Introduction
Although stroke is one of the leading contributors
to mortality and long-term disability rates among adults around the
world, innovative neuroprotective treatments for therapy following
a stroke require further investigation. An area of investigation,
which is gaining increasing attention is the use of herbal
medicines or their extracts for treating stroke. Ilexonin A, a
3β-succinyl-18-dehydro-disodic ursolate (Fig. 1), is a compound extracted from the
herb, Ilex pubescens, which shows good therapeutic effects
when used for the treatment of coronary artery disease, angina and
vasculitis (1,3). It has been confirmed that the
therapeutic effects of Ilexonin A are achieved through improving
blood circulation via its anti-thrombotic and anti-inflammatory
activities (4). As early as 1985,
Luo et al found that dysfunction induced by cerebral
ischemia was ameliorated following treatment with Ilexonin A.
Evidence obtained in subsequent studies has suggested that Ilexonin
A, which can reduce infarct volume and improve neurological
deficit, has a neuroprotective potential following cerebral
ischemia/reperfusion injury, the effect of which is partly
attributed to enhancement of the secretion of neurotrophic factors,
mitigating cerebral edema and promoting neural regeneration
(5,6).
Astrocytes and microglia cells reside in the central
nervous system (CNS) as ubiquitously distributed quiescent cell
populations, which are able to in change morphology, number and
function when activated by ischemic conditions (7,8). It
has been suggested that reactive astrocytes and microglia are
multifunctional cells, which, depending on the microenvironment,
can be either beneficial or detrimental following
ischemia/reperfusion (9,10). Previous reports have suggested that
reactive astrocytes and microglia can promote tissue integrity,
seal off injured tissue and restrict neuronal death through cell
proliferation (11), phagocytosis
of cellular debris and the secretion of neurotrophic factors
(11,12). However, they can also mediate brain
edema and inflammation by producing neurotoxic substances and
pro-inflammatory cytokines following ischemic injury (13,14).
The present study aimed to investigate the characteristics of
activated astrocytes and microglia following the post-ischemia
administration of Ilexonin A.
It has been suggested that angiogenesis and
neurogenesis occurring in the peri-infarct region are associated
with long-term recovery (15,16);
and the two processes appear to be governed by ischemia-induced
growth factors. One of the most important neurotrophic and
angiogenic factors found during stroke recovery, which is
fundamental for adult angiogenesis, is vascular endothelial growth
factor (VEGF) (17). A previous
study suggested that VEGF mediates neuroprotection and promotes
neural regeneration when it is bound by its specific receptor,
fetal liver kinase 1 (Flk-1) (18). Endogenous neurogenesis and
neovascularization are substantially activated and occur in close
proximity to the ipsilateral neocortex of the ischemic brain
(15).
The present study was performed in order to explore
the underlying protective mechanisms of Ilexonin A in rats, and to
provide a reliable scientific basis for treating ischemic
cerebrovascular disease with Ilexonin A. The current study
investigated the effects of Ilexonin A on the expression levels of
VEGF, Flk-1 and Nestin in the peri-infarct region following
transient focal cerebral ischemia.
Materials and methods
Reagents
All reagents were purchased from Beijing Zhongshan
Biotechnology Co., Ltd. (Beijing, China), unless otherwise stated.
Ilexonin A was supplied by Guangdong Boro Pioneer Pharmaceutical
Group Co., Ltd. (Guangdong, China; approval no. Z44023366).
Animal treatment and administration
A total of 108 male Sprague-Dawley rats (Grade II)
weighing 250±10 g, aged 6–8 weeks, were supplied by Shanghai SLAC
Laboratory Animal Co. Ltd (Shanghai, China; certificate no.
2007-0005). The rats were housed in temperature (22-25°C) and
humidity (40–70%) controlled conditions with a 12 h light/dark
cycle and ad libitum access to food and water. The study was
approved by the ethics committee of The Affiliated Union Hospital
of Fujian Medican University (Fuzhou, China). The rats were divided
into six groups for investigation: Normal group; Sham group,
Ischemia group, and Ilexonin A groups receiving 20, 40, and 80
mg/kg, respectively. Each group was divided into four subgroups
(n=6), based on the onset of reperfusion, set at 1, 3, 7 or 14 days
following ischemia. The normal control rats were injected with
equal volumes of saline.
Focal cerebral ischemia/reperfusion
procedure
Focal cerebral ischemia/reperfusion injury was
induced by middle cerebral artery occlusion (MCAO), based on the
method developed by Longa et al (19). The rats were anesthetized with an
intraperitoneal injection of 10% chloral hydrate (300 mg/kg;
Tianjin Kemiou Chemical Reagent Co., Ltd., Tianjin, China). The
left common carotid artery, external carotid artery (ECA) and
internal carotid artery (ICA) were isolated via blunt dissection
following a ventral midline incision of the neck. The branches of
the ECA were cut off, and the whole of the ECA was ligated and
dissociated at the distal end by electric coagulation. A
monofilament nylon suture (0.26 mm; Beijing Zhongshan Biotechnology
Co., Ltd.), with its tip rounded by paraffin (Beijing Donglin
Changsheng Biotechnology Co., Ltd., Beijing, China), was inserted
18±0.5 mm into the ICA from the ECA, to occlude the origin of the
MCA. The suture was fixed and the incision was closed. Following 2
h of ischemia, the nylon suture was withdrawn to allow reperfusion.
The rats in the normal group did not undergo surgery. The
sham-operated rats underwent identical surgery, with the exception
that the nylon suture was not inserted.
Inclusion criteria of MCAO and
neurological severity scoring of animals
Following recovery of the rats from anesthesia,
neurological severity was evaluated for the determination of MCAO
using a five point score, according to Longa's method (19): 0, no deficit; 1, failure to extend
right forelimb fully; 2, circling to the right; 3, falling down to
the right; 4 no spontaneous walking with a depressed level of
consciousness. Achievement of MCAO was confirmed using a score of
1–3 points. The rats were re-evaluated using the same method prior
to sacrifice by anaesthetic overdose using 10% chloral hydrate.
Tetrazolium chloride staining
The rats were anesthetized and received cardiac
perfusion with 100 ml saline, followed by 200 ml 4%
paraformaldehyde (Beijing Donglin Changsheng Biotechnology Co..,
Ltd.) in 0.1 M phosphate buffer (pH 7.4). The brains were carefully
removed when the liver and extremities turned white in color. The
brains were frozen at -20°C for 20 min, and then cut from the
anterior pole into five coronal slices of 2 mm thickness. The
slices were stained with 2% 2, 3, 5-triphenyltetrazolium chloride
solution (Sigma-Aldrich, St. Louis, MO, USA) in the dark at 37°C in
an incubator for 30 min, and turned over every 5 min. A 10%
buffered-formalin solution (Beijing Donglin Changsheng
Biotechnology Co.., Ltd.) was used for fixation (24 h) prior to
imaging with a digital camera (VPC-SH1; Sanyo Electric Co., Ltd.,
Osaka, Japan). The normal brain tissue was stained red, whereas the
ischemic area remained unstained.
Immunohistochemical examination
The rats were anesthetized and perfused in the same
manner as that described above. The brains were removed and
post-fixed for 24 h in the same fixative solution, and dehydrated
in a sucrose gradient (15, 20 and 30%) in 0.1 M phosphate buffer
until they sank to the bottom. Subsequently, 8 µm sections
were cut using a cryomicrotome (Microm HM525; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for use in immunohistochemical
probing. Immunohistochemistry (IHC) was performed in strict
accordance with the datasheet of the Elivision kit (Fuzhou Maixin
Biotechnology Development Co., Ltd., Fuzhou, China), with rabbit
polyclonal anti-glial fibrillary acidic protein (GFAP) antibody
(1:5,000; ab7260; Abcam, Cambridge, UK) to identify astrocytes,
goat polyclonal anti-ionized calcium-binding adapter molecule-1
(Iba-1) antibody (1:100; ab5076; Abcam) to identify microglia, and
mouse monoclonal anti-Nestin antibody (1:100; sc-33677; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) to identify neural progenitor
cells. In the negative controls, 0.01 M PBS was substituted for the
primary antibody. The results were visualized using a 3,
3′-diaminobenzidine kit (Beijing Zhongshan Golden Bridge
Biotechnology Co., Ltd
Beijing, China). A microscope (CX40; Olympus
Corporation, Tokyo, Japan) was used for image acquisition. The
positive cells in each section were evaluated using Image Pro Plus
5.0 (Media Cybernetics, Inc., Rockville, MD, USA) in five high
power microscopic fields (CX40; Olympus Corporation, Tokyo, Japan;
magnification, ×400).
Western blot analysis
The rats were infused with 100–200 ml saline
following anesthesia and the brains were removed. The peri-infarct
region was disassociated and lysed in 10 µl/µg
radioimmunoprecipitation assay lysis buffer with 0.01 M
phenylmethanesulfonylfluoride (both from Beyotime Institute of
Biotechnology, Haimen, China). The samples were homogenized by
sonication and centrifuged at 14,000 × g at 4°C for five min,
following which the supernatant was collected. A bicinchoninic acid
protein assay kit (P0010S; Beyotime Institute of Biotechnology) was
used to quantify protein concentrations. The protein samples were
boiled for 5 min, and 20 µg was loaded onto SDS-PAGE gels
(12% for Iba-1, GFAP and VEGF; 6% for Nestin and Flk-1). Following
electrophoresis, the proteins were electrically transferred onto a
nitrocellulose membrane (pore size, 0.45 µm; EMD Millipore,
Billerica, MA, USA). The membranes were blocked with 5% nonfat milk
in Tris-buffered saline with tween (TBST) containing Tris-Hcl (0.1
M; pH 7.5,) 0.9% NaCl and 0.05% Tween-20, at room temperature for 1
h on a shaking table. The membranes were subsequently incubated
with the following primary antibodies in TBST at 4°C overnight:
Rabbit polyclonal anti-GFAP, (1:20,000; ab7260), goat poly-clonal
anti-Iba-1 (1:200; ab5076; both purchased from Abcam), mouse
monoclonal anti-Nestin (1:300), mouse monoclonal anti-VEGF (1:200;
sc-7269), mouse monoclonal anti-Flk-1 (1:200; sc-6251) and mouse
monoclonal anti-β-actin (1:2,000; sc-47778) (all obtained from
Santa Cruz Biotechnology, Inc.). Following being washed with TBST,
the membranes were incubated with secondary antibodies (1:6,000;
Beijing Zhongshan Golden Bridge Biotechnology, Co., Ltd.) at room
temperature for 2 h and washed with TBST. The immunoblots were
visualized using enhanced chemiluminescence detection reagents
(KPL, Gaithersburg, MD, USA). Densitometry was performed using
ImageMaster® VDS gel imaging and analysis system
(AlphaImager 2200; Alpha Innotech Corporation Company, San Jose,
CA, USA), with β-actin as the loading control.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using one-way analysis of variance
using the SPSS 17.0 Software package (SPSS, Inc., Chicago, IL,
USA). The differences between the groups were analyzed using the
least significant difference test for homogeneity of variance and
the Games-Howell test for heterogeneity of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
Tetrazolium chloride staining
The induction of ischemic infarction by MCAO
involves the striatum and cortex in shorter time spans and other
regions at later time points (20). In the present study, brain atrophy
induced by tissue degeneration and necrosis was observed in the
ischemic core. Following treatment with Ilexonin A, the infarct
volume was decreased, particularly at 7 days, in the Ilexonin A 40
mg/kg group (Fig. 2).
Effect of Ilexonin A on
neurological deficits
Neurological symptoms, including unconsciousness,
ptosis and tonic seizures, were exhibited on day 1 in the rats
subjected to MCAO, with or without administration of Ilexonin A.
With prolongation of reperfusion, the neurological deficits
gradually disappeared, and rats treated with Ilexonin A recovered
at a faster rate, compared with those in the ischemia group
(Table I).
 | Table INeurological severity scores in each
group. |
Table I
Neurological severity scores in each
group.
| Group | 1 day
post-MCAO | 3 days post-
MCAO | 7 days
post-MCAO | 14 days
post-MCAO |
|---|
| Normal | 0 | 0 | 0 | 0 |
| Sham | 0 | 0.17±0.41 | 0 | 0 |
| Ischemia | 2.67±0.52 | 2.33±0.52 | 2.00±0.63 | 1.50±0.55a |
| IA20 mg/kg | 2.33±0.52 | 1.67±0.52a | 1.33±0.52a | 1.17±0.75 |
| IA40 mg/kg | 2.00±0.63 | 1.50±0.55 | 1.00±0.63a | 0.67±0.52a |
| IA80 mg/kg | 2.16±0.41 | 1.67±0.52a | 1.33±0.52a | 0.83±0.41a |
Effects of Ilexonin A on astrocyte
activation
In the IHC rat brain sections, GFAP-positive cells
were observed sporadically, primarily in the cortex, in the normal
and sham groups, whereas their numbers were markedly increased in
the peri-infarct region following ischemia/reperfusion (Fig. 3A). Over time, the GFAP-positive
cells in the ischemia group became markedly activated, with a
marked change in morphology, which included swelling of the soma,
darkening of the cytoplasm and growth and thickening of cell
processes, which even interlaced with each other, like a network,
at day 14. Compared with the ischemia group, an increase in
GFAP-immunoreactivity was observed from day 1 following
ischemia/reperfusion in the Ilexonin A groups, persisting until day
7 (Fig. 3B). Of note, the
GFAP-immunoreactivity decreased per contra 14 days following
ischemia/reperfusion in all Ilexonin A groups, and the morphology
was altered less, particularly in the 40 mg/kg dose Ilexonin A
group.
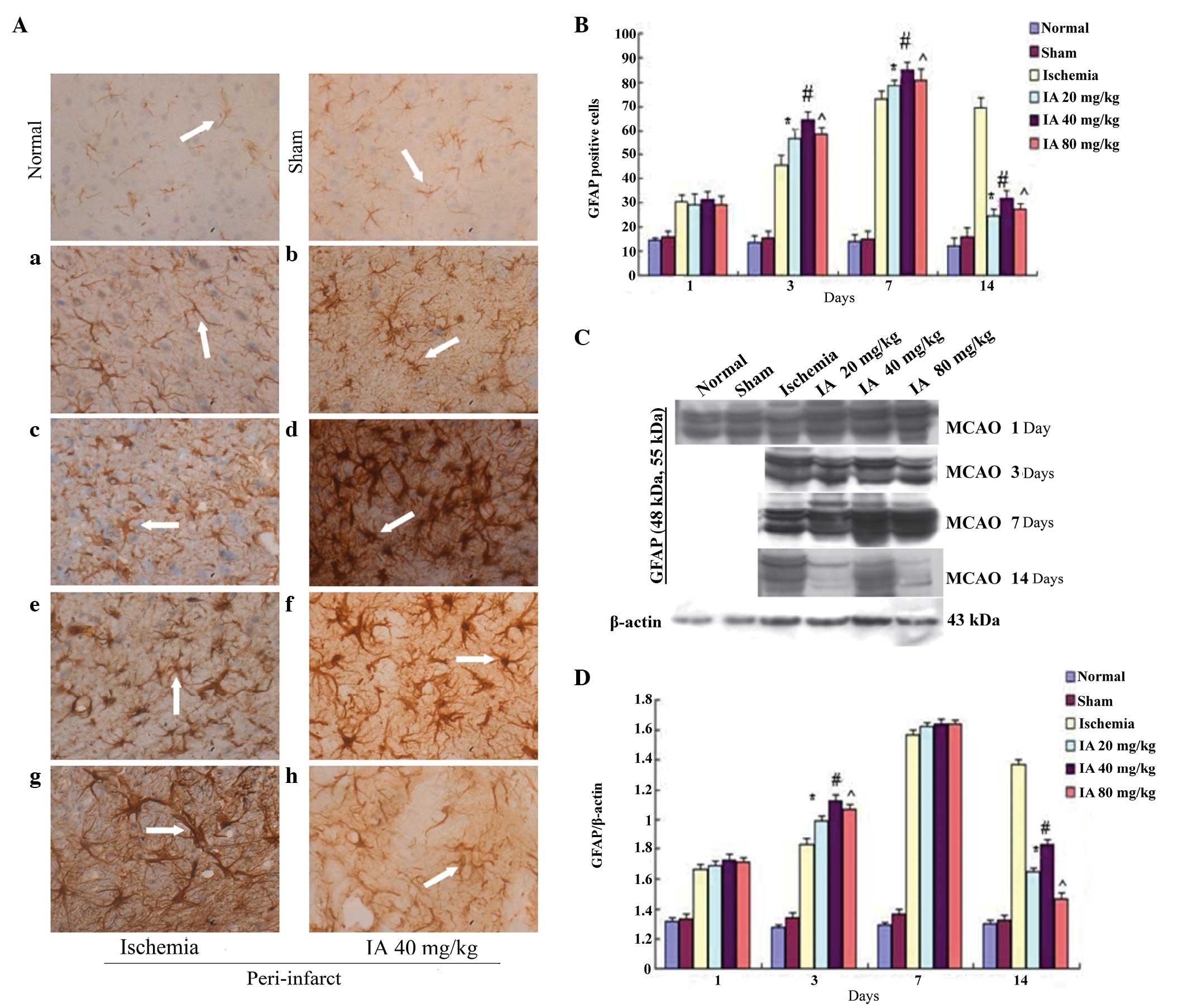 | Figure 3Effect of Ilexonin A on the
activation of astrocytes. Immunostaining for GFAP in the
peri-infarct region at different time points following
ischemia/reperfusion. (A) Ischemia group (a) 1, (c) 3, (e) 7 and
(g)14 days after MCAO; and the administration of 40 mg/kg IA (b) 1,
(d) 3, (f) 7 and (h) 14 days after MCAO. Astrocytes were stained
brown (arrow). Magnification, ×400; scale bar=50 µm. (B)
GFAP-positive cells at different doses of IA. Protein expression
levels of GFAP were detected using (C) Western blot analysis, and
(D) quantification of the results was performed using β-actin
protein as a loading control. Values are expressed as the mean ±
standard deviation (n=6). *P<0.05,
#P<0.05 and ^P<0.05, vs.
ischemia group. MCAO, middle cerebral artery occlusion; IA,
Ilexonin A; GFAP, glial fibrillary acidic protein. |
Following reperfusion, the expression level of GFAP
in the peri-infarct region was markedly increased, and reached a
peak at day 7 in the ischemia and Ilexonin A group, as demonstrated
by Western blot analysis (Fig.
3C). Compared with the ischemia group, the levels of GFAP in
the Ilexonin A group were higher on days 1, 3 and 7, however, there
were fewer GFAP-positive cells on day 14, compared with the
ischemia rats (Fig. 3D).
Effect of Ilexonin A on the
activation of microglia
The IHC results revealed no significant difference
in the numbers of Iba-1-positive cells between the normal and sham
groups, with only quiescent microglia in the peri-infarct region.
In the ischemia group, the number of Iba-1-positive cells was
increased between days 1 and 14, peaking 7 days
post-ischemia/reperfusion (Fig. 4A and
B). The quiescent microglia, which had elliptic and small soma,
and more cell processes, gradually changed into rod-like cells,
which had enlarged soma and thicker, shorter processes. These
finally changed to amoeba-like macrophages, with large rounded soma
and fewer or no cell processes. The numbers of Iba-1 positive
cells, particularly the round macrophage-like cells, in the
Ilexonin A groups were significantly reduced at each time point,
compared with the ischemia group. The change was most marked at 7
days post-ischemia/reperfusion in the 40 mg/kg Ilexonin A
group.
 | Figure 4Effect of IA on the activation of
microglia. Immunostaining for Iba-1 in the peri-infarct region at
different time points following ischemia/reperfusion. (A) Ischemia
group (a) 1, (c) 3, (e) 7 and (g) 14 days after MCAO; and the
administration of 40 mg/kg IA (b) 1, (d) 3, (f) 7 and (h) 14 days
after MCAO. (B) Iba-1-positive cells at different doses of IA.
Microglia are stained brown (arrow). Magnification, ×400; scale
bar=50 µm. Protein expression levels of Iba-1 were detected
using (C) Western blot analysis and (D) quantification of results
was performed using β-actin as a loading control. Values are
expressed as the mean ± standard deviation (n=6).
*P<0.05, #P<0.05 and
^P<0.05, vs. ischemia group. MCAO, middle
cerebral artery occlusion; IA, Ilexonin A; Iba-1, ionized
calcium-binding adapter molecule-1. |
As shown in Fig. 4,
compared with the normal and sham groups at time points >1 day,
cerebral ischemia in the rats subjected to MCAO and Ilexonin A
treatment significantly induced the expression of Iba-1, as
detected using Western blot analysis (Fig. 4C and D). The expression of Iba-1
peaked 7 days following reperfusion in the ischemia and Ilexonin A
groups. Compared with the ischemia group, Ilexonin A treatment
significantly decreased the expression of Iba-1, compared with the
group exposed to reperfusion alone, particularly in the 40 mg/kg
Ilexonin A group.
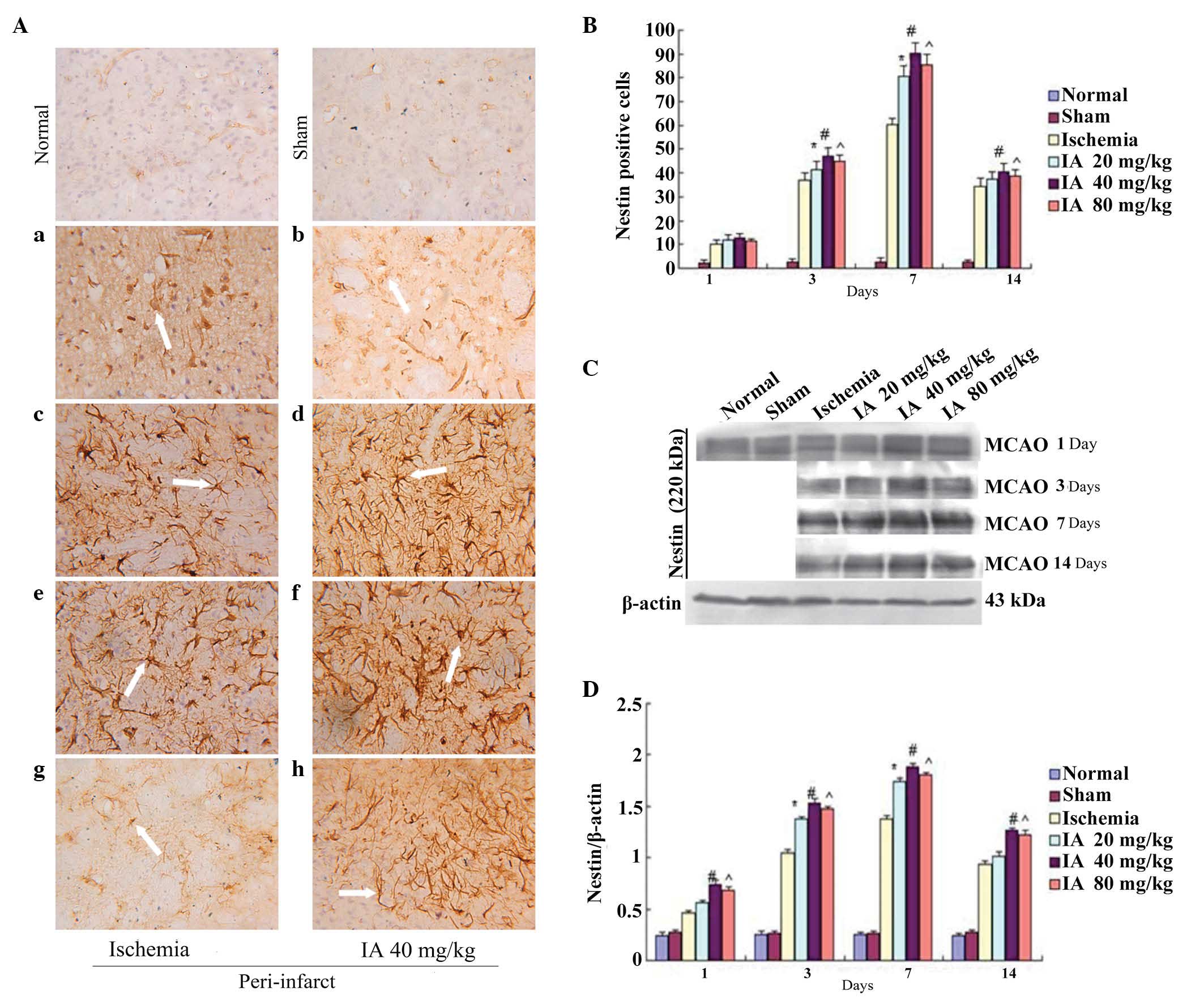
Effect of Ilexonin A on the
proliferation of neural stem cells
In the normal and sham groups, a number of small
vessels and micro-vascular endothelial cells were weakly positive
for Nestin. Following ischemia/reperfusion, the numbers of
Nestin-positive cells around the ischemia core increased
significantly on days 3, 7 and 14, with a peak at 7 days, in the
ischemia and Ilexonin A groups (Fig.
5A and B). In the rats subjected to MCAO treatment, a
significant induction of Nestin was observed in the peri-infarct
region. A further increase was observed in all Ilexonin A groups
(Fig. 5C and D).
Effect of Ilexonin A on the expression of
VEGF
Compared with the normal and sham group, the
expression of VEGF was increased at day 1
post-ischemia/reperfusion, and was highest at 7 days. The
expression of VEGF was further increased in the rats receiving
Ilexonin A at each time point, particularly at 7 days in the 80
mg/kg group (Fig. 6).
Effect of Ilexonin A on the expression of
Flk-1
The expression levels of Flk-1 were increased 3 days
following ischemia/reperfusion, and reached a peak at 7 days in the
ischemia group, compared with the normal and sham groups. Following
treatment with Ilexonin A, the expression levels of Flk-1 were
further increased at each time point, particularly at 7 days in the
80 mg/kg Ilexonin A group (Fig.
7).
Discussion
Ilexonin A is a compound, which is extracted from
the herb, Ilex pubescens, and has been shown to have
antithrombotic effects, anti-inflammatory effects and to improve
blood circulation (3). In the
present study, it was observed that Ilexonin A effectively reduced
infarct volume and improved neurological deficits, in a
dose-dependent manner, which demonstrated that Ilexonin A was a
neuroprotectant. Previous studies have suggested that the
neuroprotective effect of Ilexonin A may include enhancing the
secretion of endogenous neural trophic factors, including FGF and
GAP-43, promoting neurogenesis in the neocortical and subependymal
zones (SVZs), mitigating cerebral edema, inhibiting free radical
production and lipid peroxidation following cerebral
ischemia/reperfusion (4,5). However, the specific mechanism
underlying the neuroprotective effects of Ilexonin A remain to be
fully elucidated and requires further investigation.
The astrocyte response to ischemia in adults is
complex and remains to be fully elucidated. A previous study found
that reactive astrocytes with nuclear membranes increase cell size
and number following MCAO (21).
It is accepted that the moderate activation and proliferation of
astrocytes can have a protective effect following
ischemia/reperfusion; the protective mechanism of reactive
astrocytes may be the result of multiple aspects. Previous studies
have suggested that reactive astrocytes can acquire stem cell
properties following injury and, thus, alter cell type to initiate
repair following brain injury (22,23).
Reactive astrocytes are involved in neurovas-cular remodeling
following ischemia/reperfusion (24,25),
and they also release 17 β-estradiol; neurotrophic factors,
including glia-derived neurotrophic factor; neurotrophin receptors,
including tropomyosin receptor kinase B; and antioxidants to
promote repair (26–29). If the activation of astrocytes is
attenuated, the infarct volume expands 2–3-fold (30), and neuronal death is further
enhanced following brain ischemia (25). The axonal growth cones grow only
when reactive astrocytes survive, whereas this does not happened if
astrocytes are injured in the same area following ischemia injury
(31,32). These findings indicate that
reactive astrocytes are essential for neuronal survival and
regeneration. However, the glial scar, which is formed by the
excessive proliferation of reactive astrocytes in the later stage
following ischemia/reperfusion, rebuilds the integrity of the CNS,
but inhibits axonal regeneration (33). In the present study, Ilexonin A
either enhanced the activation of astrocytes in the early stage (1,
3 or 7 days post-MCAO) or inhibited the excessive proliferation of
astrocytes in the later stage (14 days post-MCAO) in the
peri-infarct region following ischemia/reperfusion. This dual
effect indicated that Ilexonin A promotes favorable conditions for
neuronal restoration.
Microglia are the resident immune cells in the CNS.
When there is a stimulatory signal in the brain, for example
cerebral ischemia, the microglia become activated and can undergo
phenotypic and morphological changes (10). Based on the expression of
immunocytochemical markers, the microglia become amoeba-like cells
following ischemia/reperfusion, which are morphologically
indistinguishable from blood-derived macrophages (34). Therefore, Iba-1 positive cells are
likely derived from resident microglial and blood-derived
macrophages. Novel data, based on the use of GFP radiation bone
marrow-chimeric mice, suggests that blood-derived macrophages are a
low percentage of the population of cells in the peri-infarct
region at day 2 post-ischemia, peak at day 7 and decrease
thereafter (35). By contrast,
resident microglial cells are rapidly activated at day 1
post-ischemia. Notably, even at days 4 an 7, the majority of
macrophage-like cells remain resident microglia-derived cells
(36,37). In addition, previous reports have
suggested that microglia expressing different phenotypes and
morphologies have different functions when the brain receives
different signals from different diseases; and the function
(neuroprotective or neurotoxic) may be determined by the
steady-state conditions among various microglial factors released
into the microenvironment (38–42).
A previous study also suggested that resident microglial and newly
recruited macrophages assume a ʻhealthyʼ M2 phenotype in the 7 days
following MCAO, which protects neurons against ischemic injury.
This is followed by a transition to an ʻunhealthyʼ M1 phenotype in
the peri-infarct region, which exacerbates neuronal death during
ischemic stroke (43). The dual
and opposing roles of microglial/macrophages suggest that ischemic
stroke therapy requires a focus on adjusting the balance between
beneficial and detrimental microglia and macrophage responses,
rather than simply suppressing microglia/macrophages.
In conclusion, the present study, found that the
number of amoeba-like cells significantly decreased, whereas the
number of rod-like cells increased following treatment with
Ilexonin A. This indicated that the rod-like microglia, which
appeared within the first 3 days of MCAO and were maintained by the
administration of Ilexonin A may have a protective effect against
cerebral ischemia/reperfusion injury. The neuroprotective effects
of Ilexonin A may be achieved through adjusting the balance of
microglia/macrophage responses. In previous studies, the protective
effect of microglia/macrophages were reported to occur through
reduction in the secretions of neuronal plasticity proteins and
neurotrophic factors, including IGF-1 and GDNF (42–45).
However, the evidence for this is limited, and further
investigation is required.
In the present study, the expression of Nestin was
induced on day 1 following MCAO, increased progressively until day
7, when the highest expression was observed, and was sustained for
>2 weeks. These results are consistent with those of previous
studies (15,46). The astrocyte-like Nestin-positive
cells observed around the ischemic area following 3 days of MCAO
indicated a trend for neural stem cells to predominantly
differentiate into astrocytes. By contrast, a previous study
suggested that either endogenous or transplanted neural stem cells
at the site of brain injury predominantly differentiated into
gliocytes, with the exception of SVZs and subgranular zones
(47). In addition, reactive
astrocytes express the Nestin protein and assist in neural stem
cell proliferation and differentiation (48,49).
Although neural stem cells that proliferate into the peri-infarct
region predominantly differentiate into gliocytes, a number of
newly matured neurons are observed in the striatum and cortex at
later time points post-ischemia/reperfusion (50,51).
It has been suggested that newly matured neurons in the striatum
and cortex may be associated with neuronal migration from the SVZ
(52). Our previous study
suggested that the number of newly matured neurons were increased
in the cortex, and that neurogenesis and neuronal migration from
the SVZ were increased following treatment with Ilexonin A
(6).
VEGF, one of the most important angiogenic factors,
is a double-edged sword in ischemic injury. VEGF is known as a
potent mitogen for angiogenesis in the ischemic boundary, and is a
neuroprotective factor involved in reducing infarct volume,
improving behavioral recovery, and enhancing cortical, striatal and
SVZ neurogenesis and neuromigration following cerebral ischemia in
rats (53–55). In addition, VEGF acts as an
instructive signal and induces responsive astroglial cells toward
neuronal differentiation (56).
However, VEGF is a potent vascular permeability factor, which may
mediate a harmful response through blood-brain barrier leakage,
brain edema, hemorrhage, aggravation of inflammatory responses and
aberrant systemic hemodynamics (57–59).
Therefore, a major challenge is to accelerate cerebral angiogenesis
without exacerbating edema in the brain of stroke patients.
Previous studies have suggested that the results of
VEGF administration are variable and are associated with the
following aspects of treatment: i) Dose-effectiveness, in which
there are dose-dependent effects when different doses are
administrated by different methods, and excess VEGF administration
may have a detrimental effect in functional recovery (60-62);
ii) Route dependence, in which local VEGF administration
consistently enhances neurologic recovery, whereas acute
intravenous delivery exacerbates brain infarcts due to enhanced
brain edema; suggesting exogenous VEGF can be directly neurotoxic
(63); iii) Time-dependence, in
which VEGF has a therapeutic time window within the first 3 h or
following 24 h of transient MCAO (62-64);
iv) Special receptors, in which VEGF is known to be rapidly induced
following focal cerebral ischemia and to bind to two tyrosine
kinase receptors, fms-like tyrosine kinase-1 (Flt-1) and fetal
liver kinase-1 (Flk-1), on the surface of endothelial cells. These
receptors are induced following ischemia and show a similar
temporal pattern of expression, however, the post-ischemic increase
of Flk-1 is higher than that of Flt-1 (65). A previous study suggested that
activation of Flt-1, but not Flk-1, is sufficient to induce
hyper-permeability by hypoxia and VEGF (66). The results of the present study
suggested that VEGF and Flk-1, which were progressively increased
following transient focal cerebral ischemia and administration of
Ilexonin A, predominantly exerted neuroprotective effects, which
may be dose-dependent.
According to the observation of the dose-dependent
effects of Ilexonin A, the present study found that the dose of 40
mg/kg produced the optimal protective effects on the brains of rats
following transient focal cerebral ischemia. This result is in
accordance with previously published data from Zheng and Shi
(6).
In conclusion, neurologic recovery is a complex
process, which requires the integration of numerous intrinsic and
extrinsic cues to establish intact circuits. No single factor alone
is sufficient to support long-term neurologic recovery. The results
of the present study suggested that Ilexonin A provided a favorable
microenvironment for neurological recovery through regulating the
activation of gliocytes, and promoting revascularization and
neurogenesis.
Acknowledgments
This study was supported by the Program for New
Century Excellent Talents in Fujian Province University, China
(grant no. NCETFJ-0704) and the Professorial Academic Development
Foundation of Fujian Medical University (grant no. JS09014). The
authors would like to thank Clarity Manuscript Consultants LLC for
their assistance with manuscript editing.
References
|
1
|
Fu XC, Xu Z and Chen JJ: Effects of MDQR4
on cardiac function and hemodynamics in anesthetized dogs. Zhong Xi
Yi Jie He Xin Nao Xue Guan Bing. 9:1471–1473. 2011.In Chinese.
|
|
2
|
Li XR: Overview of research progresses on
pharmacological effects Ilex phescens. Glob Trad Chin Med.
9:238–240. 2011.
|
|
3
|
Huang XW, You ZD, Chen J and Xian SX:
Effect of Radix Ilecis Pubescentis on heart function and the
miR133a expression in chronic heart failure rats. Zhong Yao Xin Yao
Yu Lin Chuang Yao Li. 25:48–92. 2014.In Chinese.
|
|
4
|
Hua HY and Li YY: The pharmacological
effects of Ilexonin A. Chin J Med. 8:137–138. 2006.
|
|
5
|
Sheng HL, Dong XL and Jiang YF: Protective
effects of Radix Ilicis Pubescentis extract on focal cerebral
ischemia reperfusion injury in rats. Zhongguo Xin Yao Za Zh.
18:1020–1022. 2009.In Chinese.
|
|
6
|
Zheng GY and Shi WQ: Influence of Ilexonin
A on the expression of bFGF, GAP-43 and neurogenesis after cerebral
ischemia-reperfusion in rats. Yao Xue Xue Bao. 46:1065–1071.
2011.In Chinese. PubMed/NCBI
|
|
7
|
Kim DH, Kim S, Jung WY, Park SJ, Park DH,
Kim JM, Cheong JH and Ryu JH: The neuroprotective effects of the
seeds of Cassia obtusifolia on transient cerebral global ischemia
in mice. Food Chem Toxicol. 47:1473–1479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee CH, Yoo KY, Hwang IK, Choi JH, Park
OK, Li H, Kang IJ, Kwon YG, Kim YM and Won MH: Hypothyroid state
does not protect but delays neuronal death in the hippocampal CA1
region following transient cerebral ischemia: Focus on oxidative
stress and gliosis. J Neurosci Res. 88:2661–2668. 2010.PubMed/NCBI
|
|
9
|
Villapol S, Gelot A, Renolleau S and
Charriaut-Marlangue C: Astrocyte responses after neonatal ischemia:
The yin and the yang. Neuroscientist. 14:339–344. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Badoer E: Microglia: Activation in acute
and chronic inflammatory states and in response to cardiovascular
dysfunction. Int J Biochem Cell Biol. 42:1580–1585. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang N, Zhang Y, Wu L, Wang Y, Cao Y, He
L, Li X and Zhao J: Puerarin protected the brain from cerebral
ischemia injury via astrocyte apoptosis inhibition.
Neuropharmacology. 79:282–289. 2014. View Article : Google Scholar
|
|
12
|
Shen Y, Sun A, Wang Y, Cha D, Wang H, Wang
F, Feng L, Fang S and Shen Y: Upregulation of mesencephalic
astrocyte-derived neurotrophic factor in glial cells is associated
with ischemia-induced glial activation. J Neuroinflammation.
9:2542012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yenari MA, Xu L, Tang XN, Qiao Y and
Giffard RG: Microglia potentiate damage to blood-brain barrier
constituents: improvement by minocycline in vivo and in vitro.
Stroke. 37:1087–1093. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Z, Zhong L, Zhong S, Xian R and Yuan
B: Hypoxia induces microglia autophagy and neural inflammation
injury in focal cerebral ischemia model. Exp Mol Pathol.
98:219–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shin HY, Kim JH, Phi JH, Park CK, Kim JE,
Kim J-H, Paek SH, Wang KC and Kim DG: Endogenous neurogenesis and
neovascularization in the neocortex of the rat after focal cerebral
ischemia. J Neurosci Res. 86:356–367. 2008. View Article : Google Scholar
|
|
16
|
Navaratna D, Guo S, Arai K and Lo EH:
Mechanisms and targets for angiogenic therapy after stroke. Cell
Adh Migr. 3:216–223. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mac Gabhann F, Qutub AA, Annex BH and
Popel AS: Systems biology of pro-angiogenic therapies targeting the
VEGF system. Wiley Interdiscip Rev Syst Biol Med. 2:694–707. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Zhang J and Liang SD: Function and
mechanism of VEGF in the nervous system. Shenjing Jiepouxue Zazhi.
26:561–563. 2010.In Chinese.
|
|
19
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
cranicetomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abedin V, Toctam S and Mahdi Z: Evaluation
the protective effect of aminoguanidine on cortex and striatum
damage in acute phase of focal cerebral ischemia in rat. Physiol
Pharmacol. 11:38–43. 2007.
|
|
21
|
Chu X, Fu X, Zou L, Qi C, Li Z, Rao Y and
Ma K: Oncosis, the possible cell death pathway in astrocytes after
focal cerebral ischemia. Brain Res. 1149:157–164. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Buffo A, Rite I, Tripathi P, Lepier A,
Colak D, Horn AP, Mori T and Götz M: Origin and progeny of reactive
gliosis: A source of multipotent cells in the injured brain. Proc
Natl Acad Sci USA. 105:3581–3586. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sirko S, Behrendt G, Johansson PA,
Tripathi P, Costa M, Bek S, Heinrich C, Tiedt S, Colak D, Dichgans
M, et al: Reactive glia in the injured brain acquire stem cell
properties in response to sonic hedgehog. Cell Stem Cell.
12:426–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hayakawa K, Nakano T, Irie K, Higuchi S,
Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K and Fujiwara
M: Inhibition of reactive astrocytes with fluorocitrate retards
neurovascular remodeling and recovery after focal cerebral ischemia
in mice. J Cereb Blood Flow Metab. 30:871–882. 2010. View Article : Google Scholar
|
|
25
|
Jing L, Mai L, Zhang JZ, Wang JG, Chang Y,
Dong JD, Guo FY and Li PA: Diabetes inhibits cerebral
ischemia-induced astrocyte activation - an observation in the
cingulate cortex. Int J Biol Sci. 9:980–988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin CH, Cheng FC, Lu YZ, Chu LF, Wang CH
and Hsueh CM: Protection of ischemic brain cells is dependent on
astrocyte-derived growth factors and their receptors. Exp Neurol.
201:225–233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tonchev AB, Boneva NB, Kaplamadzhiev DB,
Kikuchi M, Mori Y, Sahara S and Yamashima T: Expression of
neurotrophin receptors by proliferating glia in postischemic
hippocampal CA1 sector of adult monkeys. J Neuroimmunol. 205:20–24.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ding J, Yu JZ, Li QY, Wang X, Lu CZ and
Xiao BG: Rho kinase inhibitor Fasudil induces neuroprotection and
neurogenesis partially through astrocyte-derived G-CSF. Brain Behav
Immun. 23:1083–1088. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang QG, Wang R, Tang H, Dong Y, Chan A,
Sareddy GR, Vadlamudi RK and Brann DW: Brain-derived estrogen
exerts anti-inflammatory and neuroprotective actions in the rat
hippo-campus. Mol Cell Endocrinol. 389:84–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li L, Lundkvist A, Andersson D,
Wilhelmsson U, Nagai N, Pardo AC, Nodin C, Ståhlberg A, Aprico K,
Larsson K, et al: Protective role of reactive astrocytes in brain
ischemia. J Cereb Blood Flow Metab. 28:468–481. 2008. View Article : Google Scholar
|
|
31
|
Jeong HK, Jou I and Joe EH: Absence of
delayed neuronal death in ATP-injected brain: Possible roles of
astrogliosis. Exp Neurobiol. 22:308–314. 2013. View Article : Google Scholar
|
|
32
|
Takano T, Oberheim N, Cotrina ML and
Nedergaard M: Astrocytes and Ischemic Injury. Stroke. 40:8–12.
2009. View Article : Google Scholar
|
|
33
|
Davies SJ, Goucher DR, Doller C and Silver
J: Robust regeneration of adult sensory axons in degenerating white
matter of the adult rat spinal cord. J Neurosci. 19:5810–5822.
1999.PubMed/NCBI
|
|
34
|
Garcia JA, Cardona SM and Cardona AE:
Analyses of microglia effector function using CX3CR1-GFP knock-in
mice. Methods Mol Biol. 1041:307–317. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schilling M, Besselmann M, Leonhard C,
Mueller M, Ringelstein EB and Kiefer R: Microglial activation
precedes and predominates over macrophage infiltration in transient
focal cerebral ischemia: A study in green fluorescent protein
transgenic bone marrow chimeric mice. Exp Neurol. 183:25–33. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lalancette-Hébert M, Gowing G, Simard A,
Weng YC and Kriz J: Selective ablation of proliferating microglial
cells exacerbates ischemic injury in the brain. J Neurosci.
27:2596–2605. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Breckwoldt MO, Chen JW, Stangenberg L,
Aikawa E, Rodriguez E, Qiu S, Moskowitz MA and Weissleder R:
Tracking the inflammatory response in stroke in vivo by sensing the
enzyme myeloperoxidase. Proc Natl Acad Sci USA. 105:18584–18589.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakajima K and KoItsaka S: Microglia:
Neuroprotective and neurotrophic cells in the central nervous
system. Curr Drug Targets Cardiovasc Haematol Disord. 4:65–84.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim SU and de Vellis J: Microglia in
health and disease. J Neurosci Res. 81:302–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li L, Lua J, Taya SS, Moochhalab SM and
Hea BP: The function of microglia, either neuroprotection or
neurotoxicity, is determined by the equilibrium among factors
released from activated microglia in vitro. Brain Res. 1159:8–17.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ekdahl CT, Kokaia Z and Lindvall O: Brain
inflammation and adult neurogenesis: The dual role of microglia.
Neuroscience. 158:1021–1029. 2009. View Article : Google Scholar
|
|
42
|
Wang JP, Yang ZT, Liu C, Zhao YZ and Chen
YB: Activated microglia provide a neuroprotective role by balancing
glial cell-line derived neurotrophic factor and tumor necrosis
factor-α secretion after subacute cerebral ischemia. Int J Mol Med.
31:172–178. 2013.
|
|
43
|
Hu X, Li P, Guo Y, Wang H, Leak RK, Chen
S, Gao Y and Chen J: Microglia/macrophage polarization dynamics
reveal novel mechanism of injury expansion after focal cerebral
ischemia. Stroke. 43:3063–3070. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Arroba AI, Alvarez-Lindo N, van Rooijen N
and de la Rosa EJ: Microglia-mediated IGF-I neuroprotection in the
rd10 mouse model of retinitis pigmentosa. Invest Ophthalmol Vis
Sci. 52:9124–9130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Montero M, González B and Zimmer J:
Immunotoxic depletion of microglia in mouse hippocampal slice
cultures enhances ischemia-like neurodegeneration. Brain Res.
1291:140–152. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu W, Song DL, Zhang GH, Chen JW, Xie RH,
Lin HJ, et al: Nestin expression in different area of brain after
cerebral ischemia/reperfusion in rats. Zu Zhong Yu Shen Jing Ji
Bing. 16:34–36. 2009.
|
|
47
|
Song H, Stevens CF and Gage FH: Astroglia
induce neurogenesis from adult neural stem cells. Nature.
417:39–44. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shin YJ, Kim HL, Park JM, Cho JM, Kim SY
and Lee MY: Characterization of nestin expression and vessel
association in the ischemic core following focal cerebral ischemia
in rats. Cell Tissue Res. 351:383–395. 2013. View Article : Google Scholar
|
|
49
|
Kronenberg G, Gertz K, Cheung G, Buffo A,
Kettenmann H, Gotz M and Endres M: Modulation of fate determinants
Olig2 and Pax6 in resident glia evokes spiking neuroblasts in a
model of mild brain ischemia. Stroke. 41:2944–2949. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kim HJ, Leeds P and Chuang DM: The HDAC
inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic
brain. J Neurochem. 110:1226–1240. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yao RQ, Zhang L, Wang W and Li L: Cornel
iridoid glycoside promote neurogenesis and angiogenesis and
improves neurological function after focal cerebral ischemia in
rats. Brain Res Bull. 79:69–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhong J, Tang MK, Zhang Y, Xu QP and Zhang
JT: Effect of salvianolic acid B on neural cells damage and
neurogenesis after brain ischemia-reperfusion in rats. Yao Xue Xue
Bao. 42:716–721. 2007.In Chinese. PubMed/NCBI
|
|
53
|
Wang YQ, Cui HR, Yang SZ, Sun HP, Qiu MH,
Feng XY and Sun FY: VEGF enhance cortical newborn neurons and their
neurite development in adult rat brain after cerebral ischemia.
Neurochem Int. 55:629–636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang JP, Liu HJ and Liu XF: VEGF promotes
angiogenesis and functional recovery in stroke rats. J Invest Surg.
23:149–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Herz J, Reitmeir R, Hagen SI, Reinboth BS,
Guo Z, Zechariah A, ElAli A, Doeppner TR, Bacigaluppi M, Pluchino
S, et al: Intracerebroventricularly delivered VEGF promotes
contralesional corticorubral plasticity after focal cerebral
ischemia via mechanisms involving anti-inflammatory actions.
Neurobiol Dis. 45:1077–1085. 2012. View Article : Google Scholar
|
|
56
|
Li P, Huang JJ, Ni JJ and Sun FY:
WITHDRAWN: VEGF evokes reactive astroglia to convert into neuronal
cells by affecting the biological function of MeCP2 in adult rat
brain after cerebral ischemia. Neurochem Int. 2012.Epub ahead of
print. View Article : Google Scholar
|
|
57
|
Wang Y, Kilic E, Kilic U, Weber B,
Bassetti CL, Marti HH and Hermann DM: VEGF overexpression induces
post-ischaemic neuroprotection, but facilitates haemodynamic steal
phenomena. Brain. 128:52–63. 2005. View Article : Google Scholar
|
|
58
|
Chibay Y, Sasayama T, Miyake TS, Koyama J,
Kondoh T, Hosoda K and Kohmura E: Anti-VEGF receptor antagonist
(VGA1155) reduces infarction in rat permanent focal brain ischemia.
Kobe J Med Sci. 54:E136–E146. 2008.
|
|
59
|
Li Z, Wang R, Li S, Wei J, Zhang Z, Li G,
Dou W, Wei Y and Feng M: Intraventricular pre-treatment with
rAAV-VEGF induces intracranial hypertension and aggravates ischemic
injury at the early stage of transient focal cerebral ischemia in
rats. Neurol Res. 30:868–875. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Manoonkitiwongsa PS, Schultz RL, McCreery
DB, Whitter EF and Lyden PD: Neuroprotection of ischemic brain by
vascular endothelial growth factor is critically dependent on
proper dosage and may be compromised by angiogenesis. J Cereb Blood
Flow Metab. 24:693–702. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Katsumata A, Sugiu K, Tokunaga K, Kusaka
N, Watanabe K, Nishida A, Namba K, Hamada H, Nakashima H and Date
I: Optimal dose of plasmid vascular endothelial growth factor for
enhancement of angiogenesis in the rat brain ischemia model. Neurol
Med Chir (Tokyo). 50:449–455. 2010. View Article : Google Scholar
|
|
62
|
Yang JP, Guo L, Liu RC and Liu HJ:
Neuroprotective effects of VEGF administration after focal cerebral
ischemia/reperfusion: Dose response and time window. Neurochem Int.
60:592–596. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hermann DM and Zechariah A: Implications
of vascular endothelial growth factor for postischemic
neurovascular remodeling. J Cereb Blood Flow Metab. 29:1620–1643.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun Y, Jin K, Xie L, Childs J, Mao XO,
Logvinova A and Greenberg DA: VEGF-induced neuroprotection,
neurogenesis and angiogenesis after focal cerebral ischemia. J Clin
Invest. 111:1843–1851. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Choi JS, Kim HY, Cha JH, Choi JY, Chun MH
and Lee MY: Upregulation of vascular endothelial growth factor
receptors Flt-1 and Flk-1 in rat hippocampus after transient
forebrain ischemia. J Neurotrauma. 24:521–531. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vogel C, Bauer A, Wiesnet M, Preissner K,
Schaper W, Marti HH and Fischer S: Flt-1, but not Flk-1 mediates
hyperpermeability through activation of the PI3-K/Akt pathway. J
Cell Physiol. 212:236–243. 2007. View Article : Google Scholar : PubMed/NCBI
|