Introduction
Tumors are complex structures composed of malignant
cancer cells surrounded by the tumor stroma. The cells and
components of the tumor stroma have received increasing attention
due their roles in tumor development, invasion and metastasis
(1), and in the response to cancer
therapy (2,3). The tumor stroma contains various cell
types, including myeloid cell sub-populations, inflammatory cells,
immunocytes, endothelial cells, epithelial cells and fibroblasts
(4,5), which communicate between themselves
and also directly with the cancer cells through cell-cell contacts
and indirectly through paracrine/exocrine signaling, release of
proteases and modulation of the extracellular matrix (ECM).
Together, the stromal cells constitute the complex tumor
microenvironment, which has a key role in tumor development
(1,6). On this basis, identification of
stromal targets for cancer therapeutics is of great interest, and
such strategies may complement therapies directed against cancer
cells. Among the potential targets in the stroma, cancer-associated
fibroblasts (CAFs) have received intensive interest.
It is thought that the fibroblasts in the tumor
stroma acquire a modified phenotype, which can be utilized for
distinguishing them from normal tissue fibroblasts. Such
'activated' fibroblasts have been termed peritumoral fibroblasts,
reactive stromal fibroblasts, myofibroblasts or CAFs (7). The precise origin of CAFs remains to
be elucidated; however, a previous study has identified two
potential pathways: Resident fibroblasts may be converted into CAFs
through stimulation by cytokines, including transforming growth
factor-beta (TGF-β) and stromal cell-derived factor-1 (SDF-1)
(8); furthermore epithelial or
endothelial cells may transform into CAFs via
epithelial-mesenchymal transition (EMT) and
endothelial-to-mesenchymal transition (EndMT), which are also
mediated by cytokines, including fibroblast growth factor (FGF),
osteopontin (9), TGF-β and SDF-1
(10,11).
Numerous studies have provided evidence for the
cancer-promoting role of CAFs. In contrast to resting fibroblasts,
CAFs are characterized by an increased rate of proliferation and
differential expression of ECM components and growth factors,
including TGF-β, vascular endothelial growth factor (VEGF),
platelet-derived growth factor (PDGF) and fibroblast growth factor
2 (FGF-2) (12,13). These cytokines and growth factors
have all been show to have important roles in synchronizing key
events which continuously occur in the tumor microenvironment. For
instance, TGF-β promotes the infiltration of inflammatory/immune
cells and CAFs into the tumor microenvironment, directly leading to
changes in tumor cells (14). CAFs
orchestrate tumor-promoting inflammation in a nuclear factor
(NF)-κB signaling-dependent manner (15). VEGF induces microvascular
permeability, leading to the extravasation of plasma proteins such
as fibrin, which subsequently attracts CAFs, inflammatory immune
cells and endothelial cells, leading to tumor angiogenesis
(16). While CAFs and inflammatory
cells are the principal sources of host-derived VEGF (16), PDGF and FGF-2 also have significant
roles in angiogenesis. Therefore, CAFs are key factors that can
promote tumor growth by inducing angiogenesis, recruiting
inflammatory/immune cells and remodeling the ECM.
Compared to normal fibroblasts, extensive changes in
the expression of genes that encode certain extracellular matrix
proteins and proteases have been observed in CAFs in numerous types
of carcinoma (17–19); among these, fibroblast activation
protein (FAP), a type II membrane-bound serine protease, has
recently gained attention. FAP has been shown to possess dipeptidyl
peptidase- (20) and
collagenase-like (21) activity
in vitro, and has been implicated in ECM remodeling.
However, the in vivo substrates of FAP remain to be
identified. Due to its tightly regulated pattern of expression in
the stroma of malignant solid tumors (22,23),
FAP has been classified as a candidate protein for targeting CAFs.
The present study hypothesized that FAP inhibition may be useful
for cancer therapy. FAP belongs to the post-proline dipeptidyl
aminopeptidase family and has the highest similarity to dipeptidyl
peptidase IV (DPPIV/CD26) (24).
The catalytic sites of CD26/DPP-IV and FAP contain the
characteristic catalytic triad of Ser630/624,
Asp708/702, His740/734 (the residues are
numbered according to human CD26/DPP-IV and FAP, respectively), and
the active serine is situated in a nucleophilic elbow motif within
the sequence Gly-Trp-Ser-Tyr-Gly (13–15)
(25,26). The aminoboronic dipeptide and
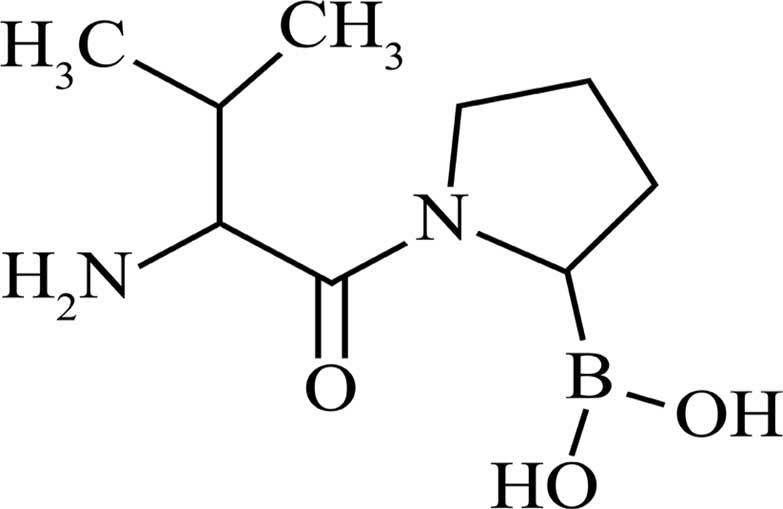
Val-boro-Pro (PT-100; Fig. 1)
appear to be interesting drug candidates for the post-proline
dipeptidyl aminopeptidase family. PT-100 competitively inhibits the
DPP activity of FAP and CD26/DPP-IV, and forms a high-affinity
interaction with the catalytic sites due to the formation of a
complex between Ser630/624 and the boron atom of PT-100
(27).
Oxaliplatin is recognized as one of the standard
drugs for chemotherapy of colorectal cancer in clinical practice;
however, there is a certain risk of drug resistance and tumor
recurrence in patients treated with oxaliplatin (28,29),
and a previous study demonstrated that these events may be
associated with changes in CAFs (30). In order to further provide insight
into the mechanisms by which CAFs contribute to tumor progression
and resistance to chemotherapy, the present study investigated the
combined effects of oxaliplatin and PT-100 in the treatment of CT26
colorectal cancer cell-derived tumors in a murine xeno-graft model,
and observed the combined effects of oxaliplatin and PT-100 on the
tumor microenvironment.
Materials and methods
Animals and cell lines
A total of 40 female BALB⁄c mice (6–8 weeks old and
weighing 20 g) were purchased from Beijing HFK Bioscience Co. Ltd.,
(Beijing, China) and maintained in a under a specific pathogen-free
environment at the State Key Laboratory of Biotherapy (Chengdu,
China) with controlled temperature (20–26°C), humidity (40–70%) and
a 12-h light/dark cycle.
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Sichuan University
(Chengdu, China). CT26, a BALB/c-derived murine colon carcinoma
cell line, was purchased from the American Type Culture Collection
(Manassas, VA, USA). CT26 cells were cultured in RPMI-1640
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) in a humidified atmosphere
containing 5% CO2 at 37°C.
PT-100
PT-100 was purchased from Shanghai Speed Chemical
Co. Ltd., (Shanghai, China). The purity was >97%. The molecular
weight of PT-100 is 246.1 g/mol and its structure is shown in
Fig. 1.
Murine tumor xenograft model
The flanks of the mice were shaved and
subcutaneously inoculated with 1×106 CT26 cells. The
tumor-inoculated mice were divided into four groups: The saline
vehicle group; the oxaliplatin group, which was treated with 5
mg/kg oxaliplatin (Jiangsu Hengrui Medicine Co., Ltd., Lianyungang,
China) three times a week; the PT-100 group, which was treated with
20 µg PT-100 per mouse daily; and a combined oxaliplatin and
PT-100 group. All drugs were dissolved in saline and administered
by intraperitoneal injection. The day of tumor inoculation was
defined as day 0, the treatment began on day 8 and animals were
treated for 14 days. Tumor growth was monitored every 3 days by
measurement of the length (L) and the width (W) of the xenograft
tumors using Vernier calipers (Shanghai Taihai Measuring Tools Co.,
Ltd., Shanghai, China). The tumor volume was calculated using the
following formula: Volume (cm3) = W2×0.5L.
The xenograft experiment was performed twice with 5 mice per group
in each experiment.
Tissue preparation, histology and
immunostaining
At the appropriate time-points, for paraffin
embedding, the tumors were excised and post-fixed in 4%
paraformaldehyde overnight. For cryopreservation, the tumors were
excised and stored in optimum cutting temperature compound (OCT;
Leica Microsystems GmbH, Wetzlar, Germany) at −20°C.
FAP expression was assessed by immunohistochemistry
in the CT26 colon cancer cell-derived xenograft tumors from female
BALB⁄c mice. Tissues were cut into 5-µm sections, incubated
with 3% H2O2 at 4°C for 20 min, washed using
phosphate-buffered saline (PBS) containing 3.74 g 12H2O.
Na2HPO4, 0.44 g
NaH2PO4.2H2O and 7.2 g NaCl in 1l distilled
water (PH 7.4) and incubated with serum at 37°C for 20 min.
Subsequently, the sections were incubated with the following
primary antibodies at 4°C overnight: Polyclonal anti-FAP (1:300;
ab28244) (v/v), monoclonal anti-vimentin (1:500;
ab92547) (v/v) and monoclonal anti-CD31 (1:50;
ab7388; all Abcam, Cambridge, MA, USA) (v/v).
Following washing with PBS, the sections were incubated with a
biotin-streptavidin-horseradish peroxidase (HRP) detection system
(SP-9000; OriGene Technologies, Inc., Beijing, China) at 37°C for 1
h, washed with PBS and subsequently incubated with
3,3′-diaminobenzidine (DAB) for 2 min and washed using PBS. The
HRP-conjugated secondary antibody and the DAB were included in the
detection system (SP-9000; ZSGB-Bio Origene Co, Ltd., Beijing,
China), which was not diluted and was used according to the
manufacturer's instructions. To assess the levels of apoptosis in
CT26 tumors, in situ terminal deoxynucleotidyl
transferase-mediated deoxy-UTP nick end labeling (TUNEL) staining
was performed on the tumor sections using the the Deadend™
Fluorometric TUNEL system (G3250; Promega, Madison, WI, USA)
according to the manufacturer's instructions.
For histological analysis, the paraffin-embedded
tumor tissues were de-paraffinized in xylene, re-hydrated in 100,
95, 85 and then 75% ethanol, immersed in PBS (pH 7.4) and stained
with hematoxylin (H3136-25G) and eosin (E4009-5G; both
Sigma-Aldrich, St. Louis, MO, USA), according to the manufacturer's
instructions. A Leica DM 2500 microscope was used for visualization
(Leica Microsystems GmbH).
Western blot analysis
On day 22, the tumors were excised and proteins were
extracted using radioimmunoprecipitation assay buffer (Beyotime
Institute of Biotechnology, Inc., Haimen, China) supplemented with
protease inhibitor cocktail (100:1; Sigma-Aldrich). Tumor tissues
were homogenized prior to western blot and PCR analysis in liquid
nitrogen using a mortar and pestle. Total protein concentrations in
the supernatant were determined via the Bicinchoninic Acid assay
(P0013B; Beyotime Institute of Biotechnology, Inc.). A total of 30
µg protein was loaded per lane for western blot analysis.
Protein samples were separated by 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). After blocking with 5% skimmed milk in TBS containing 0.1%
Tween 20 (TBST) for 2 h at 37°C, the membranes were incubated with
anti-FAP (1:500; 28244; Abcam) or anti-β-actin (1:1,000; sc-130657;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA) primary polyclonal
antibodies at 4°C overnight. After washing four times with TBST for
10 min, the membranes were incubated with the goat anti-rabbit
HRP-conjugated secondary antibody (1:5,000; sc-2054; Santa Cruz
Biotechnology, Inc.) for 1 h at 37°C. After washing with TBST as
described above, the bands were visualized using enhanced
chemiluminescence reagents (WBKLS0100 EMD Millipore).
Flow cytometry
On day 22, the tumors were excised, dissected into
small pieces using a scalpel, digested in collagenase digestion
buffer (1% collagenase in RPMI-1640; Gibco; Thermo Fisher
Scientific, Inc.) for 2 h at 37°C with agitation and the cells in
the resulting suspension were counted. Cells were passed through a
70 µm cell strainer (BD Biosciences, Franklin Lakes, NJ,
USA) following disaggregation A total of 1×106 cells in
100 µl were stained. The cells were labeled using rat
anti-mouse CD11b-allophycocyanine (1:50; cat no. 553312; BD
Biosciences), hamster anti-mouse CD11c-fluorescein isothiocyanate
(1:100; cat no. 553801; BD Biosciences), or rat anti-mouse
F4/80-A488 (1:50; cat no. MCA497A488; Serotec, Bio-Rad
Laboratories, Inc., Hercules, CA, USA) antibodies on ice for 30
min, followed by analysis by flow cytometry using FACSCalibur or
LSR II flow cytometers (BD Biosciences); data were analyzed using
CellQuest 6.0 software (BD Biosciences).
Reverse-transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the xenograft tumors
using the AxyPrep™ Multisource Total RNA Miniprep kit (Axygen
Biosciences, Union City, CA, USA) in accordance with the
manufacturer's instructions. The PrimeScript™ RT reagent kit with
gDNA Eraser (Takara Bio Inc., Otsu, Japan) was used to synthesize
cDNA, and RT-qPCR analysis was performed using SsoAdvanced™ SYBR
Green Supermix (Bio-Rad Laboratories, Inc.) following the
manufacturer's instructions. Transcript expression was determined
relative to GAPDH. The primers used were purchased from
BGI-Shenzhen (Shenzhen, China) and were as follows: GAPDH,
5′-ACCCAGAAGACTGTGGATGG-3′ (forward) and 5′-TCTAGACGGCAGGTCAGGTC-3′
(reverse); TGF-β, 5′-AAGTGGGTCCATGAACCTAA-3′ (forward) and
5′-GCTACATTTACAAGAC66CAC-3′ (reverse); FGF-2,
5′-GGCTGCTGGCTTCTAAGTGT-3′ (forward) and 5′-CCGTTTTGGATCCGAGTTTA-3′
(reverse); and osteopontin, 5′-TGCACCCAGATCCTATAGCC-3′ (forward)
and 5′-CTCCATCGTCATCATCATC-3′ (reverse). The 20 µl PCR
system contained 2 µl genomic DNA, 2 µl dNTPs, 10
µl buffer, 0.5 µl Rox reference dye and 5 pmol of
each primer. Thermal cycling was performed as follows: Denaturing
at 95°C for 3 min, then 30 cycles of 95°C for 30 sec, 55°C for 30
sec and 72°C for 30 sec, with elongation at 72°C for 10 min using a
CFX96 Real-Time C1000 thermocycler (Bio-Rad Laboratories, Inc.).
PCR products were electrophoresed on 1% agarose gel (800669;
Schwarz/Mann Biotech, Cleveland, OH, USA) at 200 mA until
separation was achieved. DNA fragments were visualized using a long
wave UV light box and images were captured using a Universal Hood
II Gel Doc™ XR camera (1708170) and were analyzed using Image Lab
software (both Bio-Rad Laboratories, Ltd.).
Statistical analysis
SPSS version 11.5 (SPSS, Inc., Chicago, IL, USA) was
used for statistical analyses. Values are expressed as the mean ±
standard error of the mean. One-way analysis of variance was used
to assess statistical significance. Survival curves were compared
using the log-rank test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Oxaliplatin induces accumulation of CAFs
in the tumor microenvironment
Chemotherapy is widely used for the treatment of
colorectal cancer; however, drug resistance has become problematic
due to its increasing frequency. Several mechanisms of drug
resistance of cancers have been proposed (31,32);
however, a consensus has not yet been reached and further
elucidation is required. The present study hypothesized that CAFs
may have an important role in drug resistance of cancers. To test
this hypothesis, Balb/C mice (n=5 per group) were inoculated with
CT26 colon carcinoma cells (day 0) and then treated with the saline
vehicle or oxaliplatin for 14 days (from days 8–22). Subsequently,
the mice were sacrificed via cervical dislocation and the tumor
xenografts were excised, sectioned and stained using an anti-FAP
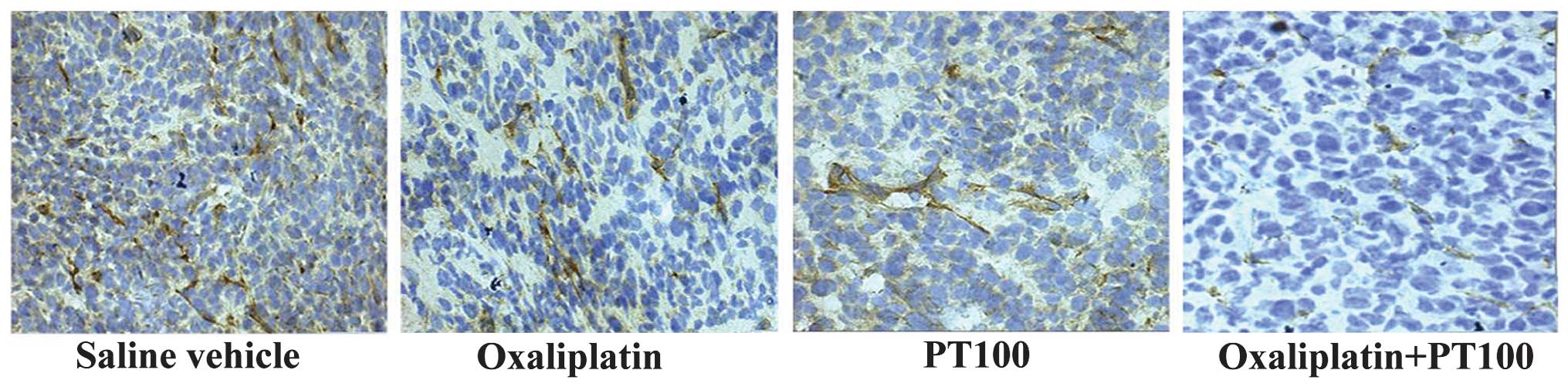
and anti-vimentin antibodies. As shown in Fig. 2, treatment with oxaliplatin
markedly increased the amount of FAP and vimentin, which are
specific markers of CAFs, expressed in the stroma of the tumor
tissues, indicating that oxaliplatin increased the accumulation of
CAFs in the xenograft tumors.
Combined treatment with oxaliplatin and a
pharmacological inhibitor of CAFs reduces tumor growth in vivo
In order to evaluate whether the ability of
oxaliplatin to increase the accumulation of CAFs in the xenograft
tumors was clinically relevant, the present study combined
pharmacological inhibition of CAFs by PT-100 with the
chemotherapeutic drug oxaliplatin, to which CT26 cells are
partially sensitive, in a xenograft model. Mice (n=5 per group)
were subcutaneously injected with CT26 cells on day 0 and then
treated with saline vehicle, PT-100 alone, oxaliplatin alone, or
PT-100 combined with oxaliplatin from days 8–32. As shown in
Fig. 3A, PT-100 combined with
oxaliplatin suppressed tumor growth more significantly than
treatment with either oxaliplatin or PT-100 alone (P<0.05).
PT-100 combined with oxaliplatin also significantly increased the
survival of the mice compared to that in the other three treatment
groups, particularly the mice treated with saline vehicle, which
died after 45–60 days (Fig.
3B).
Consistent with the results shown in Fig. 2, FAP, a specific marker for CAFs,
was most highly expressed in the stroma of the tumors of the mice
treated with oxaliplatin alone, indicating that oxaliplatin
increased the accumulation of CAFs in the xenograft tumors.
However, PT-100 combined with oxaliplatin markedly reduced the
expression of FAP and therefore prevented the accumulation of CAFs
in the stroma of the tumor tissues. In addition, TUNEL staining was
performed on the xenograft tumor sections, which revealed that
PT-100 combined with oxaliplatin increased the number of apoptotic
tumor cells compared with that in the three other treatment groups
(Fig. 3C–F).
Next, to investigate how oxaliplatin induced the
accumulation of CAFs, RT-qPCR analysis was performed to assess the
expression of a number of cytokines which are associated with the
accumulation of CAFs in the xenograft tumor tissues. CAFs are
hypothesized to accumulate in the tumor microenvironment by the
following major mechanisms: Resident fibroblasts transforming into
CAFs, the EMT and the EndMT (8,10,11);
these processes are mediated by several common cytokines, including
TGF-β3, FGF-2 and osteopontin (9,12).
Thus, the present study quantitatively measured the abundance these
cytokines in the xenograft tumor tissues. As shown in Fig. 3G, oxaliplatin increased the
expression of TGF-β3 and FGF-2 (basic FGF) by 1.79-
and 2.63-fold, respectively, compared to those in the tumors of the
saline vehicle-treated animals. Furthermore, the expression of
TGF-β3 and FGF-2 in the tumors of animals treated
with PT-100 alone or with PT-100 combined with oxaliplatin was
lower than that in the saline vehicle-treated animals. Although
oxaliplatin treatment did not significantly increase the expression
of osteopontin mRNA, its expression was significantly decreased in
the tumors of the animals treated with PT-100 only or with PT-100
combined with oxaliplatin (0.28- and 0.16-fold, respectively, of
that in the saline vehicle-treated group). These results indicated
that pharmacological inhibition of CAFs using PT-100 reduced the
tumor expression levels of cytokines known to promote the
accumulation of CAFs.
Combed treatment with oxaliplatin and a
pharmacological inhibitor of CAFs reduces the recruitment of
tumor-associated macrophages (TAMs) and dendritic cells
The tumor micro-environment can affect the malignant
potential of the tumor; tumor-associated macrophages and dendritic
cells have an important role in this process (33,34).
Macrophages constitute an extremely heterogeneous population,
including M2 (or alternatively activated) macrophages, which are
now generally accepted to be TAMs. TAMs mostly exert pro-tumor
functions by promoting tumor-cell survival, proliferation and
dissemination. Tumor-associated dendritic cells (DCs) are another
immune-regulatory cell population. A variety of sub-populations of
tumor-associated DCs are known, among which CD11c+ DCs are able to
promote tumorigenesis (35). In
order to test whether CAFs interact with M2 macrophages or immature
dendritic cells (imDCs) and tumor-infiltrating DCs (TIDCs), tumor
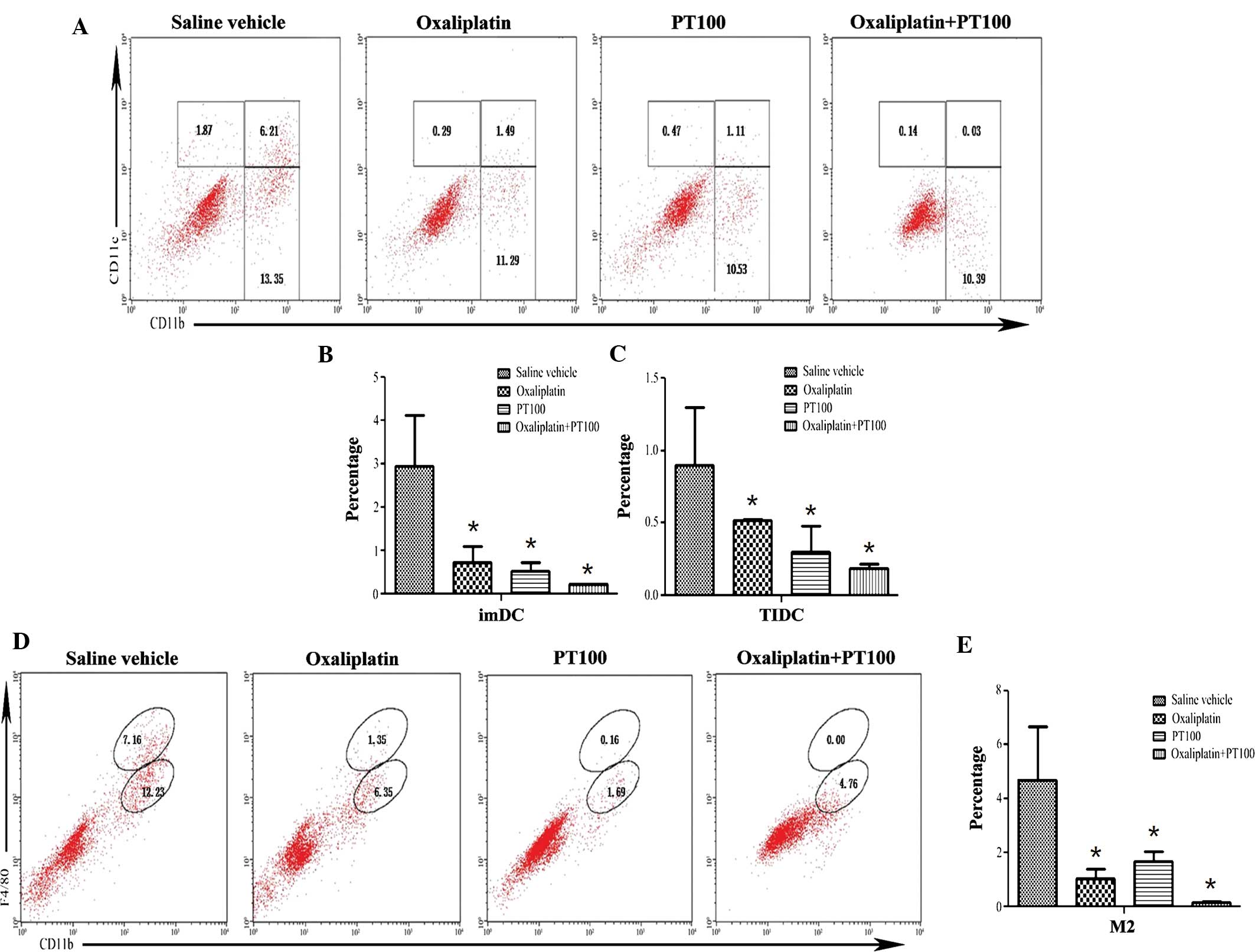
cells from mice at day 22 were analyzed by flow cytometry. As shown
in Fig. 4, the number of M2
macrophages (F4/80+) was highest in the saline vehicle-treated
group and lowest in the xenograft tumors of animals treated with
PT-100 combined with oxaliplatin; furthermore, tumors of animals
treated with PT-100 combined with oxaliplatin contained
significantly lower amounts of M2 macrophages compared with those
in the groups treated with saline vehicle, oxaliplatin alone and
PT-100 alone (P<0.01). In addition, the number of imDCs and
TIDCs was highest in the saline vehicle-treated tumors, while the
tumors of animals treated with PT-100 combined with oxaliplatin
contained a significantly lower number of imDCs and TIDCs, as
compared with the groups treated with saline vehicle, oxaliplatin
only and PT-100 only (P<0.05). These results indicated that
pharmacological inhibition of CAFs using PT-100 in combination with
oxaliplatin significantly reduced the recruitment of M2 macrophages
and CD11c+ DCs to the xenograft tumors.
Combined treatment of oxaliplatin with a
pharmacological inhibitor of CAFs reduces tumor angiogenesis
Angiogenesis, a key event required for tumor
progression, is dependent on ECM remodeling (1). As an important source of ECM
components, CAFs are able to promote angiogenesis during tumor
growth and metastasis (1,5). In order to test whether
pharmacological inhibition of CAFs had any effect on angiogenesis
in the tumor xenografts, immunohistochemical analysis was used to
determine the density of CD31+ endothelial cells in the xenograft
tumors at day 22. The tumors treated with PT-100 combined with
oxaliplatin had a markedly lower density of CD31+ cells compared to
that in the groups treated with saline vehicle, oxaliplatin alone
and PT-100 alone (Fig. 5). These
results indicated that pharmacological inhibition of CAFs using
PT-100 in combination with oxaliplatin significantly reduced tumor
angiogenesis.
Discussion
Chemotherapy has been a basic, vital and widespread
means of treating colorectal cancer for numerous years; however,
chemoresistance has become a significant obstacle. A number of
studies have investigated the mechanisms of drug resistance to
chemotherapeutic agents; it is mainly attributed to gene mutations,
gene amplification, epigenetic changes that influence the uptake,
metabolism or export of drugs from single cells (36,37),
or alterations to the signaling pathways which protect tumor stem
cells (31). The tumor
microenvironment has become a research hot spot, as it may be
involved in the resistance of solid tumors to chemotherapy
(38).
The present study demonstrated that the
chemotherapeutic drug oxaliplatin increased the accumulation of
CAFs in colon cancer xenograft tumor tissues, which may, to a
certain extent, be responsible for drug resistance. The mechanisms
by which oxaliplatin increased the accumulation of CAFs may be due
to the upregulation of cytokines associated with the accumulation
of CAFs. Studies have demonstrated that chemotherapy can increase
hypoxia in the tumor microenvironment, and that such hypoxic
conditions can induce a molecular response that drives the
activation of a key transcription factor, namely the
hypoxia-inducible factor (HIF). HIF regulates a variety of genes
encoding cytokines that influence the growth, progression and
metastasis of tumors (39,40). In the present study oxaliplatin may
possibly have induced hypoxia in the tumors, which may explain for
the upregulation of TGF-β3, FGF-2 and osteopontin observed in the
xenograft tumors. TGF-β is a multifunctional cytokine that
regulates tissue morphogenesis and differentiation by influencing
cell proliferation, differentiation, apoptosis and ECM production
(8,11). FGF-2, a prototypical pro-angiogenic
factor, regulates a variety of important intracellular
signal-transduction pathways, which mediate a series of cellular
and molecular changes associated with the EMT (9). Osteopontin is a soluble ECM protein
with known effector functions in tumor growth, angiogenesis and
metastasis (12). All of these
cytokines have been associated with the accumulation of CAFs
(8,9). As a result of the increased
accumulation of CAFs, the secretion of these cytokines is elevated
and eventually accelerates tumor growth (10–12).
Although the importance of CAFs in tumor progression
has been recognized (41), the
understanding of the mechanisms by which CAFs influence
tumorigenesis remains limited. One generally accepted hypothesis is
that an increase in the accumulation of CAFs, a vital component of
the structure of the tumor microenvironment, may increase fibrosis
in the tumor microenvironment, which may reduce drug uptake by the
tumor cells (30). However, the
present study suggested that CAFs induce the recruitment of
inflammatory immune cells, which may facilitate tumor growth and
progression. Tumor-associated macrophages and dendritic cells have
been reported to produce matrix remodeling enzymes, reactive oxygen
species and other bioactive molecules that influence cancer-cell
proliferation, angiogenesis, invasion and metastasis (35,42).
In the present study, pharmacological inhibition of CAFs markedly
suppressed the recruitment of pro-tumorigenic immune cells in
vivo, which indicated that CAFs are able to influence the vital
signaling pathways associated with the recruitment of
pro-tumorigenic immune cells. It has been reported that CAFs
orchestrate signaling processes associated with tumor-promoting
inflammation in incipient neoplasia via a NF-κB signaling-dependent
mechanism (15), while CAFs also
modulated the tumor immune microenvironment in a 4T1 murine breast
cancer model (43). In addition,
as mentioned above, hypoxic tissues, in which the accumulation of
CAFs is enhanced, secrete cytokines that undermine normal
anti-tumor surveillance by macrophages, turning the macrophages
into accomplices and facilitators of invasion and angiogenesis
(44). However, in contrast to
previous studies on tumor-associated immune cells (30,43),
no significant differences between the number of CD4+/CD8+ T cells
were identified among the four experimental groups of the present
study. Therefore, the role of CAFs as tumor immune modulators in
tumorigenesis requires further study.
Apart from recruiting inflammatory immune cells, the
pro-tumor function of CAFs may also be associated with the
induction of angiogenesis within the tumor stroma. Angiogenesis,
the formation of new blood vessels, is required for malignant tumor
growth and metastasis (45). The
findings of the present study demonstrated that pharmacological
inhibition of CAFs simultaneously suppressed angiogenesis in the
tumor xenografts. CAFs may affect angiogenesis at several levels.
CAFs secret growth factors, includings VEGF and FGF-2, which are
known to be important for endothelial cell migration (13). Moreover, CAFs are capable of
remodeling connective tissue, and interact with epithelial cells
and other cell types in connective tissues, which may promote
angiogenesis (46). In addition,
inflammatory immune cells may also affect tumor angiogenesis
(15,42). The present study demonstrated that
pharmacological inhibition of CAFs reduced the recruitment of
inflammatory immune cells, which indicated that CAFs may promote
tumor angiogenesis by recruiting inflammatory immune cells. It has
recently been reported that vascular endothelial cells and CAFs
express similar genes (47). Thus,
it is necessary to study the similarities and differences between
CAFs and tumor endothelial cells in order to accurately guide the
clinical application of anti-angiogenic agents.
In conclusion, the present study demonstrated that
chemotherapy with oxaliplatin increased the accumulation of CAFs in
the stroma of colon tumors in vivo, whereas pharmacological
inhibition of CAFs alongside oxaliplatin treatment reduced the
accumulation of CAFs, enhanced the response to oxaliplatin
chemotherapy as well as reduced the expression of cytokines
associated with the accumulation of CAFs, the recruitment of
pro-tumorigenic immune cells and angiogenesis. Chemotherapy
combined with treatments which inhibit the recruitment, formation
or activity of CAFs may represent a novel method of improving the
tumor response to chemotherapy and help to overcome resistance to
chemotherapeutic agents.
Acknowledgments
The present study was supported by grants from the
National Science Foundation of China (grant nos. 81123003 and
81201787).
References
|
1
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: Regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck
M, Sung M, Schwartz M, Divino CM, Pan PY and Chen SH: The novel
role of tyrosine kinase inhibitor in the reversal of immune
suppression and modulation of tumor microenvironment for
immune-based cancer therapies. Cancer Res. 69:2514–2522. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Swartz MA, Lida N, Roberts EW, Sangaletti
S, Wong MH, Yull FE, Coussens LM and DeClerck YA: Tumor
microenvironment complexity: Emerging roles in cancer therapy.
Cancer Res. 72:2473–2480. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Albini A and Sporn MB: The tumour
microenvironment as a target for chemoprevention. Nat Rev Cancer.
7:139–147. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zi F, He J, He D, Li Y, Yang L and Cai Z:
Fibroblast activation protein α in tumor microenvironment: recent
progression and implications (review). Mol Med Rep. 11:3203–3211.
2015.PubMed/NCBI
|
|
6
|
Whiteside TL: The tumor microenvironment
and its role in promoting tumor growth. Oncogene. 27:5904–5912.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mueller MM and Fusenig NE: Friends or foes
- bipolar effects of the tumour stroma in cancer. Nat Rev Cancer.
4:839–849. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kojima Y, Acar A, Eaton EN, Mellody KT,
Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg
RA and Orimo A: Autocrine TGF-beta and stromal cell-derived
factor-1(SDF-1) signaling drives the evolution of tumor-promoting
mammary stromal myofibroblasts. Proc Natl Acad Sci USA.
107:20009–20014. 2010. View Article : Google Scholar
|
|
9
|
Billottet C, Tuefferd M, Gentien D,
Rapinat A, Thiery JP, Broët P and Jouanneau J: Modulation of
several waves of gene expression during FGF-1 induced
epithelial-mesenchymal transition of carcinoma cells. J Cell
Biochem. 104:826–839. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Potenta S, Zeisberg E and Kalluri R: The
role of endothelial-to-mesenchymal transition in cancer
progression. Br J Cancer. 99:1375–1379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J, Lamouille S and Derynck R:
TGF-beta-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anderberg C, Li H, Fredriksson L, Andrae
J, Betsholtz C, Li X, Eriksson U and Pietras K: Paracrine signaling
by platelet-derived growth factor-CC promotes tumor growth by
recruitment of cancer-associated fibroblasts. Cancer Res.
69:369–378. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hughes CC: Endothelial-stromal
interactions in angiogenesis. Curr Opin Hematol. 15:204–209. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang L, Pang Y and Moses HL: TGF-beta and
immune cells: An important regulatory axis in the tumor
microenvironment and progression. Trends Immunol. 31:220–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Erez N, Truitt M, Olson P, Arron ST and
Hanahan D: Cancer-Associated Fibroblasts Are Activated in Incipient
Neoplasia to Orchestrate Tumor-Promoting Inflammation in an
NF-kappaB-Dependent Manner. Cancer Cell. 17:135–147. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fukumura D, Xavier R, Sugiura T, Chen Y,
Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK and Seed B:
Tumor induction of VEGF promoter activity in stromal cells. Cell.
94:715–725. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Infante JR, Matsubayashi H, Sato N,
Tonascia J, Klein AP, Riall TA, Yeo C, Iacobuzio-Donahue C and
Goggins M: Peritumoral fibroblast SPARC expression and patient
outcome with resectable pancreatic adenocarcinoma. J Clin Oncol.
25:319–325. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Micke P, Kappert K, Ohshima M, Sundquist
C, Scheidl S, Lindahl P, Heldin CH, Botling J, Ponten F and Ostman
A: In situ identification of genes regulated specifically in
fibroblasts of human basal cell carcinoma. J Invest Dermatol.
127:1516–1523. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsujino T, Seshimo I, Yamamoto H, Ngan CY,
Ezumi K, Takemasa I, Ikeda M, Sekimoto M, Matsuura N and Monden M:
Stromal myofibroblasts predict disease recurrence for colorectal
cancer. Clin Cancer Res. 13:2082–2090. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aertgeerts K, Levin I, Shi L, Snell GP,
Jennings A, Prasad GS, Zhang Y, Kraus ML, Salakian S, Sridhar V, et
al: Structural and kinetic analysis of the substrate specificity of
human fibroblast activation protein alpha. J Biol Chem.
280:19441–19444. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aggarwal S, Brennen WN, Kole TP, Schneider
E, Topaloglu O, Yates M, Cotter RJ and Denmeade SR: Fibroblast
activation protein peptide substrates identified from human
collagen I derived gelatin cleavage sites. Biochemistry.
47:1076–1086. 2008. View Article : Google Scholar
|
|
22
|
Edosada CY, Quan C, Wiesmann C, Tran T,
Sutherlin D, Reynolds M, Elliott JM, Raab H, Fairbrother W and Wolf
BB: Selective inhibition of fibroblast activation protein protease
based on dipeptide substrate specificity. J Biol Chem.
281:7437–7444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ostermann E, Garin-Chesa P, Heider KH,
Kalat M, Lamche H, Puri C, Kerjaschki D, Rettig WJ and Adolf GR:
Effective immunoconjugate therapy in cancer models targeting a
serine protease of tumor fibroblasts. Clin Cancer Res.
14:4584–4592. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niedermeyer J, Enenkel B, Park JE, Lenter
M, Rettig WJ, Damm K and Schnapp A: Mouse fibroblast-activation
protein: Conserved Fap gene organization and biochemical function
as a serine protease. Eur J Biochem. 254:650–654. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cheng JD, Dunbrack RL Jr, Valianou M,
Rogatko A, Alpaugh RK and Weiner LM: Promotion of tumor growth by
murine fibroblast activation protein, a serine protease in an
animal model. Cancer Res. 62:4767–4772. 2002.PubMed/NCBI
|
|
26
|
Rasmussen HB, Branner S, Wiberg FC and
Wagtmann N: Crystal structure of human dipeptidyl peptidase IV/CD26
in complex with a substrate analog. Nat Struct Biol. 10:19–25.
2003. View
Article : Google Scholar
|
|
27
|
Jones B, Adams S, Miller GT, Jesson MI,
Watanabe T and Wallner BP: Hematopoietic stimulation by a
dipeptidyl peptidase inhibitor reveal a novel regulatory mechanism
and therapeutic treatment for blood cell deficiencies. Blood.
102:1641–1648. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Diaz-Rubio E, Sastre J, Zaniboni A,
Labianca R, Cortés-Funes H, de Braud F, Boni C, Benavides M,
Dallavalle G and Homerin M: Oxaliplatin as single agent in
previously untreated colorectal carcinoma patients: A phase II
multicentric study. Ann Oncol. 9:105–108. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Raymond E, Chaney SG, Taamma A and
Cvitkovic E: Oxaliplatin: A review of preclinical and clinical
studies. Ann Oncol. 9:1053–1071. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loeffler M, Krüger JA, Niethammer AG and
Reisfeld RA: Targeting tumor-associated fibroblasts improves cancer
chemotherapy by increasing intratumoral drug uptake. J Clin Invest.
116:1955–1962. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dean M: ABC transporters, drug resistance,
and cancer stem cells. J Mammary Gland Biol Neoplasia. 14:3–9.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Redmond KM, Wilson TR, Johnston PG and
Longley DB: Resistance mechanisms to cancer chemotherapy. Front
Biosci. 13:5138–5154. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma Y, Shurin GV, Gutkin DW and Shurin MR:
Tumor associated regulatory dendritic cells. Semin Cancer Biol.
22:298–306. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cubillos-Ruiz JR, Engle X, Scarlett UK,
Martinez D, Barber A, Elgueta R, Wang L, Nesbeth Y, Durant Y and
Gewirtz AT: Polyethylenimine-based siRNA nanocomplexes reprogram
tumor-associated dendritic cells via TLR5 to elicit therapeutic
antitumor immunity. J Clin Invest. 119:2231–2244. 2009.PubMed/NCBI
|
|
36
|
Perugorria MJ, Castillo J, Latasa MU, Goñi
S, Segura V, Sangro B, Prieto J, Avila MA and Berasain C: Wilms'
tumor 1 gene expression in hepatocellular carcinoma promotes cell
dedifferentiation and resistance to chemotherapy. Cancer Res.
69:1358–1367. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Plumb JA, Strathdee G, Sludden J, Kaye SB
and Brown R: Reversal of drug resistance in human tumor xenografts
by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene
promoter. Cancer Res. 60:6039–6044. 2000.PubMed/NCBI
|
|
38
|
Trédan O, Galmarini CM, Patel K and
Tannock IF: Drug resistance and the solid tumor microenvironment. J
Natl Cancer Inst. 99:1441–1454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brahimi-Horn MC, Chiche J and Pouysségur
J: Hypoxia and cancer. J Mol Med (Berl). 85:1301–1307. 2007.
View Article : Google Scholar
|
|
40
|
Pouysségur J, Dayan F and Mazure NM:
Hypoxia signalling in cancer and approaches to enforce tumour
regression. Nature. 441:437–443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Condeelis J and Pollard JW: Macrophages:
Obligate partners for tumor cell migration, invasion and
metastasis. Cell. 124:263–266. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liao D, Luo Y, Markowitz D, Xiang R and
Reisfeld RA: Cancer associated fibroblasts promote tumor growth and
metastasis by modulating the tumor immune microenvironment in a 4T1
murine breast cancer mode. PLoS One. 4:e79652009. View Article : Google Scholar
|
|
44
|
DeClerck K and Elble RC: The role of
hypoxia and acidosis in promoting metastasis and resistance to
chemotherapy. Front Biosci (Landmark Ed). 15:213–225. 2010.
View Article : Google Scholar
|
|
45
|
Saharinen P, Eklund L, Pulkki K, Bono P
and Alitalo K: VEGF and angiopoietin signaling in tumor
angiogenesis and metastasis. Trends Mol Med. 17:347–362. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Desmoulière A, Guyot C and Gabbiani G: The
stroma reaction myofibroblast: A key player in the control of tumor
cell behavior. Int J Dev Biol. 48:509–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeisberg EM, Potenta S, Xie L, Zeisberg M
and Kalluri R: Discovery of endothelial to mesenchymal transition
as a source for carcinoma-associated fibroblasts. Cancer Res.
67:10123–10128. 2007. View Article : Google Scholar : PubMed/NCBI
|