Introduction
Idiopathic pulmonary fibrosis (IPF) is a
progressive, fibrotic, interstitial lung disease of unknown cause,
with a median survival time of ~3 years. The estimated incidence of
IPF is 10 cases per 100,000, with a prevalence of 30 cases per
100,000 (1). Progressive
interstitial fibrosis is a hallmark of IPF, which results in a
decline in lung function. Due to the unknown pathogenesis of this
disease, patients with IPF have few treatment options. China is
estimated to have the largest number of patients with IPF, which is
due, at least in part, to its large population. Published
epidemiological data for IPF in China are limited; however, known
major risk factors include cigarette smoking and environmental
exposure (2–4). Smoking strongly increases the
occurrence of IPF, and China has one of the largest populations of
smokers worldwide (~350 million). Furthermore, as a rapidly
developing nation, Chinese individuals are exposed to high levels
of vehicle exhaust, industrial emissions, and metal, wood and coal
dusts, all of which can increase the likelihood of developing IPF.
Fog and haze have rapidly increased in the North China Plain since
1954 (5), and airborne particulate
matter (PM2.5) has become the focus of major public interest in
China. Therefore, it is not surprising that the incidence of IPF is
higher in China than it is in many other countries, and studies on
the epidemiology and pathogenesis of IPF are critical and urgently
required.
IPF is considered the most common and severe form of
pulmonary fibrosis, which is characterized by the accumulation of
extracellular matrix (ECM) proteins within the interstitium and
alveolar space of the lungs. The pathology associated with
pulmonary fibrosis is a consequence of the disturbance of two
physiologically-balanced processes: The proliferation and apoptosis
of fibroblasts, and the accumulation and breakdown of the ECM
(6). Fibroblasts and
fibroblast-derived myofibroblasts are the key effector cells in
fibrogenesis, since they are major producers of ECM proteins. An
overabundance of these cell types may contribute to the progression
of pulmonary fibrosis. Furthermore, an increased number of
fibroblastic foci, which are small aggregates of proliferating
fibroblasts and myofibroblasts, has been reported to be associated
with an increased risk of mortality in patients with IPF (7). To inhibit pulmonary fibrosis and to
slow the progression of IPF, it is important to suppress the
myofibroblast differentiation of fibroblasts and the subsequent ECM
production.
Phosphatase and tensin homolog deleted on chromosome
10 (PTEN) is a tumor suppressor, the expression of which is lost in
numerous types of human cancer. PTEN is able to strongly suppress
cell migration (8), promote
apoptosis (9), and inhibit cell
growth (10). Recent studies have
focused on the role of PTEN in IPF pathogenesis, due to the
presence of some similarities between IPF and cancer, including IPF
phenotype (11), global
methylation patterns (12), and
underlying disease mechanisms (13). White et al (14) reported that PTEN expression and
phosphatase activity are diminished in fibroblasts isolated from
patients with IPF. In addition, inhibition of PTEN in vivo
promotes fibrosis, and PTEN inhibits myofibroblast differentiation
in vitro (14,15). PTEN is the primary negative
regulator of the cell survival signaling pathway initiated by
phosphatidylinositol 3-kinase (PI3K). Reduced PTEN expression and
Akt hyperactivation have been detected in the alveolar epithelial
cells and fibroblastic foci of human IPF lungs; additionally, low
levels of PTEN expression/phosphatase activity have been shown to
facilitate aberrant activation of the PI3K/Akt pathway and
pathological proliferation of fibroblasts (16–19).
Therefore, altered PTEN/PI3K/Akt signaling is considered to have a
crucial role in IPF pathogenesis, thus suggesting that this pathway
may be a potential therapeutic target for IPF.
Transforming growth factor (TGF)-β is a cytokine
with an important role in fibrosis. It promotes the differentiation
of fibroblasts into myofibroblasts and enhances ECM protein
synthesis (20–22). By binding to its receptor, TGF-β
triggers the activation of numerous signaling pathways, which
contribute to progressive fibrosis, including the Smad pathway, the
PI3K/Akt pathway, and the glycogen synthase kinase
(GSK)-3β/β-catenin pathway (22–25).
TGF-β suppresses PTEN expression (19,26),
and inhibition of PTEN activity is necessary for TGF-β-induced
expression of α-smooth muscle actin (α-SMA), which is a marker of
myofibroblasts (15). However, the
exact mechanism by which PTEN regulates the TGF-β-mediated
transition of fibroblasts into myofibroblasts remains unclear.
PTEN has been implicated in the pathogenesis of IPF.
Therefore, the present study hypothesized that there may be
differences in PTEN serum levels between individuals with IPF and
healthy individuals. If so, altered PTEN expression may serve as a
diagnostic marker for IPF. In addition, we further hypothesized
that altered PTEN expression may influence the production of α-SMA
and ECM proteins in TGF-β-treated lung fibroblasts, and that the
PI3K/Akt signaling pathway may be associated with these
PTEN-mediated effects.
A total of 42 Chinese patients with IPF and 42
healthy controls were included in the present study. Serum levels
of PTEN were measured by enzyme-linked immunosorbent assay (ELISA),
and comparisons between patients with IPF and healthy controls were
made. To evaluate the role of PTEN in lung fibrosis, human
embryonic lung fibroblasts were treated with TGF-β1, in order to
mimic pulmonary fibrosis. PTEN was overexpressed in TGF-β1-treated
human embryonic lung fibroblasts, and proliferation, apoptosis and
migration were assayed. Western blotting was performed to examine
PI3K/Akt and TGF-β/Smad3 signaling, as well as the expression of
downstream proteins, including the myofibroblast marker α-SMA and
matrix metalloproteinases (MMPs). The results of the present study
suggested that low serum levels of PTEN may be used to diagnose
IPF. Furthermore, PTEN overexpression was shown to inhibit
TGF-β1-induced fibroblast differentiation.
Materials and methods
Subjects
A total of 42 patients with IPF who were admitted to
Fujian Province Hospital between June 2013 and April 2014 were
enrolled in the present study. The clinical diagnosis of IPF was
made by high-resolution computed tomography (HRCT) and pulmonary
function tests, according to the guidelines presented by
ATS/ERS/JRS/ALAT in 2011 (27).
Patients were included in the study if they met the following two
criteria: i) Absence of other known causes of interstitial lung
disease (e.g., domestic and occupational environmental exposures,
connective tissue disease and drug toxicity); ii) detection of
usual interstitial pneumonia pattern by HRCT exam. Patients who
were <18 years of age or who had other diseases, including
phthisis, bronchogenic carcinoma, or other malignant tumors, were
excluded from the study. A total of 42 healthy subjects were
included as controls, based on the following criteria: i) Men and
women who were ≥18 years of age; ii) with no lung diseases.
Subjects were excluded from participating as healthy controls if
they were <18 years of age or if they presented with a malignant
tumor. The present study was approved by the Medical Ethics
Committee of Fujian Province Hospital (Fuzhou, China). Written
informed consent was obtained from all patients and healthy
subjects prior to inclusion in the study.
Pulmonary function tests
The following pulmonary function tests were
conducted on all patients: Forced vital capacity (FVC); diffusing
capacity of the lungs for carbon monoxide (DLCO), using a
JAEGER® MasterScreen™ Diffusion Pulmonary Function
Analyzer (CareFusion Germany, Höchberg, Germany); and arterial
partial pressure of oxygen (PaO2), using an ABL800 BASIC
Arterial Blood Gas Analyzer (Radiometer Medical ApS, Brønshøj,
Denmark). FVC and DLCO measurements are expressed as percentages of
predicted values.
Measurement of D-dimer levels
Plasma concentrations of D-dimer were measured using
a latex-enhanced immuno turbidimetric assay. Cells were removed
from plasma by centrifugation at 2,000 × g for 10 min and samples
were prepared according to the manufacturer's protocol (Liatest
D-Di 113837; Diagnostica Stago, Asnières, France). Sample values
were measured using a STA-R Evolution Automated Blood Coagulation
Analyzer (Stago, Paris, France).
Detection of serum PTEN levels by
ELISA
Venous blood samples (5 ml) were collected and PTEN
ELISA assays (Colorfulgene Biotechnology, Inc., Wuhan, China) were
performed using the Multiskan MK3 enzyme-labeled instrument (Thermo
Fisher Scientific, Inc., Vantaa, Finland). Briefly, the test
samples were thawed to room temperature, and 100-µl aliquots
of each sample were added into monoclonal antibody-coated 96-well
plates. Following incubation at 37°C for 60 min, the closure plate
membrane was uncovered and the liquid was discarded and
subsequently dried by manually swinging the plates in the air.
Subsequently, each well was supplemented with washing buffer, left
for 30 sec then drained. This was repeated five times and the
plates were dried by patting. Each well was supplemented with 50
µl horseradish peroxidase (HRP)-conjugated anti-PTEN
antibody and shaken prior to incubation at 37°C for 60 min, and the
washing step outlined above was repeated. Subsequently, 100
µl chromogen solution was added to each well and incubated
for 15 min, prior to the addition of 50 µl termination
solution per well. The plates were shaken on a shaker for 5 sec in
order to thoroughly mix the substrate and termination solutions.
The absorbance of the reaction mixture was measured at 450 nm
wavelength (A value) using a Multiskan MS 252 microplate reader
(Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cell culture
The HFL-I human embryonic lung fibroblast cell line
was purchased from Cell Bank of the Chinese Academy of Sciences
(Beijing, China). The cells were maintained in F12K medium
(Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Beijing Solarbio Science & Technology
Co., Ltd., Beijing, China).
Plasmid construction and
transfection
To generate the PTEN expression vector
pcDNA3.1-PTEN, the PTEN coding region was amplified from cDNA by
PCR using the following primers: Forward,
5′-CGGAATTCCATGACAGCCATCATCAAAGAG-3′ and reverse,
5′-CCCTCGAGGTCAGACTTTTGTAATTTGTGT-3′ (Shanghai Generay Biotech Co.,
Ltd., Shanghai, China). The PCR product was digested using
EcoRI and XhoI (both Thermo Fisher Scientific, Inc.)
and cloned into the pcDNA3.1(+) green fluorescent protein vector
(JRDUN Biotechnology Co., Ltd., Shanghai, China).
One day prior to transfection, HFL-1 cells were
plated in 6-well dishes at 5×105 cells/well. The cells
were transfected with either 2 µg PTEN overexpression vector
(PTEN) or with a control vector (Vec; Addgene, Inc., Cambridge, MA,
USA); an untransfected control group (Con) was also included. Cells
were harvested with trypsin (Kilton Biological Technology Co.,
Ltd.) 48 h post-transfection, and total RNA was extracted from the
cells.
TGF-β1 stimulation was performed 48 h
post-transfection. TGF-β1 (5 ng/ml; cat. no. PHG9204; Invitrogen;
Thermo Fisher Scientific, Inc.) was added to the cells of all
transfection groups: PTEN overexpression (P + T), control vector (V
+ T), or untransfected cells (TGF). A total of 24 h
post-stimulation, the cells were harvested and subjected to
analysis by apoptosis assay and western blotting.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Shanghai, China) and was reverse transcribed using an RT kit
(Thermo Fisher Scientific, Inc., Shanghai, China), according to the
manufacturer's protocol. SYBR Green PCR kit (Thermo Fisher
Scientific, Inc., Shanghai, China) was used to conduct RT-qPCR. The
following sequence-specific primers were used: PTEN, forward
5′-TCAGGCGAGGGAGATGAGAG-3′ and reverse 5′-CGAAGAGGAGGCGAGAAACG-3′;
and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), forward
5′-CACCCACTCCTCCACCTTTG-3′ and reverse 5′-CCACCACCCTGTTGCTGTAG-3′
(Shanghai Generay Biotech Co., Ltd.). qPCR was performed on an
ABI-7300 Real-Time PCR instrument (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) with the following
thermal cycling conditions: 95°C for 5 min, followed by 40 cycles
of 95°C for 15 sec and 60°C for 45 sec, 95°C for 15 sec, 60°C for 1
min, 95°C for 15 sec and 60°C for 15 sec. Relative differences in
gene expression between the groups were calculated using the
quantification cycle (Cq) method. Data analysis was performed using
the 2−ΔΔCq method (28), and GAPDH mRNA was used for
normalization. All experiments were repeated three times.
Cell viability assay
Cell viability was assessed using a Cell Counting
kit (CCK)-8 assay (Dojindo Molecular Technologies Co., Ltd.,
Kumamoto, Japan). The cells were seeded in 96-well plates
supplemented with F12K serum-free medium (Sigma-Aldrich) at a
density of 1–5×104 cells per well. CCK-8 and medium were
mixed at a ratio of 10:100 µl per well in sterile Eppendorf
tubes. Medium was aspirated, and the cells were washed once with
phosphate-buffered saline (PBS). Subsequently, premixed CCK-8 and
medium were added to each well and incubated at 37°C for 0.5–1 h.
Absorbance values at 450 nm were obtained using an automatic
LabSystems 352 Multiskan MS microplate reader (Thermo Fisher
Scientific, Inc.).
Apoptosis assay
Apoptosis of HFL-1 cells was determined using flow
cytometry. The cells were scraped, washed twice with PBS, and
centrifuged at 1,000 × g for 5 min at 4°C. Pelleted cells were
incubated in 1X binding buffer containing fluorescein
isothiocyanate (FITC)-Annexin V and propidium iodide (BD
Biosciences, San Jose, CA, USA) in the dark for 15 min. Apoptotic
cells were examined using a FACSCalibur flow cytometer (BD
Biosciences) and analyzed with FlowJo software (version 7.6.1;
FlowJo, LLC, Ashland, OR, USA). A minimum of 1×104
events per sample were acquired and analyzed. Three independent
experiments were performed. Values are expressed as the mean ±
standard error of the mean in three separate experiments.
Cell migration assay
HFL-1 cells were left untransfected or transfected
with either a PTEN overexpression vector or a control vector. A
total of 48 h post transfection, Transwell migration assays were
performed. Following trypsinization, cells in the logarithmic
growth phase were suspended in F12K serum-free medium containing
0.5% FBS. A 100 µl cell suspension (1.0×105
cells/ml) was added to the upper chamber of the Transwell system,
and 500 µl medium containing 10% FBS was added to the lower
chamber. Following 16 h culture, the upper chamber was removed, and
washed three times with PBS. The cells of the upper chamber were
gently wiped with a cotton swab and cells that had migrated to the
lower chamber were fixed with 4% paraformaldehyde for 30 min and
subsequently stained with crystalline violet for 15 min. Four
fields (magnification, ×200) were randomly selected to quantify the
number of cells that had successfully migrated to the lower
chamber, using an Olympus CX41 (Olympus Corporation, Tokyo,
Japan).
Western blot analysis
Western blotting was performed to detect both total
and phosphorylated forms of numerous proteins of interest. Proteins
were extracted from the cells using RAPI protein extraction medium
(Solarbio R0010; Beijing Solarbio Science & Technology Co.,
Ltd.). Protein concentration was determined by Bradford assay using
the Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad
Laboratories, Inc. Hercules, CA, USA), according to the
manufacturer's protocol. Total cell protein extracts (25 µg)
were separated by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and were then transferred to nitrocellulose
membranes (EMD Millipore, Billerica, MA, USA). The blots were
blocked with 5% nonfat milk overnight, and were then incubated
overnight at 4°C with the following primary antibodies: Anti-PTEN
(ab32199), anti-α-SMA (both 1:500; ab5694; both Abcam, Cambridge,
MA, USA), anti-β-catenin (1:1,000; 8814S; Cell Signaling
Technology, Inc., Beverly, MA, USA), anti-Smad3 (1:400; ab28379),
anti-phosphorylated (p)-Smad3 (1:500; ab63403; both Abcam),
anti-Akt (9272S), anti-p-Akt (4058S; both Cell Signaling
Technology, Inc.), anti-MMP-2 (1948S; Epitomics, Inc., Burlingame,
CA, USA), anti-MMP-9 (all 1:1,000; ab38898; Abcam), and anti-GAPDH
(1:1,500; 5471; Cell Signaling Technology, Inc.). The following
day, membranes were incubated at room temperature for 1 h with
HRP-conjugated goat anti-rabbit (A0208) or anti-mouse
immunoglobulin G secondary antibodies (both 1:1,000; A0216; both
Beyotime Institute of Biotechnology, Shanghai, China). After
washing with PBS, the blots were visualized using Pierce Enhanced
Chemiluminescence Western blotting substrate (Pierce Protein
Biology; Thermo Fisher Scientific, Inc., Rockford, IL, USA),
according to the manufacturer's protocol. Densitometric analysis of
immunoblots was performed using TotalLab Software, version 2.00
(TotalLab Limited, Newcastle upon Tyne, UK). The densitometry
values of the proteins of interest were normalized to those of
GAPDH. All experiments were repeated three times.
Statistical analysis
Statistical analysis was performed using SPSS 19.0
software (SPSS IBM, Armonk, NY, USA). All data are presented as the
mean ± standard deviation. One-factor analysis of variance was used
for multiple comparisons, and the least significant difference test
was used for intra-group comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
Baseline clinical characteristics and
serum expression of PTEN in patients with IPF
Baseline characteristics, including age, gender,
smoking status, diabetic status and hypertensive status, were
similar between patients with IPF and healthy controls (Table I). Although each of these baseline
characteristics was higher in the IPF patient group, there were no
significant differences when compared to the controls. Conversely,
the prevalence of coronary heart disease was significantly higher
in patients with IPF, as compared with in healthy subjects
(P<0.05), which is consistent with an earlier report (29).
 | Table IBaseline clinical characteristics of
study subjects. |
Table I
Baseline clinical characteristics of
study subjects.
| Characteristic | IPF patients
(n=42) | Healthy subjects
(n=42) | P-value |
|---|
| Age (years) | 67.9±11.7 | 65.5±8.9 | 0.296 |
| Gender
(men/women) | 31/11 | 28/14 | 0.474 |
| Smoking (%) | 15/42 (35.71) | 13/42 (30.95) | 0.643 |
| Pulmonary function
tests | | | |
| FVC (% pred) | 67±10 | - | |
| DLCO (% pred) | 54±17 | - | |
| PaO2
(mmHg) | 73.26±11.65 | - | |
| Diabetes (%) | 8/42 (19.05) | 7/42 (16.67) | 0.776 |
| Hypertension
(%) | 17/42 (40.48) | 15/42 (35.71) | 0.653 |
| Coronary heart
disease (%) | 15/42 (35.71) | 3/42 (7.14) | 0.001 |
| D-dimer
(µg/ml) | 1.23±1.02 | - | |
| Serum PTEN
(ng/ml) | 8.11±0.24 | 9.37±0.23 | <0.001 |
To evaluate the expression levels of PTEN in each of
the two study groups, serum samples were collected and analyzed by
ELISA. Serum PTEN protein expression levels were significantly
reduced in patients with IPF, as compared with in healthy subjects
(P<0.01; Table I).
Effects of PTEN overexpression on
TGF-β1-induced lung fibroblasts
The role of PTEN in fibrosis was examined by
overexpressing PTEN in human embryonic lung fibroblasts. Following
transfection of the cells with PTEN, overexpression was confirmed
at both the mRNA and protein level (Fig. 1).
Subsequently, TGF-β1-stimulated HFL-1 cells were
used to examine the effects of PTEN overexpression on lung
fibrosis. Fibroblast proliferation is a primary event in fibrosis;
therefore, HFL-1 cell viability was assessed. As shown in Fig. 2, HFL-1 cell proliferation was
significantly enhanced following 24 or 48 h of TGF-β1 stimulation
(P<0.05). These effects on proliferation are consistent with the
effects of TGF-β1 on the induction of fibrosis. Overexpression of
PTEN significantly reduced HFL-1 cell proliferation at both 24 and
48 h (P<0.01), and attenuated TGF-β1-mediated proliferation at
the 48 h time point (P<0.05).
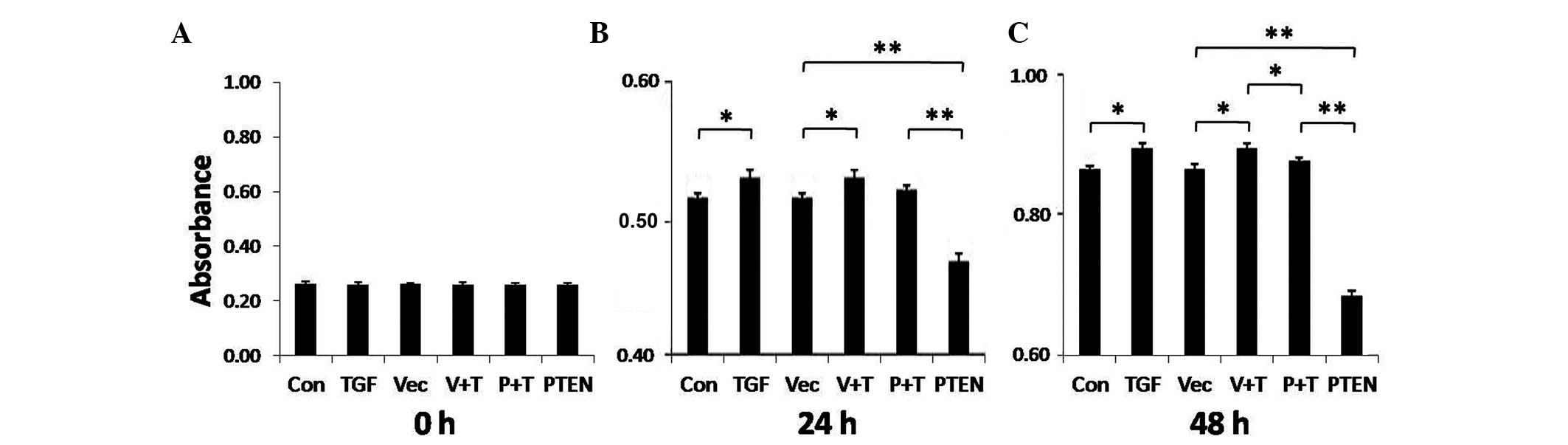 | Figure 2Cell viability assay of transforming
growth factor (TGF)-β1-stimulated fibroblasts after phosphatase and
tensin homolog deleted on chromosome 10 (PTEN) overexpression.
HFL-1 cells were transfected with a PTEN overexpression vector, a
control vector, or remained untransfected. A total of 48 h
post-transfection, the cells were treated with 5 ng/ml TGF-β1 or
were left untreated for (A) 0 h, (B) 24 h, or (C) 48 h. Cell
proliferation was analyzed by Cell Counting kit-8 assay. The
experiment was repeated three times. Data are presented as the mean
± standard deviation (n=3). *P<0.05,
**P<0.01. Con, control; TGF, TGF-β1 stimulation; Vec,
vector control; V + T, vector control + TGF-β1 stimulation; P + T,
PTEN overexpression vector + TGF-β1 stimulation; PTEN, PTEN
overexpression vector. |
An apoptosis analysis was also performed following
PTEN overexpression and TGF-β1 stimulation. Treatment with TGF-β1
decreased apoptosis of HFL-1 cells; conversely, overexpression of
PTEN enhanced apoptosis of the cells both in the absence or
presence of TGF-β1 stimulation (Fig.
3). These results indicate that PTEN may inhibit fibrosis by
both inhibiting proliferation and promoting apoptosis of
fibroblasts.
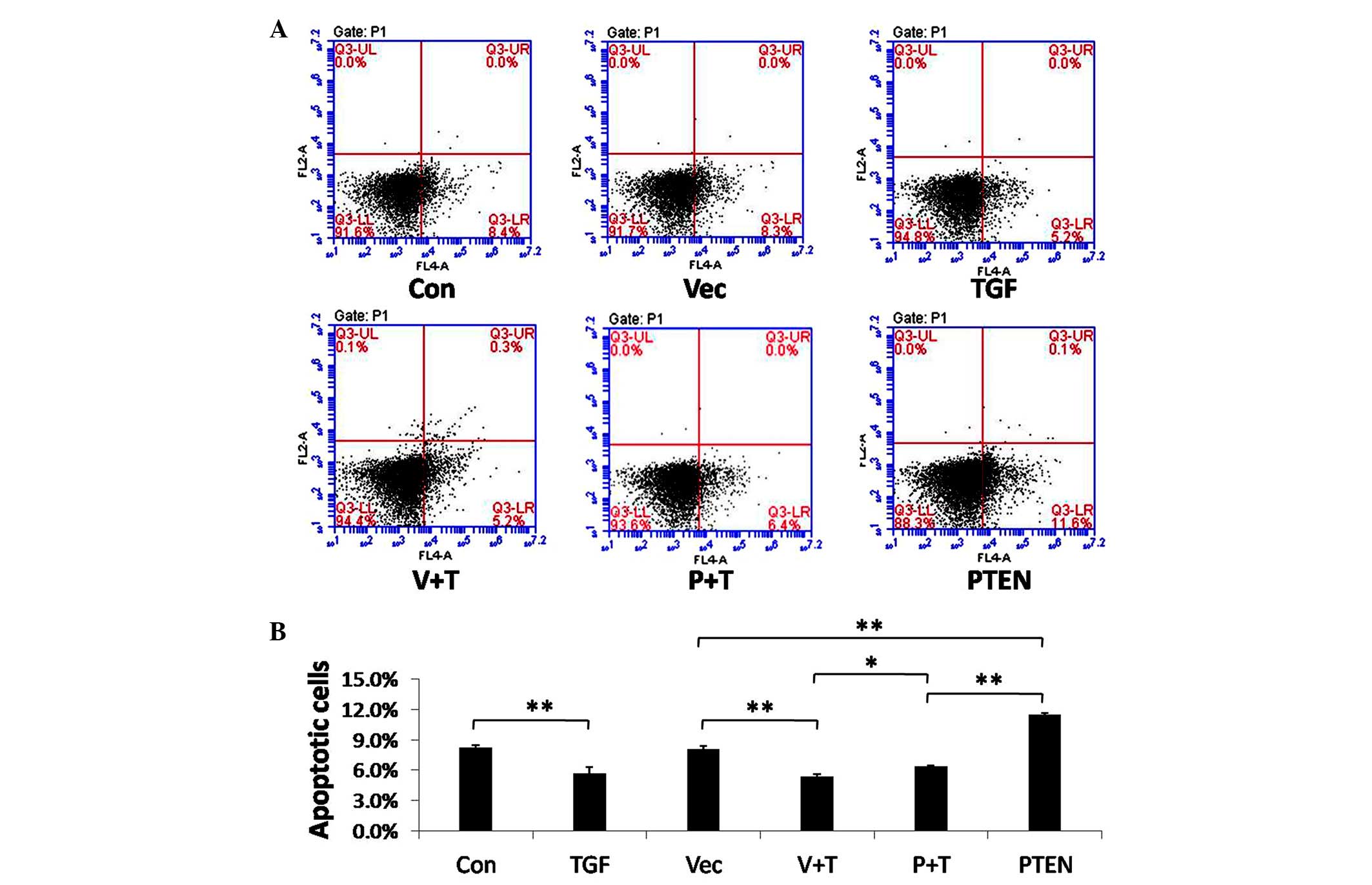 | Figure 3Apoptosis analysis of transforming
growth factor (TGF)-β1-stimulated fibroblasts after phosphatase and
tensin homolog deleted on chromosome 10 (PTEN) overexpression.
HFL-1 cells were transfected with a PTEN overexpression vector, a
control vector, or remained untransfected. A total of 48 h
post-transfection, the cells were treated with 5 ng/ml TGF-β1 or
were left untreated for 24 h. (A) Apoptosis was measured by surface
Annexin V staining and flow cytometry. (B) Data represent the
results of three independent experiments. Data are presented as the
mean ± standard error of the mean. *P<0.05,
**P<0.01. Con, control; TGF, TGF-β1 stimulation; Vec,
vector control; V + T, vector control + TGF-β1 stimulation; P + T,
PTEN overexpression vector + TGF-β1 stimulation; PTEN, PTEN
overexpression vector. |
The present study examined the role of PTEN in
fibroblast migration by performing a Transwell migration assay
following PTEN overexpression. As shown in Fig. 4, TGF-β1 stimulation significantly
increased the migration of HFL-1 cells, as compared with the
control groups (P<0.01). Conversely, overexpression of PTEN
significantly suppressed cell migration both in the presence or
absence of TGF-β1 stimulation (P<0.01).
 | Figure 4Phosphatase and tensin homolog
deleted on chromosome 10 (PTEN) inhibits transforming growth factor
(TGF)-β1-induced fibroblast migration. HFL-1 cells were transfected
with a PTEN overexpression vector, a control vector, or remained
untransfected. A total of 24 h post-transfection, the cells were
plated in Transwell inserts and were incubated for 24 h. Cells were
then treated with 5 ng/ml TGF-β1 or were left untreated for 24 h.
(A) Migratory cells were fixed, stained and images were captured
(magnification, ×100). (B) Cells from three different fields were
counted, and averages of three independent experiments were
plotted. Data are presented as the mean ± standard deviation.
**P<0.01. Con, control; TGF, TGF-β1 stimulation; Vec,
vector control; V + T, vector control + TGF-β1 stimulation; P + T,
PTEN overexpression vector + TGF-β1 stimulation; PTEN, PTEN
overexpression vector. |
These results suggest that PTEN overexpression
enhances apoptosis and suppresses proliferation and migration of
TGF-β1-induced fibroblasts. Therefore, PTEN may function as an
inhibitor of fibroblast differentiation.
Effects of PTEN overexpression on p-Akt,
p-Smad3, β-catenin, α-SMA, MMP-2 and MMP-9
To further evaluate the role of PTEN in
TGF-β1-induced fibroblasts, the effects of PTEN overexpression were
determined on several signaling pathways. As shown in Fig. 5, phosphorylation of Akt and Smad3
was significantly increased following TGF-β1 stimulation
(P<0.01); notably, these increases were attenuated by PTEN
overexpression (P<0.01). The expression levels of several
TGF-β1-induced proteins, including α-SMA, MMP-2 and MMP-9, were
also significantly attenuated by PTEN overexpression (P<0.01).
The protein expression levels of β-catenin were not significantly
altered in TGF-β1-stimulated HFL-1 cells (data not shown). These
data indicate that PTEN overexpression may suppress TGF-β1-induced
target protein expression by inhibiting phosphorylation of Akt and
Smad3, two downstream targets of TGF-β1 signaling.
 | Figure 5Overexpression of phosphatase and
tensin homolog deleted on chromosome 10 (PTEN) suppresses
phosphorylation (p-) of Akt and Smad3, and the expression of
α-smooth muscle actin (α-SMA), matrix metalloproteinase (MMP)-2 and
MMP-9 in transforming growth factor (TGF)-β1-induced fibroblasts.
HFL-1 cells were transfected with a PTEN overexpression vector, a
control vector, or remained untransfected. A total of 48 h
post-transfection, cells were treated with 5 ng/ml TGF-β1 or were
left untreated for 24 h. (A) Cell lysates were collected and
subjected to western blotting. (B) Densitometry data from three
independent experiments are plotted. Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) served as a loading control. Data are
presented as the mean ± standard deviation (n=3).
*P<0.05, **P<0.01. Con, control; TGF,
TGF-β1 stimulation; Vec, vector control; V + T, vector control +
TGF-β1 stimulation; P + T, PTEN overexpression vector + TGF-β1
stimulation; PTEN, PTEN overexpression vector. |
Discussion
The present study examined the expression levels of
PTEN in clinical cases of IPF, and determined the effects of PTEN
overexpression on a TGF-β1-induced fibroblast model of IPF
pathogenesis. PTEN serum levels were significantly lower in
patients with IPF, as compared with in healthy control subjects,
thus suggesting that PTEN may be a useful diagnostic marker for
IPF. Treatment of human fibroblasts with TGF-β1 promotes their
differentiation into myofibroblasts. The results of the present
study demonstrated that PTEN overexpression inhibits fibroblast
proliferation and migration, and enhances apoptosis. In addition,
PTEN may inhibit TGF-β1-induced fibrosis via suppressing the
phosphorylation of downstream targets of TGF-β1, including Akt and
Smad3. Consistent with this, several TGF-β1 target proteins,
including α-SMA, MMP-2 and MMP-9, were revealed to be
downregulated. These data establish an important inhibitory role
for PTEN in pulmonary fibrosis. PTEN may prove to be a useful
diagnostic marker for IPF, and components of the PTEN/PI3K/Akt
signaling pathway may serve as potentially novel therapeutic
targets for the treatment of IPF.
PTEN, which is a tumor suppressor that functions as
a dual protein/lipid phosphatase, negatively regulates the cell
survival signaling pathway initiated by PI3K. As an important
modulator of apoptosis, cell growth, migration and invasion, PTEN
deficiency suppresses apoptosis, and enhances cell proliferation
and migration (8–10). The present study detected reduced
serum levels of PTEN in patients with IPF. Overexpression of PTEN
in TGF-β1-induced fibroblasts resulted in increased apoptosis,
decreased proliferation and migration, and reduced expression of
α-SMA. These findings are consistent with the findings of a
previous study in IPF fibroblasts and pten−/−
fibroblasts (15). Taken together,
PTEN may have a critical role in IPF pathogenesis via the
inhibition of fibroblast differentiation into myofibroblasts.
Previous studies have highlighted the importance of
the PTEN/PI3K/Akt pathway in lung injury and fibrosis (16,18).
For example, reduced PTEN expression/activity and Akt
hyperactivation have been described in IPF fibroblasts; findings
such as this help establish the importance of PTEN/PI3K/Akt axis in
IPF pathogenesis (30). In
addition, immunohistochemical analysis of IPF lung tissue
identified high Akt activity and inactive forkhead box O3a (a
transcription factor downstream of the PI3K/Akt pathway) in
patients with IPF (31). TGF-β is
believed to be a key mediator of tissue fibrosis as a consequence
of ECM accumulation in pathological states (32). It has been demonstrated that
inhibition of PTEN activity is necessary for TGF-β-induced α-SMA
expression (15). However, the
mechanisms underlying PTEN-inhibited differentiation of
TGF-β-induced fibroblasts remain elusive. In the present study,
PTEN was overexpressed in human lung fibroblasts, and the activity
of Akt was examined following TGF-β1 stimulation. PTEN
overexpression was able to suppress TGF-β1-induced Akt
phosphorylation. Inhibition of PI3K has also been shown to prevent
proliferation and differentiation of TGF-β-treated human lung
fibroblasts into myofibroblasts (33). Wilkes et al (34) reported that the PI3K/Akt pathway
can be activated by TGF-β via the phosphorylation of Akt, and that
this signaling cascade has an important role in TGF-β-mediated
fibroblast proliferation and morphological transformation. This
activation appears to be independent of Smad2/3 activation
(34). Notably, the present study
demonstrated that overexpression of PTEN suppressed TGF-β1-mediated
phosphorylation of Smad3. TGF-β1/Smad3 has been shown to be a major
pathway regulating myofibroblast differentiation (35). Therefore, PTEN may suppress
TGF-β1-induced myofibroblast differentiation by inhibiting both the
PI3K/Akt pathway and the TGF-β/Smad3 pathway.
Aberrant activation of Wnt/β-catenin signaling has
been detected in patients with IPF (36), and signaling via GSK-3β/β-catenin
is associated with TGF-β1-mediated differentiation of human lung
fibroblasts into myofibroblasts (24). The present study also examined the
expression levels of β-catenin; however, PTEN overexpression had no
significant effect on β-catenin levels in TGF-β1-induced
fibroblasts. Therefore, in contrast to previous reports (24,37,38),
β-catenin was not upregulated by TGF-β1 in the present study. This
may be due to differences in human embryonic lung fibroblasts and
lung fibroblasts derived from patients with IPF. Further studies
are required to explore the role of Wnt/β-catenin signaling in the
pathogenesis of IPF.
IPF is associated with the deposition of ECM
components into the lung interstitium. MMPs are known to have a
central role in ECM remodeling. MMP-2 and MMP-9, collectively known
as the gelatinases, are highly expressed in IPF (39), and both were significantly
upregulated by TGF-β1 stimulation in the present study.
Overexpression of PTEN in TGF-β1-induced fibroblasts resulted in
inhibition of MMP-2 and MMP-9 expression, suggesting a role for
PTEN in ECM remodeling. Certain limitations of our study should be
noted. Due to the relatively small sample size (n=42), there is
limited statistical power to detect putative associations between
patients with IPF and healthy subjects. For example, smoking status
and diabetic status, which have previously been shown to be
strongly associated with IPF (2,4,15),
were not significantly different between cases and controls in the
present study. This limitation will be addressed in future studies,
in which we plan to enroll larger numbers of individuals. A second
potential limitation is that the studies were performed in only
human embryonic lung fibroblasts. Additional in vivo studies
or studies using lung fibroblasts from patients with IPF are
required to further investigate PTEN function in IPF.
In conclusion, the present study demonstrated that
PTEN expression is reduced in patients with IPF, as compared with
in healthy individuals. In addition, PTEN overexpression inhibits
TGF-β1-mediated myofibroblast differentiation of fibroblasts by
attenuating signaling via the PI3K/Akt and TGF-β/Smad3 pathways.
These results suggested that PTEN may be a useful diagnostic marker
for IPF and that the PI3K pathway may be a target for therapeutic
intervention for the disease. Notably, a previous study recently
described a translational preclinical animal model of IPF (40), which may be a useful model for the
exploration of anti-PI3K/Akt therapies in vivo.
Acknowledgments
The present study was supported by the Project of
Natural Science Fund of Fujian Province (grant no. 2012J01321).
References
|
1
|
Gay SE, Kazerooni EA, Toews GB, Lynch JP
III, Gross BH, Cascade PN, Spizarny DL, Flint A, Schork MA, Whyte
RI, et al: Idiopathic pulmonary fibrosis: Predicting response to
therapy and survival. Am J Respir Crit Care Med. 157:1063–1072.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baumgartner KB, Samet JM, Stidley CA,
Colby TV and Waldron JA: Cigarette smoking: A risk factor for
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
155:242–248. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iwai K, Mori T, Yamada N, Yamaguchi M and
Hosoda Y: Idiopathic pulmonary fibrosis. Epidemiologic approaches
to occupational exposure. Am J Respir Crit Care Med. 150:670–675.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taskar VS and Coultas DB: Is idiopathic
pulmonary fibrosis an environmental disease? Proc Am Thorac Soc.
3:293–298. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Quan J, Zhang Q, He H, Liu J, Huang M and
Jin H: Analysis of the formation of fog and haze in North China
Plain (NCP). Atmos Chem Phys. 11:8205–8214. 2011. View Article : Google Scholar
|
|
6
|
Todd NW, Luzina IG and Atamas SP:
Molecular and cellular mechanisms of pulmonary fibrosis.
Fibrogenesis Tissue Repair. 5:112012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
King TE Jr, Schwarz MI, Brown K, Tooze JA,
Colby TV, Waldron JA Jr, Flint A, Thurlbeck W and Cherniack RM:
Idiopathic pulmonary fibrosis: Relationship between histopathologic
features and mortality. Am J Respir Crit Care Med. 164:1025–1032.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tamura M, Gu J, Matsumoto K, Aota S,
Parsons R and Yamada KM: Inhibition of cell migration, spreading,
and focal adhesions by tumor suppressor PTEN. Science.
280:1614–1617. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stambolic V, Suzuki A, de la Pompa JL,
Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM,
Siderovski DP and Mak TW: Negative regulation of PKB/Akt-dependent
cell survival by the tumor suppressor PTEN. Cell. 95:29–39. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tamura M, Gu J, Takino T and Yamada KM:
Tumor suppressor PTEN inhibition of cell invasion, migration, and
growth: Differential involvement of focal adhesion kinase and
p130Cas. Cancer Res. 59:442–449. 1999.PubMed/NCBI
|
|
11
|
Vancheri C, Failla M, Crimi N and Raghu G:
Idiopathic pulmonary fibrosis: A disease with similarities and
links to cancer biology. Eur Respir J. 35:496–504. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rabinovich EI, Kapetanaki MG, Steinfeld I,
Gibson KF, Pandit KV, Yu G, Yakhini Z and Kaminski N: Global
methylation patterns in idiopathic pulmonary fibrosis. PLoS One.
7:e337702012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vancheri C: Common pathways in idiopathic
pulmonary fibrosis and cancer. Eur Respir Rev. 22:265–272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
White ES, Thannickal VJ, Carskadon SL,
Dickie EG, Livant DL, Markwart S, Toews GB and Arenberg DA:
Integrin alpha4beta1 regulates migration across basement membranes
by lung fibroblasts: A role for phosphatase and tensin homologue
deleted on chromosome 10. Am J Respir Crit Care Med. 168:436–442.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
White ES, Atrasz RG, Hu B, Phan SH,
Stambolic V, Mak TW, Hogaboam CM, Flaherty KR, Martinez FJ, Kontos
CD and Toews GB: Negative regulation of myofibroblast
differentiation by PTEN (phosphatase and tensin homolog deleted on
chromosome 10). Am J Respir Crit Care Med. 173:112–121. 2006.
View Article : Google Scholar :
|
|
16
|
Miyoshi K, Yanagi S, Kawahara K, Nishio M,
Tsubouchi H, Imazu Y, Koshida R, Matsumoto N, Taguchi A, Yamashita
S, et al: Epithelial Pten controls acute lung injury and fibrosis
by regulating alveolar epithelial cell integrity. Am J Respir Crit
Care Med. 187:262–275. 2013. View Article : Google Scholar
|
|
17
|
Xia H, Khalil W, Kahm J, Jessurun J,
Kleidon J and Henke CA: Pathologic caveolin-1 regulation of PTEN in
idiopathic pulmonary fibrosis. Am J Pathol. 176:2626–2637. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xia H, Diebold D, Nho R, Perlman D,
Kleidon J, Kahm J, Avdulov S, Peterson M, Nerva J, Bitterman P and
Henke C: Pathological integrin signaling enhances proliferation of
primary lung fibroblasts from patients with idiopathic pulmonary
fibrosis. J Exp Med. 205:1659–1672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fagone E, Conte E, Gili E, Fruciano M,
Pistorio MP, Lo Furno D, Giuffrida R, Crimi N and Vancheri C:
Resveratrol inhibits transforming growth factor-β-induced
proliferation and differentiation of ex vivo human lung fibroblasts
into myofibroblasts through ERK/Akt inhibition and PTEN
restoration. Exp Lung Res. 37:162–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Desmoulière A, Geinoz A, Gabbiani F and
Gabbiani G: Transforming growth factor-beta 1 induces alpha-smooth
muscle actin expression in granulation tissue myofibroblasts and in
quiescent and growing cultured fibroblasts. J Cell Biol.
122:103–111. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sime PJ and O'Reilly KM: Fibrosis of the
lung and other tissues: New concepts in pathogenesis and treatment.
Clin Immunol. 99:308–319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leask A and Abraham DJ: TGF-beta signaling
and the fibrotic response. FASEB J. 18:816–827. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kulkarni AA, Thatcher TH, Olsen KC,
Maggirwar SB, Phipps RP and Sime PJ: PPAR-γ ligands repress
TGFβ-induced myofibroblast differentiation by targeting the
PI3K/Akt pathway: Implications for therapy of fibrosis. PLoS One.
6:e159092011. View Article : Google Scholar
|
|
24
|
Caraci F, Gili E, Calafiore M, Failla M,
La Rosa C, Crimi N, Sortino MA, Nicoletti F, Copani A and Vancheri
C: TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK
activation in the transition of human lung fibroblasts into
myofibroblasts. Pharmacol Res. 57:274–282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gauldie J, Bonniaud P, Sime P, Ask K and
Kolb M: TGF-beta, Smad3 and the process of progressive fibrosis.
Biochem Soc Trans. 35:661–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li DM and Sun H: TEP1, encoded by a
candidate tumor suppressor locus, is a novel protein tyrosine
phosphatase regulated by transforming growth factor beta. Cancer
Res. 57:2124–2129. 1997.PubMed/NCBI
|
|
27
|
Raghu G, Collard HR, Egan JJ, Martinez FJ,
Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et
al ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis: An
official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis:
Evidence-based guidelines for diagnosis and management. Am J Respir
Crit Care Med. 183:788–824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
29
|
Nathan SD, Basavaraj A, Reichner C,
Shlobin OA, Ahmad S, Kiernan J, Burton N and Barnett SD: Prevalence
and impact of coronary artery disease in idiopathic pulmonary
fibrosis. Respir Med. 104:1035–1041. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nho RS: Current concept for the
pathogenesis of idiopathic pulmonary fibrosis (IPF). Clin Res
Pulmono. 1:1006–1008. 2013.
|
|
31
|
Nho RS, Hergert P, Kahm J, Jessurun J and
Henke C: Pathological alteration of FoxO3a activity promotes
idiopathic pulmonary fibrosis fibroblast proliferation on type I
collagen matrix. Am J Pathol. 179:2420–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ihn H: Pathogenesis of fibrosis: Role of
TGF-beta and CTGF. Curr Opin Rheumatol. 14:681–685. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Conte E, Fruciano M, Fagone E, Gili E,
Caraci F, Iemmolo M, Crimi N and Vancheri C: Inhibition of PI3K
prevents the proliferation and differentiation of human lung
fibroblasts into myofibroblasts: The role of class I P110 isoforms.
PLoS One. 6:e246632011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wilkes MC, Mitchell H, Penheiter SG, Doré
JJ, Suzuki K, Edens M, Sharma DK, Pagano RE and Leof EB:
Transforming growth factor-beta activation of phosphatidylinositol
3-kinase is independent of Smad2 and Smad3 and regulates fibroblast
responses via p21-activated kinase-2. Cancer Res. 65:10431–10440.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gu L, Zhu YJ, Yang X, Guo ZJ, Xu WB and
Tian XL: Effect of TGF-beta/Smad signaling pathway on lung
myofibroblast differentiation. Acta Pharmacol Sin. 28:382–391.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chilosi M, Poletti V, Zamò A, Lestani M,
Montagna L, Piccoli P, Pedron S, Bertaso M, Scarpa A, Murer B, et
al: Aberrant Wnt/beta-catenin pathway activation in idiopathic
pulmonary fibrosis. Am J Pathol. 162:1495–1502. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Amini Nik S, Ebrahim RP, Van Dam K,
Cassiman JJ and Tejpar S: TGF-beta modulates beta-Catenin stability
and signaling in mesenchymal proliferations. Exp Cell Res.
313:2887–2895. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Baarsma HA, Spanjer AI, Haitsma G,
Engelbertink LH, Meurs H, Jonker MR, Timens W, Postma DS, Kerstjens
HA and Gosens R: Activation of WNT/β-catenin signaling in pulmonary
fibroblasts by TGF-β1 is increased in chronic
obstructive pulmonary disease. PLoS One. 6:e254502011. View Article : Google Scholar
|
|
39
|
Pardo A and Selman M: Matrix
metalloproteases in aberrant fibrotic tissue remodeling. Proc Am
Thorac Soc. 3:383–388. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jarman ER, Khambata VS, Yun Ye L, Cheung
K, Thomas M, Duggan N and Jarai G: A translational preclinical
model of interstitial pulmonary fibrosis and pulmonary
hypertension: mechanistic pathways driving disease pathophysiology.
Physiol Rep. 2:e121332014. View Article : Google Scholar : PubMed/NCBI
|















