Introduction
Hyperuricemia is one of the most common and
extensive metabolic diseases worldwide, characterized by high serum
uric acid levels, resulting in the accumulation of urate crystals
in the joints and kidneys (1,2).
Hyperuricemia is also well known as a severe risk factor for gouty
arthritis, uric acid nephrolithiasis and cardiovascular and renal
diseases, particularly hypertension (3–5). In
humans, the level of serum uric acid is determined primarily by the
ratio of urate production as an end product of the purine
metabolism in liver (for which the liver enzyme xanthine oxidase is
necessary) vs. elimination from the biliary and urinary tracts
(6). In the urate elimination
process, the organic anion transporter (OAT)-1 is hypothesized to
be an essential molecule that mediates the release of urate from
the blood to the basolateral membrane of renal proximal tubule
(7).
The OAT family consists of 10 transmembrane proteins
that are part of the solute carrier subfamily (8). Although previous studies have focused
on the kidney, the OATs are localized in the barrier epithelia of
the entire body, endothelium and other cells (9,10).
Their function is to regulate transcellular movement of numerous
anionic organic molecules across the epithelium and between body
fluid compartments. OAT1 is a secondary active transporter, whereby
OAT-mediated influx involves exchange/counter transport of another
solute involving a different transporter, such as the
Na+-K+-ATPase (8,9).
OAT1 is a p-aminohippuric acid transporter that is
responsible for the transport of several anionic drugs, toxins,
metabolites and signaling molecules (11–15).
Along with OAT3, OAT1 mediates the uptake of a wide range of
relatively small and hydrophilic organic anions from the plasma
into the cytoplasm of the proximal tubular cells of the kidneys
(15,16). OAT-mediated transport may also lead
to renal injury by virtue of their location near the proximal
tubule (17). From there, these
substrates are transported into the lumen of the nephrons of the
kidneys for excretion. Thus, it is suggested that OAT1 serves an
important role in the regulation of urate metabolism
homeostasis.
As previously reported, it has been established that
a high purine diet induced a uric acid nephropathy (UAN) rat model
that exhibited an elevated serum uric acid level in addition to
kidney damage (18). Considering
hyperuricemia is a key cause of UAN, and OAT1 regulates the urate
metabolism and is involved in the subsequent hyperuricemia, the
current study aimed to investigate whether OAT1 is involved in high
purine diet-induced hyperuricemia and nephropathy in addition to
the potential mechanisms.
Folic acid, which is an oxidized form of folate, is
highly stable and has been used widely as a nutritional supplement.
Folate deficiency is also associated with several pathologies,
including neural tube defects, atherosclerosis and various types of
cancer (19,20). It has been demonstrated that folic
acid inhibits endothelial cell proliferation through activation of
the cSrc/extracellular signal-related kinase-2/nuclear
factor-κB/p53 pathway, which is mediated by the folic acid receptor
(21). Hou et al
demonstrated that folic acid-induced cSrc activation inhibited Ras
homolog family member A (RhoA) activity via the activation of
p190RhoGAP (22). The results
demonstrated that folic acid could relocate OAT1 back to the cell
membrane by inhibiting RhoA activity.
Materials and methods
Animals
A total of 40 male Sprague-Dawley rats (250.2±1.9 g;
6 weeks-old) were obtained from the Experimental Animal Center of
the Sun Yat-sen University (Guangzhou, China). All rats were housed
in plastic cages at 25°C under a 12-h light-dark cycle and were
provided with rodent chow and water. They were housed for one week
to adapt to their environment prior to the start of the study. The
study was conducted in accordance with the Guidelines for Human
Treatment of Animals set by the Association of Laboratory Animal
Sciences and the Center for Laboratory Animal Sciences at the Sun
Yat-sen University. The current study was approved by the Committee
of Biomedical Ethics of the Sun Yat-sen University
[IACUC-2013-0604].
Reagents and plasmids
Allopurinol tablets were purchased from Guangzhou
Kanghe Pharmaceutical Co., Ltd. (Guangzhou, China), diluted with
distilled water to a final concentration of 5 mg/ml. Adenine
tablets were purchased from Amresco LLC (Solon, OH, USA) and
diluted with 0.15% sodium carboxymethylcellulose (CMC-Na) to a
final concentration of 3%. Hematoxylin (DF001) was purchased from
Guangzhou Dingguo Bio-technology Co., Ltd. (Guangzhou, China).
TRIzol, reverse transcription buffer, SYBR green I polymerase chain
reaction (PCR) buffer, dNTPs, MMLV and Taq DNA polymerase were
purchased from Takara Biotechnology, Co., Ltd. (Dalian, China).
Rabbit polyclonal OAT1 antibody purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). OAT1 constructs were
purchased from OriGene Technologies, Inc. (Beijing, China) in
wild-type form and inserted a myc tail in the C terminal by routine
sub-cloning (23). The RhoA
construct was also purchased from OriGene Technologies, Inc. and
the constitutively active RhoA G14V, and dominant-negative RhoA
T19N site mutations were produced using the QuikChange
Site-Directed Mutagenesis kit (with PfuUltra High-Fidelity DNA
Polymerase; Agilent Technologies, Inc., Santa Clara, CA, USA) and
followed by DpnI (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) digestion of the template DNA. The surface
protein extraction kit was purchased from BioVersion Life Sciences
(Dehradun, India), sulfo-NHS-SS-biotin and Avidin Agarose beads
were purchased from Thermo Fisher Scientific, Inc. MSU crystals
were produced as previously described (24).
Animal model and experimental
procedures
The 40 Sprague-Dawley rats were randomly assigned
into 4 groups. All rats were treated by intra-gastric
administration. Group 1 was allocated as the control and was fed 5
ml CMC-Na (adenine dissolving buffer), while in the remaining 3
groups, rats were fed with adenine (30 mg/kg body weight) for 18
days to establish the UAN model. On the 19th day, 3 rats were
randomly selected, anesthetized using 10% chloral hydrate (0.4
ml/100 g; Sigma-Aldrich, St. Louis, MO, USA), sacrificed using
cervical dislocation and kidney samples were collected.
Hematoxylin-eosin (H&E) staining of the kidney tissue was then
conducted to assess whether the UAN model was successfully
established. The 4 groups of rats were treated as follows: Group 1,
blank control, rats were fed with the same amount of saline buffer;
group 2, disease model control, rats were administered with the
same amount of saline buffer as in group 1; group 3, allopurinol
group, rats were treated with allopurinol (5 mg/kg body
weight/day); group 4, folic acid group, rats were treated with
folic acid (2 mg/250 g body weight/day). In all groups, rats were
administered with the drug or saline treatment for 23 days. During
the process, no rats died. Samples were collected 18 days
subsequent to model establishment, and 5 ml peripheral blood
(fasting for 12 h) was collected for detection of blood urea
nitrogen (BUN), serum creatinine (Scr) and serum uric acid (UA). At
the end of the 42-day experimental procedure, rats were fasted for
12 h, and then sacrificed under 20% urethane anesthesia
(Sigma-Aldrich). Peripheral blood was collected to detect BUN, Scr
and UA, and the left kidney was resected for further
investigation.
H&E staining
Kidney sections were stained with hematoxylin for 2
min and then eosin for 30 sec. Subsequent to washing, the slides
were dehydrated and sealed with Entellan (Merck Millipore,
Darmstadt, Germany) prior to examination under a light microscope
(model BX43/53; Olympus Corporation, Tokyo, Japan). Quantification
of cell densities was conducted using AlphaEaseFC software version
4.0 (Alpha Innotech Corporation, San Leandro, CA, USA). Results
were analyzed using Image Pro Plus software, version 6.0 (Media
Cybernetics, Inc., Rockville, MD, USA), and in each section 10
fields were selected (magnification, ×200).
Tissue surface protein extraction, cell
surface biotinylation and western blotting
Kidney tissues from rats in the different groups
were frozen and stored at −20°C immediately subsequent to
dissection. The tissues were mechanically dissociated and
homogenized (model AH96; Omni International, Kennesaw, GA, USA)
following the manufacturer's instructions to obtain the surface
protein. For HEK cell (American Type Culture Collection, Manassas,
VA, USA) surface biotinylation, OAT1-stable HEK cells were treated
with monosodium urate (MSU) for 24 h and washed twice with cold
phosphate-buffered saline (PBS). OAT1-stable HEK cells without MSU
treatment were used as a control. The cells were then incubated
with PBS supplemented with 0.5 mg/ml sulfo-NHS-SS-biotin for 1 h
with gentle shaking, and excess biotin was quenched with 50 mM
Tris-PBS buffer (Sigma-Aldrich) for 20 min. Cells were lysed in
radioimmunoprecipitation assay (RIPA) buffer and then were
subjected to streptavidin-agarose beads at 4°C for an additional 3
h. For the HEK cell internalization assay, surface biotinylated
cells were incubated at 37°C for 30 min, subsequent to quenching,
to allow internalization, then were washed with cold cleavage
buffer [50 mM glutathione, 90 mM NaCl, 1.25 mM CaCl2
dihydrate, 1.25 mM MgSO4 (all from Sigma-Aldrich), and
0.2% endotoxin-free bovine serum albumin (BSA; Cell Signaling
Technology, Inc., Danvers, MA, USA), pH 8.6] for 20 min with gentle
shaking. Cells were lysed in RIPA buffer, then were subjected to
streptavidin-agarose beads at 4°C for another 3 h. Protein samples
were boiled in Laemmli buffer (Amresco LLC), separated by 12%
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(ProSpec, Ness-Ziona, Israel), then transferred to polyvinylidene
difluoride membranes (Hybond-C-extra; GE Healthcare Life Sciences,
Chalfont, UK). Membranes were blocked using 5% BSA in Tris-buffered
saline with Tween-20 (TBST; Sigma-Aldrich) and then were incubated
overnight at 4°C with mouse monoclonal anti-OAT1 (1:100; cat. no.
ab118346; Abcam, Cambridge, MA, USA) or rabbit monoclonal
anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:8,000;
cat. no. ab181603; Abcam) antibodies. The membranes were then
washed in TBST and incubated with peroxidase-conjugated goat
anti-rabbit or anti-mouse antibodies (1:4,000; Jackson
ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 1 h.
Protein bands were developed using Enhanced Chemiluminescence plus
reagent (Western Lightning; PerkinElmer, Inc., Waltham, MA,
USA).
Detection of OAT1 mRNA expression by
reverse transcription-quantitative PCR (RT-qPCR)
Total RNA (2 µg) was extracted with TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and converted to cDNA
(1 µg) using the reverse transcription kit (Takara
Biotechnology, Co., Ltd.) according to the manufacturer's
instructions. RT-qPCR was conducted according to the following
procedure: 94°C for 3 min, followed by 40 cycles of 94°C for 30
sec, 55°C for 45 sec and 72°C for 1 min. OAT1, F
5′-CAGCCAAGGAGGCTGCTGTC-3′ and R 5′-AGTCAAACCTTTTAATGA TG-3′;
GAPDH, F 5′-TGGTCTACATGTTCCAGTATGACT-3′ and R
5′-CCATTTGATGTTAGCGGGATCTC-3′. Data were presented as the
normalized OAT1 mRNA level/GAPDH mRNA level. Results are expressed
as the mean ± standard error of three independent experiments.
Surface and internalization OAT1
staining
OAT1 stable HEK cells were treated with or without
MSU for 24 h and live cells were used for staining the OAT1
extracellular region at 4°C for 1 h. Cells were then transferred
back to the 37°C incubator for 30 min to allow for internalization.
Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich) and
surface stained using the Alexa Fluor 488 donkey anti-goat
polyclonal antibody (1:200; cat. no. ab150129; Abcam) for 1 h.
Subsequently, the cells were permeabilized with 0.25% Triton X-100
(Sigma-Aldrich) for 5 min and incubated with the mouse monoclonal
anti-Myc antibody (1:1,000; cat. no. ab23; Abcam) at 4°C overnight.
On the second day, cells were stained with the Alexa Fluor 594
donkey anti-goat antibody (1:300; cat. no. ab150132; Abcam) for
internalized OAT1 and Alexa Fluor 405 goat anti-mouse polyclonal
antibody (1:200; cat. no. ab175660; Abcam) for total OAT1.
Representative images were captured using an Olympus FV1000
confocal microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
Quantitative data was expressed as the mean ±
standard error. Comparisons of the means among multiple groups were
performed using a one-way analysis of variance followed by
Dunnett's or Tukey-Kramer's post hoc tests using GraphPad Prism
software, version 4.0 (GraphPad Software, Inc., La Jolla, CA, USA).
Quantification of images were conducted using Image J software
(National Institutes of Health, Bethesda, MD, USA). Asterisks were
used to indicate the following: *P<0.05,
**P<0.01 and ***P<0.001. P<0.05 was
considered to indicate a statistically significant difference.
Results
UAN model rats exhibited a reduction in
surface OAT1 expression
Compared with the controls, serum UA, BUN and
creatinine levels in adenine-treated rats (UAN model rats) were
significantly increased, suggesting that the UAN model had been
successfully established. Treatment with allopurinol led to a
reduction in serum UA, BUN and creatinine levels compared with the
UAN model group, however, serum BUN and creatinine levels of
allopurinol-treated rats remained significantly greater than those
of the control group (Fig. 1A–C).
Using H&E staining, it was additionally identified that UAN
model rats exhibited kidney damage (14) (renal interstitial tissue was
characterized by edema, inflammatory cell infiltration, granuloma
hyperplasia, glomeruli exhibiting partial atrophy and tubular cells
with diffuse swelling), while treatment with allopurinol attenuated
these kidney impairments (Fig.
1D). The above-mentioned data indicate that the model was
successful as previously reported (18).
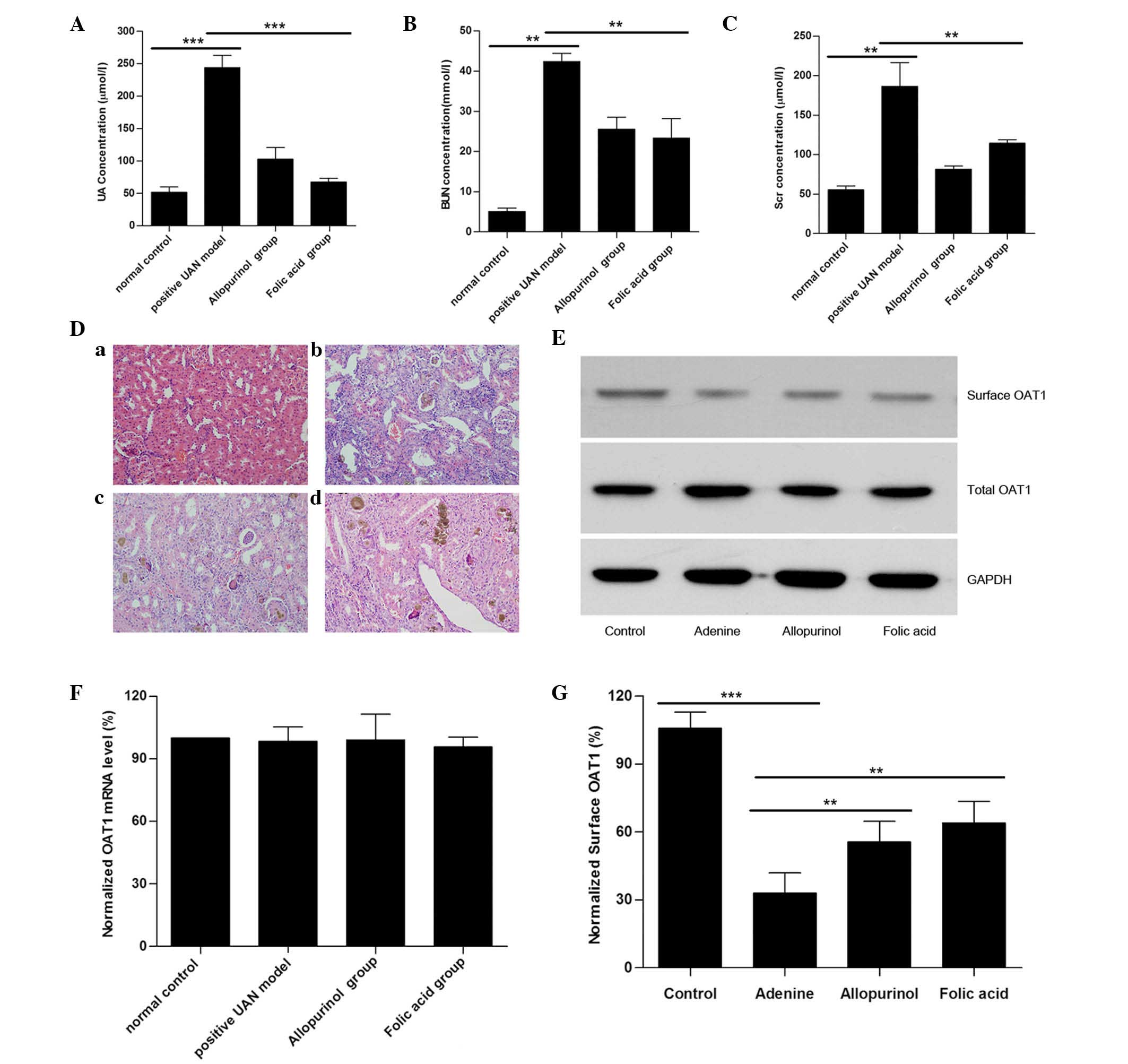 | Figure 1UAN model rats exhibited a reduction
in surface OAT1 levels in kidney tissues. Histograms for the serum
(A) UA, (B) BUN, and (C) Scr levels in the UAN rats are presented.
(D) Light microscopy images of kidney tissue sections with H&E
staining (magnification, ×200): (a) Control, (b) UAN model, (c)
allopurinol and (d) folic acid. (E) Representative western blot of
surface OAT1 and total OAT1 from rat kidney tissues. GAPDH was used
as a loading control. (F) Reverse transcription-quantitative
polymerase chain reaction results of OAT-1 mRNA expression levels
from various groups. (G) Histogram of quantification of OAT1
expression levels, normalized to GAPDH. Data represent the mean ±
standard error (**P<0.01, ***P<0.001).
UAN, uric acid nephropathy; OAT1, organic anion transporter-1; UA,
serum uric acid; BUN, blood urea nitrogen; Scr, serum creatinine;
GAPDH, glyceraldehyde 3-phosphate dehydrogenase. |
To further investigate the role of OAT1 in UAN, the
OAT1 protein expression levels were measured. As presented in
Fig. 1E, no significant different
in the total OAT1 level was observed between the different groups,
and the mRNA levels of OAT1 were not altered (Fig. 1F). As OAT1 is a membrane protein,
the total plasma membrane proteins were extracted, and the surface
OAT1 levels were measured. It was identified that although total
OAT1 expression was not altered, there was a significant reduction
in the level of surface OAT1 in the UAN group (Fig. 1G). The results indicated that high
uric acid generation may reduce the surface OAT1 levels.
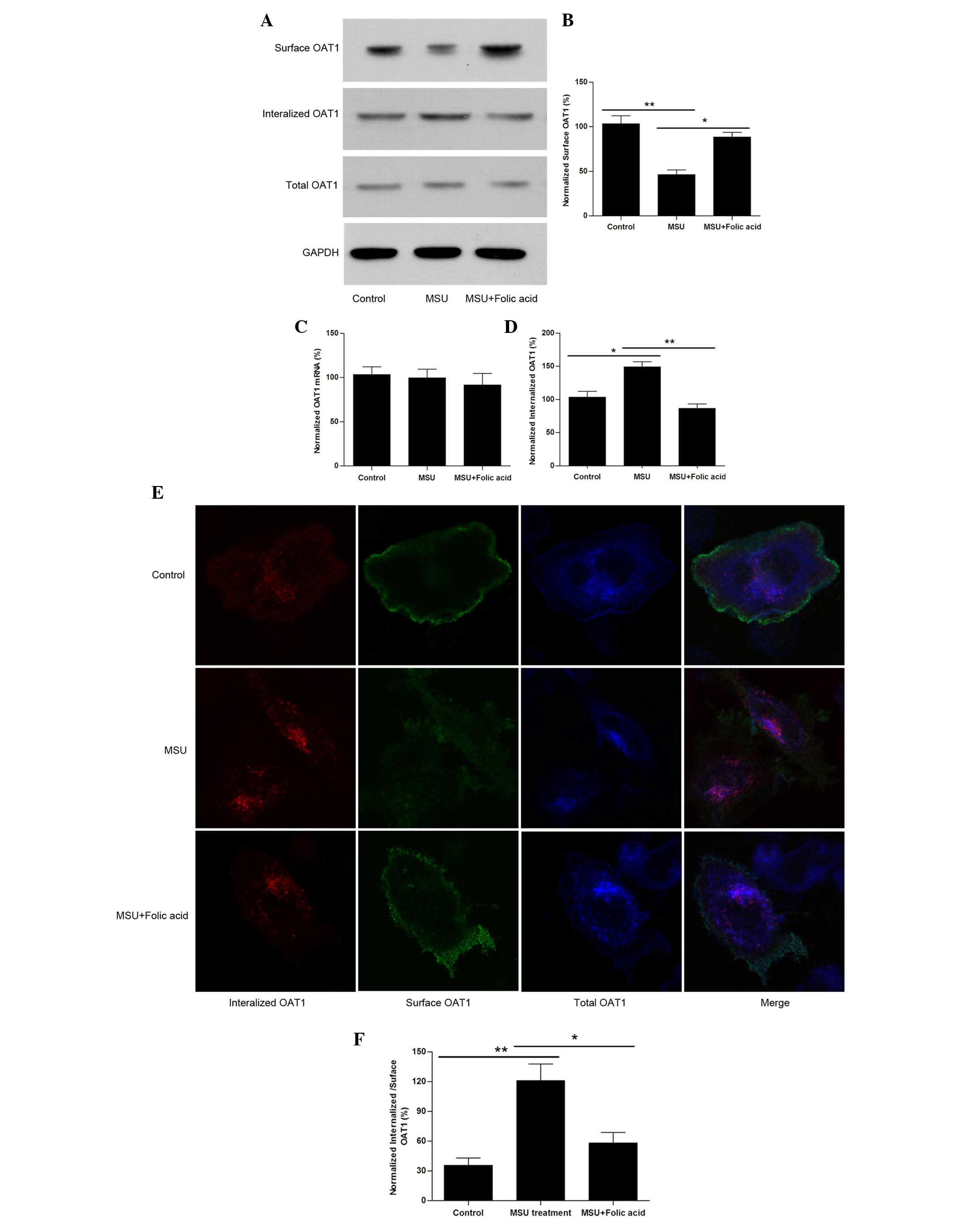
Uric acid crystals reduce surface levels
of OAT1 by facilitating its internalization
To further investigate the detailed mechanism of how
uric acid regulates OAT1 expression or trafficking, OAT1 stable
expressing HEK cells were used and they were treated with or
without MSU crystals (1 µM) for 24 h. The results
demonstrated that treatment with MSU crystals resulted in a
reduction in surface OAT1 levels however not in the total protein
(Fig. 2A) or mRNA levels (Fig. 2B). The internalized OAT1 levels
were then measured, and it was identified that following treatment
with MSU crystals, OAT1 internalization was increased (Fig. 2C and D). Notably, it was observed
that treatment of HEK cells exposed to MSU with folic acid led to a
reversal of these effects (Fig.
2A–D). Using confocal imaging it was also confirmed that
although total OAT1 was not significantly altered (Fig. 2E), the reduction in surface
expression levels corresponded with an increase in OAT1
internalization, while treatment with folic acid reversed this
effect (Fig. 2F).
Uric acid crystals activate RhoA and
induce OAT1 internalization
How the OAT1 trafficking process is regulated was
then investigated. Considering that the cytoskeleton is a major
component that controls vesicular movement, the experiments focused
upon the small guanosine triphosphatase, RhoA, which is known to
regulate actin-stress fiber formation and facilitates endocytosis
following activation (24,25). HEK cells were treated with uric
acid crystals, and it was identified that although total RhoA
levels were not significantly altered, the guanosine triphosphate
bound form of RhoA (active form) was increased significantly
(Fig. 3A B, C and D). Notably,
treatment with folic acid led to a reversal in RhoA levels. To
further investigate the effects of RhoA on OAT1 internalization,
constitutively active RhoA G14V and dominant-negative RhoA T19N
site mutation constructs were produced. By overexpression of these
constructs in OAT1-HEK cells, RhoA V14 was identified to lead to an
increase in OAT1 internalization without crystal stimulation, while
RhoA N19 inhibited uric acid-induced OAT1 internalization (Fig. 3E and F).
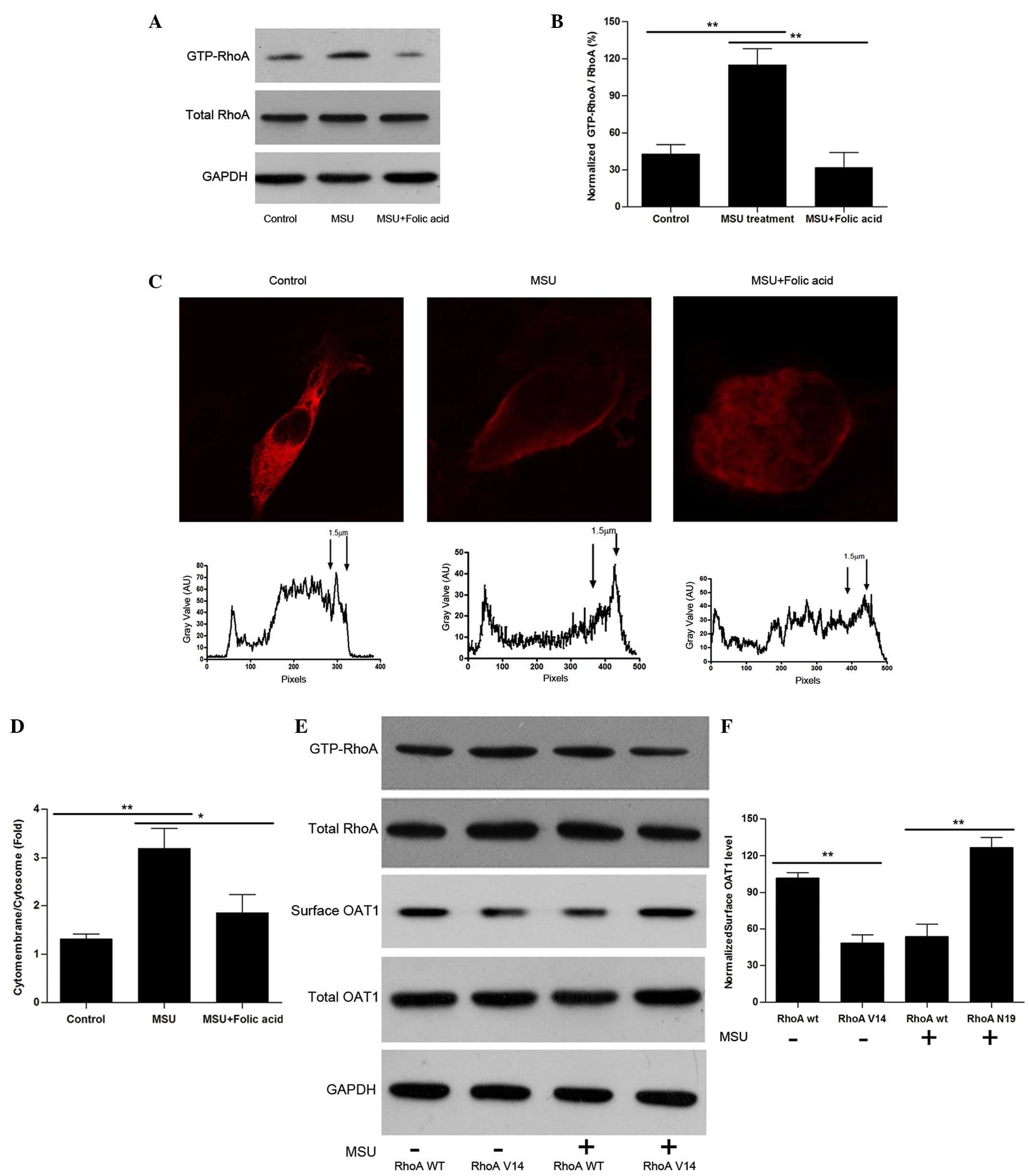 | Figure 3Uric acid crystals regulate OAT1
trafficking through the activation of RhoA. (A) Western blot
analysis of active and total RhoA in OAT1 stable HEK cells with or
without MSU and folic acid treatment. (B) Quantification of active
RhoA expression levels, normalized to total RhoA. (C) Confocal
images of plot profile analysis for RhoA distribution in OAT1
stable HEK cells with or without MSU and folic acid treatment. (D)
Quantification of cytomembrance RhoA expression levels, normalized
to cytosome RhoA. (E) Western blot analysis of GTP-RhoA and total
RhoA, surface and total OAT1 expression in RhoA V14 and N19
mutation co-treated with or without MSU. (F) Quantification of the
ratio of surface and internalized OAT1, normalized to GAPDH. Data
are presented as the mean ± standard error (*P<0.05,
**P<0.01). OAT1, organic anion transporter-1; RhoA,
Ras homolog gene family member A; MSU, monosodium urate; GTP,
guanosine triphosphate; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase; WT, wild-type. |
Folic acid was able to rescue OAT1
internalization by inhibiting RhoA activity
As it has been previously reported that folic acid
was able to inhibit RhoA activity in cell models (13), the current study aimed to
demonstrate whether folic acid possesses benefits on high uric
acid-induced kidney damage. The results demonstrated that daily
supplements of folic acid were able to rescue uric acid induced
kidney damage and repress serum uric acid, BUN and creatinine
levels by restoring the levels of surface OAT1 back to normal
levels in vivo (Fig. 1).
Treatment with MSU-treated HEK cells with or without folic acid (10
µM) demonstrated that folic acid was able to rescue OAT1
membrane distribution by inhibiting RhoA activation (Figs. 2 and 3).
Discussion
In the present study, it was suggested that urate
transporter OAT1 may be involved in high purine-induced kidney
damage in rats. The results of the current study suggested that a
purine rich diet may lead to kidney damage through high serum uric
acid, and that serum uric acid could markedly reduce surface OAT1
expression levels. In addition, folic acid, which could restore
OAT1 surface expression back to normal, was able to rescue uric
acid-induced kidney damage. In HEK cells, it was identified that
high concentrations of uric acid were able to reduce cell surface
OAT1 expression levels, however not the total OAT1 protein amount
nor its mRNA level. As OAT1 serves an important role in uric acid
release, the results here indicated that the reduction in surface
OAT1 may be involved in high serum uric acid-induced kidney
impairments as a second step following serum uric acid
hyper-production. This in turn may lead to aggravation of the urate
metabolism dyshomeostasis and serum uric acid accumulation.
Furthermore, it was identified that uric acid stimulation may
facilitate OAT1 internalization through RhoA activation, and that
folic acid was able to rescue surface OAT1 distribution by
inhibiting RhoA activation.
Rho proteins, including Cdc42, Rac1 and RhoA, have
been best characterized for their effects on the cytoskeleton and
cell adhesion. Subsequent to activation, RhoA would be recruited
from the cytosome to the membrane region, and then activate
downstream signals through phosphorylation. Activation of RhoA is
recognized to mediate the formation of stress fibers and focal
contacts that firmly anchor cells to their substrata, permitting
retraction of cellular rear ends by mediating integrin endocytosis
(20,21). In addition to integrin, active RhoA
was able to mediate the endocytosis of numerous other proteins,
such as Toll-like receptor-4, β-arrestin-1 or the Kv1.2 potassium
channel (16–18). The mechanism may involve regulating
actin dynamic polymerization and selected docking of transport
vesicles. Although no clear reports have elucidated how uric acid
may activate RhoA, a previous study demonstrated that MSU treatment
could relocate RhoA to the membrane region (19), which indicated that uric acid may
activate RhoA through currently unidentified pathways.
Folic acid, an important daily nutritional
supplement which has functions in pregnancy, cancer, stroke or
heart disease, has been identified to suppress RhoA activation
through the folic acid receptor/cSrc/p190RhoGAP-signaling pathway
(15). The current study
identified that folic acid was also able to inhibit uric
acid-induced RhoA activation, and that this may regulate RhoA
activity through the same pathway by altering the phosphorylation
status of the RhoA upstream regulator, p190RhoGAP.
In summary, the present study demonstrated, for the
first time to the best of our knowledge, that in UAN, uric acid
stimulation can reduce OAT1 membrane expression by facilitating its
endocytosis, which was regulated by activated RhoA, and this could
be rescued by folic acid treatment. A potential mechanism by which
high concentration of serum uric acid would induce UAN was
described, which suggests that folic acid may have potential
therapeutic value.
Acknowledgments
The current study was supported by the Natural
Science Foundation of Guangdong Province (grant no.
2011B080701007).
References
|
1
|
Klemp P, Stansfield SA, Castle B and
Robertson MC: Gout is on the increase in New Zealand. Ann Rheum
Dis. 56:22–26. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Arromdee E, Michet CJ, Crowson CS,
O'Fallon WM and Gabriel SE: Epidemiology of gout: Is the incidence
rising? J Rheumatol. 29:2403–2406. 2002.PubMed/NCBI
|
|
3
|
Li Y, Stamler J, Xiao Z, Folsom A, Tao S
and Zhang H: Serum uric acid and its correlates in Chinese adult
populations, urban and rural, of Beijing. The PRC-USA collaborative
study in cardiovascular and cardiopulmonary epidemiology. Int J
Epidemiol. 26:288–296. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Johnson RJ, Kang DH, Feig D, Kivlighn S,
Kanellis J, Watanabe S, Tuttle KR, Rodriguez-Iturbe B,
Herrera-Acosta J and Mazzali M: Is there a pathogenetic role for
uric acid in hypertension and cardiovascular and renal disease?
Hypertension. 41:1183–1190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tseng CH: Independent association of uric
acid levels with peripheral arterial disease in Taiwanese patients
with Type 2 diabetes. Diabet Med. 21:724–729. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kai H, Kaneyuki M, Shihara M, Toyama Y,
Mitsutake Y, Umei H, Kusaba K, Ueda T, Adachi H and Imaizumi T;
MAPPY Study Investigators: Reduction in morning blood pressure is a
key factor for ameliorating urinary albumin excretion in patients
with morning hypertension irrespective of treatment regimen. Circ
J. 77:1551–1557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sekine T, Cha SH and Endou H: The
multispecific organic anion transporter (OAT) family. Pflugers
Arch. 440:337–350. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koepsell H: The SLC22 family with
transporters of organic cations, anions and zwitterions. Mol
Aspects Med. 34:413–435. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sweeney DE, Vallon V, Rieg T, Wu W,
Gallegos TF and Nigam SK: Functional maturation of drug
transporters in the developing, neonatal, and postnatal kidney. Mol
Pharmacol. 80:147–154. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ulmius M, Johansson-Persson A, Krogh M,
Olsson P and Önning G: An oat bran meal influences blood insulin
levels and related gene sets in peripheral blood mononuclear cells
of healthy subjects. Genes Nutr. 6:429–439. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pritchard JB: Coupled transport of
p-aminohippurate by rat kidney basolateral membrane vesicles. Am J
Physiol. 255:F597–F604. 1988.PubMed/NCBI
|
|
12
|
Sweet DH, Wolff NA and Pritchard JB:
Expression cloning and characterization of ROAT1. The basolateral
organic anion transporter in rat kidney. J Biol Chem.
272:30088–30095. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Burckhardt G: Drug transport by organic
anion transporters (OATs). Pharmacol Ther. 136:106–130. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pavlova A, Sakurai H, Leclercq B, Beier
DR, Yu AS and Nigam SK: Developmentally regulated expression of
organic ion transporters NKT (OAT1), OCT1, NLT (OAT2), and Roct. Am
J Physiol Renal Physiol. 278:F635–F643. 2000.PubMed/NCBI
|
|
15
|
VanWert AL, Gionfriddo MR and Sweet DH:
Organic anion transporters: Discovery, pharmacology, regulation and
roles in pathophysiology. Biopharm Drug Dispos. 31:1–71. 2010.
|
|
16
|
Kim S, Lee CH, Kang CM and Kim GH: Effects
of increased uric acid intake on the abundance of urate-anion
exchanger and organic anion transporter proteins in the rat kidney.
Electrolyte Blood Press. 5:62–67. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yarlagadda SG and Perazella MA:
Drug-induced crystal nephropathy: An update. Expert Opin Drug Saf.
7:147–158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Liu L, Xie H, Liao J, Zhou X, Wan J,
Yu K, Li J and Zhang Y: Tanshinone IIA prevents uric acid
nephropathy in rats through NF-κB inhibition. Planta Med.
78:866–873. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim YI: Will mandatory folic acid
fortification prevent or promote cancer? Am J Clin Nutr.
80:1123–1128. 2004.PubMed/NCBI
|
|
20
|
Choi SW and Mason JB: Folate status:
Effects on pathways of colorectal carcinogenesis. J Nutr. 132(Suppl
8): S2413–S2418. 2002.
|
|
21
|
Lin SY, Lee WR, Su YF, Hsu SP, Lin HC, Ho
PY, Hou TC, Chou YP, Kuo CT and Lee WS: Folic acid inhibits
endothelial cell proliferation through activating the cSrc/ERK
2/NF-κB/p53 pathway mediated by folic acid receptor. Angiogenesis.
15:671–683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hou TC, Lin JJ, Wen HC, Chen LC, Hsu SP
and Lee WS: Folic acid inhibits endothelial cell migration through
inhibiting the RhoA activity mediated by activating the folic acid
receptor/cSrc/p190RhoGAP-signaling pathway. Biochem Pharmacol.
85:376–384. 2013. View Article : Google Scholar
|
|
23
|
Fromme T and Klingenspor M: Rapid single
step subcloning procedure by combined action of type II and type
IIs endonucleases with ligase. J Biol Eng. 1:1–3. 2007. View Article : Google Scholar
|
|
24
|
Shi Y, Evans JE and Rock KL: Molecular
identification of a danger signal that alerts the immune system to
dying cells. Nature. 425:516–521. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar : PubMed/NCBI
|