ratcliffe PJ: Induction of
hypoxia-inducible factor-1, erythropoietin, vascular endothelial
growth factor, and glucose transporter-1 by hypoxia: Evidence
against a regulatory role for Src kinase. Blood. 89:503–509.
1997.PubMed/NCBI
|
|
25
|
Mabjeesh NJ and Amir S: Hypoxia-inducible
factor (HIF) in human tumorigenesis. Histol Histopathol.
22:559–572. 2007.PubMed/NCBI
|
|
26
|
Wu XY, Fu ZX and Wang XH: Effect of
hypoxia-inducible factor 1-α on Survivin in colorectal cancer. Mol
Med Rep. 3:409–415. 2010.
|
|
27
|
Piret JP, Lecocq C, Toffoli S, Ninane N,
Raes M and Michiels C: Hypoxia and CoCl2 protect HepG2
cells against serum deprivation- and t-BHP-induced apoptosis: A
possible anti-apoptotic role for HIF-1. Exp Cell Res. 295:340–349.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Greijer AE and van der Wall E: The role of
hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J
Clin Pathol. 57:1009–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen ZJ, Le HB, Zhang YK, Qian LY, Sekhar
KR and Li WD: Lung resistance protein and multidrug resistance
protein in non-small cell lung cancer and their clinical
significance. J Int Med Res. 39:1693–1700. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Linn SC, Pinedo HM, van Ark-Otte J, van
der Valk P, Hoekman K, Honkoop AH, Vermorken JB and Giaccone G:
Expression of drug resistance proteins in breast cancer, in
relation to chemotherapy. Int J Cancer. 71:787–795. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
O'Driscoll L, Walsh N, Larkin A, Ballot J,
Ooi WS, Gullo G, O'Connor R, Clynes M, Crown J and Kennedy S:
MDR1/P-glycoprotein and MRP-1 drug efflux pumps in pancreatic
carcinoma. Anticancer Res. 27:2115–2120. 2007.PubMed/NCBI
|
|
32
|
Erler JT, Cawthorne CJ, Williams KJ,
Koritzinsky M, Wouters BG, Wilson C, Miller C, Demonacos C,
Stratford IJ and Dive C: Hypoxia-mediated down regulation of Bid
and Bax in tumors occurs via hypoxia-inducible factor 1-dependent
and -independent mechanisms and contributes to drug resistance. Mol
Cell Biol. 24:2875–2889. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
An WG, Kanekal M, Simon MC, Maltepe E,
Blagosklonny MV and Neckers LM: Stabilization of wild-type p53 by
hypoxia-inducible factor 1alpha. Nature. 392:405–408. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bruick RK: Expression of the gene encoding
the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad
Sci USA. 97:9082–9087. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yin CP, Guan SH, Zhang B, Wang XX and Yue
SW: Upregulation of HIF-1α protects neuroblastoma cells from
hypoxia-induced apoptosis in a RhoA-dependent manner. Mol Med Rep.
12:7123–7131. 2015.PubMed/NCBI
|
|
36
|
Wang X, Li J, Wu D, Bu X and Qiao Y:
Hypoxia promotes apoptosis of neuronal cells through
hypoxia-inducible factor-1α-microRNA-204-B-cell lymphoma-2 pathway.
Exp Biol Med (Maywood). 241:177–183. 2016. View Article : Google Scholar
|
|
37
|
Pettersson F, Dalgleish AG, Bissonnette RP
and Colston KW: Retinoids cause apoptosis in pancreatic cancer
cells via activation of RAR-gamma and altered expression of
Bcl-2/Bax. Br J Cancer. 87:555–561. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Letai A, Bassik MC, Walensky LD,
Sorcinelli MD, Weiler S and Korsmeyer SJ: Distinct BH3 domains
either sensitize or activate mitochondrial apoptosis, serving as
prototype cancer therapeutics. Cancer Cell. 2:183–192. 2002.
View Article : Google Scholar : PubMed/NCBI
|