Introduction
Atherosclerotic plaque destabilization and rupture
are thought to account for the majority of acute coronary
syndromes. Rupture-prone unstable plaques possess the following
histological features: Lipid core formation, fibrous cap thinning,
inflammatory cell infiltration of the fibrous cap and adventitia
(1). Plaque destabilization may be
induced by external factors, including increased blood pressure and
shear stress, and/or by factors within the atherosclerotic plaque,
including inflammation and intraplaque hemorrhage. Macrophages in
atherosclerotic plaques have a marked impact on atherogenesis and
plaque destabilization. During the development of atherosclerotic
plaques, macrophages can initiate lesion progression,
destabilization and rupture via the production and release of
various cytokines and proteases, including tumor necrosis factor
(TNF)-α and matrix metalloproteinases (MMPs) (2). Furthermore, exacerbated macrophage
apoptosis may contribute to necrotic core expansion and fibrous cap
thinning during advanced stages of the disease (3).
It has previously been suggested that endoplasmic
reticulum (ER) stress may have a role in the progression of plaque
vulnerability, and the occurrence of acute complications associated
with coronary atherosclerosis (4).
ER stress is chronically activated in atherosclerotic lesional
cells, particularly in advanced lesional macrophages and
endothelial cells. In vitro and in vivo studies have
demonstrated that prolonged ER stress may result in
macrophage-derived foam cell formation and proatherogenic cytokine
expression in advanced atherosclerotic lesions (5,6).
Atherosclerotic-relevant inducers of ER stress, including modified
forms of low-density lipoprotein (LDL) and hyperhomocysteinemia
(HHcy), have an important role in inflammation and cell apoptosis
in atherosclerotic lesions (7,8).
HHcy-induced ER stress has been reported to initiate and accelerate
atherosclerosis via the upregulation of genes associated with lipid
biosynthesis and uptake, inflammation, collagen synthesis and
apoptosis (8). Due to its
indispensable role in the progression of atherosclerosis, ER stress
is therefore considered an important molecular target for the
treatment of HHcy.
Atorvastatin, which is a 3-hydroxy-3-methylglutaryl
coenzyme A reductase inhibitor, exerts antioxidant and
anti-inflammatory functions independent of its lipid-lowering
abilities. Atorvastatin has previously been shown to attenuate
macrophage foam cell formation by regulating scavenger receptor
expression, oxidized-LDL uptake and cholesterol efflux (9,10).
In addition, atorvastatin is able to alter the cytokine balance in
the microenvironment of atherosclerotic plaques (11). In our previous studies, it was
demonstrated that atorvastatin could antagonize homocysteine
(Hcy)-induced endothelial dysfunction by increasing the viability
of endothelial progenitor cells, and suppressing the apoptosis of
endothelial cells (12,13). To further define the underlying
mechanisms by which atorvastatin inhibits HHcy-induced injury, the
present study aimed to test the hypothesis that the plaque
stabilizing effects of atorvastatin are partly attributed to
inhibition of ER stress. Initially, we aimed to confirm whether
atorvastatin was able to suppress ER stress activation in the
vessel walls of hyperhomocysteinemic apolipoprotein E-deficient
(ApoE−/−) mice. Subsequently, the ability of
atorvastatin to inhibit ER stress in macrophages was
investigated.
Materials and methods
Animal experiments
The 6-week-old male ApoE−/− mice were
obtained from Peking University Health Science Center (Beijing,
China). ApoE−/− mice were chosen since they enabled the
generation of a HHcy atherosclerotic model within 2 months,
following the administration of 1 ml 2% (w/v) methionine
(Sigma-Aldrich, St. Louis, MO, USA) per day by gastric gavage. The
mice were housed in a temperature-controlled environment with a
12-h dark-light cycle and ad libitum access to food and
water. The mice were divided into three groups: The methionine
group, ApoE−/− mice were administered methionine (n=15);
the atorvastatin group, ApoE−/− mice were administered
atorvastatin (5 mg·kg−1·d−1; National
Institute for the Control of Pharmaceutical and Biological
Products, Beijing, China) suspended in 1 ml 2% methionine (n=15);
and the control group, ApoE−/− mice were administered
saline (n=20). After 2 months, the mice were anesthetized with 3%
pentobarbital (30 mg/kg; China National Medicines Corporation,
Ltd., Shanghai, China) and then sacrificed by exsanguination. The
maximum amount of blood was obtained from the abdominal aorta and
the intact circulation was flushed with phosphate-buffered saline
(PBS). The aortic roots were carefully dissected and adherent
connective tissue was removed. The animal experiments were
conducted according to the recommendations of the Guidelines of
National Animal Care and Use Committees, and the National Animal
Welfare Law (14). The study was
approved by the ethics committee of Soochow University (Suzhou,
China).
Measurements of plasma Hcy
Following a 12-h fast and the induction of
anesthesia, blood samples were obtained, anticoagulated with
ethylenediaminetetraacetic acid and placed on ice. Plasma was
quickly separated by centrifugation at 1566 × g for 10 min at room
temperature. The plasma was then stored at −20°C until further
analysis. Total Hcy concentrations were measured using
high-performance liquid chromatography following reduction of the
plasma samples (15).
Histological analysis
The aortic roots were carefully dissected and
cleaned of adherent connective tissue under a dissecting
microscope. The aortic roots were then embedded in paraffin and
were serially sectioned at 4 μm. For quantification of
atherosclerotic lesions, the sections were collected and stained
with hematoxylin and eosin (H&E) and Masson's trichrome
(Sigma-Aldrich). Images of staining were captured and quantified
using an Axioplan 2 imaging microscope system (Carl Zeiss GmbH,
Jena, Germany).
Immunohistochemical (IHC) analysis
For IHC analysis, the paraffin-embedded sections
were deparaffinized by immersion in xylene, followed by a series of
alcohol treatments. Endogenous peroxidase activity was quenched by
immersing the slides in 0.3% hydrogen peroxide in methanol for 15
min. The sections were rinsed three times in PBS (5 min/wash) and
were blocked with 5% normal serum (Cell Signaling Technology, Inc.,
Danvers, MA, USA). The sections were then incubated with primary
antibodies (1:1,000) overnight at 4°C. Rabbit polyclonal
phosphorylated (p)-protein kinase RNA-like endoplasmic reticulum
kinase (PERK) antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA; cat. no. sc-32577); rabbit
monoclonal antibodies against glucose-regulated protein 78 (GRP78;
cat. no. 3177) and p-eukaryotic initiation factor 2α (eIF2α; cat.
no. 3398) were obtained from Cell Signaling Technology, Inc.
Following rinsing in PBS with Tween 20, the sections were incubated
with labeled polymer-horseradish peroxidase (HRP) goat anti-rabbit
IgG (Cell Signaling Technology, Inc.; cat. no. 7074) at 37°C for 1
h, and then incubated with diaminobenzidine chromogen. Following
the final wash, the sections were counterstained with hematoxylin.
The intensity of IHC staining was measured using the Axioplan 2
imaging analysis system (Carl Zeiss GmbH). The average value in
each group was calculated from random observation of five high
power microscopic views of entire sections.
Cell culture
The RAW264.7 murine macrophages were obtained from
the Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). Cells were cultured in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin (Beyotime
Institute of Biotechnology, Haimen, China). The cultures were
maintained at 37°C in a humidified incubator containing 5%
CO2 until subconfluent. Cells received treatments in
serum-free medium. A total of 1 and 10 μmol/l atorvastatin
(National Institute for the Control of Pharmaceutical and
Biological Products, Beijing, China), in the presence or absence of
thapsigargin (2 μmol/l; Sigma-Aldrich) was added 30 min
prior to 500 μmol/l Hcy (Sigma-Aldrich) stimulation for 24
h.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
According to the manufacturer's protocol, RNA was
extracted from macrophages or homogenized aortic roots using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). The quality of the isolated RNA was determined using agarose
gel electrophoresis, and RNA concentration was determined by
measuring optical density at 260 and 280 nm using a BioPhotometer
(Eppendorf, Hamburg, Germany). A reverse transcription kit (Takara
Bio Inc., Otsu, Japan) was used for reverse transcription. The
following primer sequences were designed (Invitrogen; Thermo Fisher
Scientific, Inc.): TNF-α, forward 5′-TTC TAT GGC CCA GAC CCT CA-3′,
reverse 5′-ACT TGG TGG TTT GCT ACG ACG-3′; MMP-9, forward 5′-GTC
CCA CTA TAC CTC CCACG-3′, reverse 5′-ATT GCA AGG ATT GTC TGC CG-3′;
and β-actin, forward 5′-ATG GTG GGA ATG GGT CAG AA-3′ and reverse
5′-GTC ACG CAC GAT TTC CCT CT-3′. RT-qPCR was performed using
SYBR® Premix Ex Taq™ (Perfect Real Time) kit
(Takara Bio Inc., Otsu, Japan) and an Applied Biosystems 7300
Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). PCR was performed using an initial step of denaturation at
95°C for 30 sec, 40 cycles of amplification, denaturation at 95°C
for 5 sec and annealing at 60°C for 30 sec. All reactions were
performed in a 20-μl volume in triplicate. The relative
amounts of mRNA in untreated and treated cells were compared using
the comparative cycle quantification (2ΔΔCq) method
(16), with β-actin mRNA as the
internal standard.
Western blot analysis
The cells were collected and
radioimmunoprecipitation assay lysis buffer containing protease
inhibitors (Beyotime Institute of Biotechnology) was used for
extraction of total cellular protein. Protein concentrations were
determined using a protein assay kit (Pierce Protein Biology;
Thermo Fisher Scientific, Inc.). After boiling for 5 min, equal
quantities of the denatured protein sample (40 μg) were
separated by 9% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and then protein was transferred
electrophoretically to polyvinylidene fluoride membranes
(Millipore, Bedford, MA, USA). The resulting membranes were blocked
using 5% non-fat milk in Tris-buffered saline containing Tween (10
mmol/l Tris-HCl, pH 7.4; 150 mmol/l NaCl; 0.05% Tween-20) for 1 h,
and hybridized with primary antibodies (antibody against p-PERK,
1:400 dilution; cat. no. sc-32577; Santa Cruz Biotechnology, Inc.;
antibodies against GRP78 and p-eIF2α; cat. nos. 3177 and 3398; Cell
Signaling Technology, Inc.; 1:1,000 dilution) overnight at 4°C.
Following incubation with the appropriate HRP-conjugated goat
anti-rabbit IgG secondary antibody (1:1,000 dilution; cat. no.
7074; Cell Signaling Technology, Inc.), the immune complexes were
detected using an enhanced chemiluminescence detection system (GE
Healthcare Life Sciences, Chalfont, UK). Staining was quantified by
scanning densitometry (Microtek Scanwizard 5; Informer
Technologies, Inc., Walnut, CA, USA) with β-actin used as an
internal standard.
Statistical analysis
The obtained data are presented as the mean ±
standard deviation. One-way analysis of variance was used to
statistically analyze the data, using SPSS 13.0 software (SPSS,
Inc., Chicago, IL, USA). P<0.05 (two-tailed) was considered to
indicate a statistically significant difference.
Results
Effects of atorvastatin on plasma Hcy
levels in ApoE−/− mice
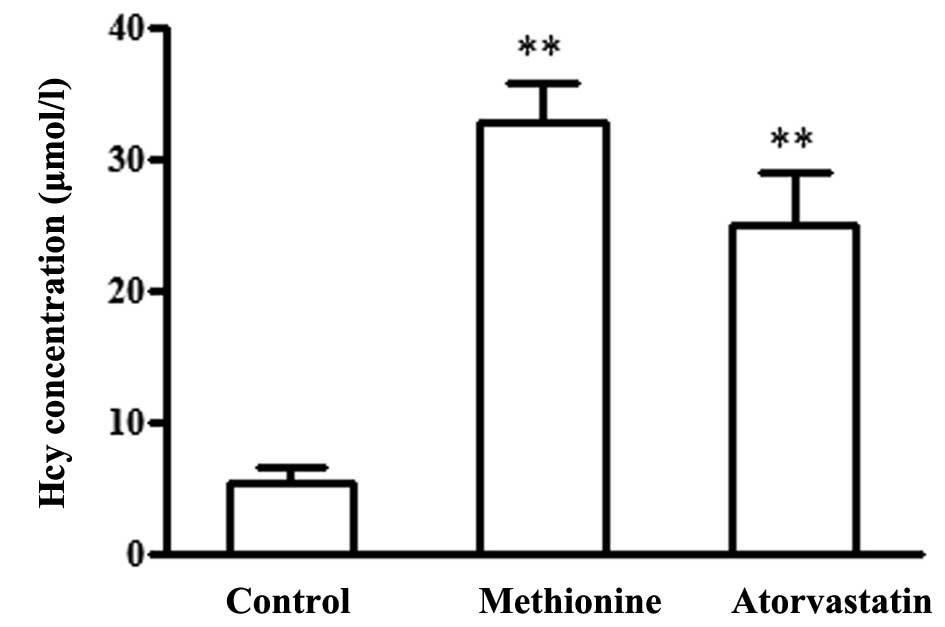
The methionine group were administered 1 ml 2% (w/v)
methionine, and the atorvastatin group were administered
atorvastatin (5 mg·kg−1·d−1) suspended in 1
ml 2% methionine daily for 2 months. After 2 months, plasma Hcy
levels in the methionine group were significantly increased, as
compared with in the control ApoE−/− mice (32.8±3.0 vs.
5.3±1.2 μmol/l, P<0.01). In addition, plasma Hcy levels
were upregulated in the atorvastatin group (25.0±3.9 μmol/l)
compared with the control group; however, there was no statistical
difference between the atorvastatin and methionine groups (Fig. 1).
Atorvastatin inhibits vulnerable plaque
formation in the aortic roots of HHcy mice
The present study analyzed large necrotic core area
and collagen content, in order to investigate plaque stability.
Paraffin sections from the aortic roots of ApoE−/− mice
were stained with H&E to assess lesion growth and histological
morphology (Fig. 2A). To determine
whether atorvastatin was able to reduce necrotic core formation,
both the number and the relative size of necrotic cores was
analyzed. Treatment with atorvastatin resulted in fewer necrotic
cores, as compared with the methionine group; 30.83±7.91 vs.
44.2±13.12% of the sections covering the entire lesion that
contained a necrotic core (P<0.05). Furthermore, the relative
size of the necrotic cores decreased from 26.42±8.23 to 19.41±7.05%
of the plaque surface area (P<0.05). Collagen is the main
stabilizing component of plaques. Masson's trichrome staining
detected a 45% increase in the relative amount of collagen in the
plaques following atorvastatin treatment, as compared with that in
the methionine group (17.75±5.25 vs. 12.21±4.28%, P<0.05)
(Fig. 2B).
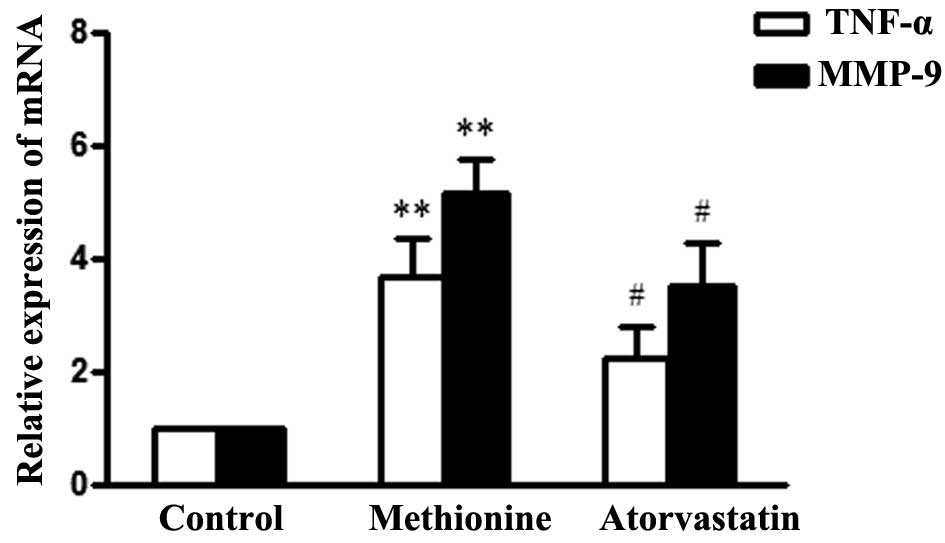
Atorvastatin reduces TNF-α and MMP-9 mRNA
expression in the aortic roots of HHcy mice
To explore the effects of atorvastatin on the
HHcy-induced inflammatory response in aortic roots of
ApoE−/− mice, TNF-α and MMP-9 mRNA expression levels
were detected by RT-qPCR. TNF-α and MMP-9 expression levels were
significantly upregulated in the methionine group, as compared with
the control group, whereas treatment with atorvastatin
significantly decreased the expression levels in atherosclerotic
lesions (Fig. 3).
Atorvastatin prevents ER stress
activation in aortic lesions of HHcy ApoE−/− mice
To further determine the mechanisms underlying
statin-induced plaque stabilization, the present study investigated
whether atorvastatin affected HHcy-induced ER stress in aortic root
lesions (Fig. 4). The IHC analysis
detected a significant decrease in p-PERK immunostaining in the
atorvastatin group, as compared with in the methionine group.
Furthermore, p-eIF2α and GRP78 immunostaining were significantly
downregulated following treatment with atorvastatin, as compared
with following methionine treatment only. These results indicate
that atorvastatin may inhibit ER stress activation in aortic
lesions of HHcy mice. The plaque stabilizing effects of
atorvastatin may be due to ER stress inhibition in HHcy mice.
Atorvastatin inhibits Hcy-induced ER
stress in macrophages
To determine the effects of atorvastatin on
Hcy-induced ER stress in macrophages, various concentrations of
atorvastatin (1 and 10 μmol/l) were added 30 min prior to
500 μmol/l Hcy stimulation. As shown in Fig. 5, 500 μmol/l Hcy induced ER
stress activation in macrophages, as determined by the increased
phosphorylation of PERK and its substrate, eIF2α, and the increased
expression of the chaperone GRP78. Compared with the Hcy group,
Hcy-induced ER stress was inhibited in response to 10 μmol/l
atorvastatin treatment. Furthermore, thapsigargin, an ER stress
inducer, attenuated the inhibitory effects of atorvastatin against
Hcy-induced ER stress.
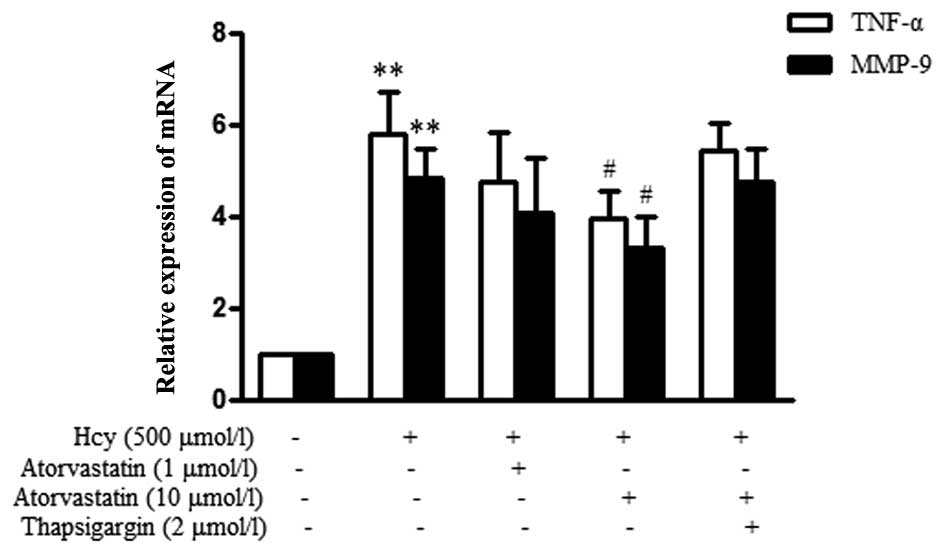
Atorvastatin suppresses the Hcy-induced
inflammatory response in macrophages
The present study also evaluated the effects of
atorvastatin on Hcy-induced TNF-α and MMP-9 mRNA expression. As
shown in Fig. 6, the relative mRNA
expression levels of TNF-α were significantly upregulated in
response to Hcy stimulation. However, treatment with 10
μmol/l atorvastatin could evidently inhibit Hcy-induced
TNF-α mRNA expression. MMP-9 expression was also significantly
upregulated in the Hcy group, as compared with in the control
group, whereas treatment with 10 μmol/l atorvastatin
significantly decreased its expression. Furthermore, thapsigargin
attenuated the inhibitory effects of atorvastatin against
Hcy-induced TNF-α and MMP-9 upregulation. These results strongly
suggest that ER stress pathways may participate in the reaction,
and atorvastatin inhibits the Hcy-induced inflammatory response via
suppressing ER stress in macrophages.
Discussion
The present study demonstrated that HHcy promoted
the development of atherosclerotic plaques and plaque instability.
Atorvastatin was able to downregulate Hcy-induced ER stress
activation in atherosclerotic lesions and macrophages, which may
suppress inflammatory responses and improve the stability of
atherosclerotic plaques.
Alterations in atherosclerotic plaque composition
can impact plaque stability, and determine the clinical course of
atherosclerosis. Previous studies have suggested that the
inflammatory cytokines have an important role in the disruption of
vascular function, and the resultant development of vascular
diseases (17,18). In addition, epidemiological studies
have reported that TNF-α is markedly elevated in the plasma and
arteries of human patients with vascular complications (19,20).
Elevated circulating levels of matrix biomarkers are a key feature
of atherosclerotic plaque development, vascular remodeling and
plaque rupture. MMP-9, which contributes to the formation of
unstable plaques, has been suggested as an atherosclerotic
inflammatory marker (21). In the
present study, methionine treatment enabled the generation of a
HHcy atherosclerotic ApoE−/− mouse model.
Atherosclerotic plaques in the aortic arteries of the mice appeared
to possess several key histological features of unstable plaques,
including large necrotic cores, reduced collagen content and
increased inflammatory cytokine infiltration.
The pathophysiological environment in HHcy mice was
able to activate prolonged ER stress in the arterial wall. The
present study demonstrated that the expression of p-PERK, p-eIF2α
and GRP78 were predominantly situated within the atherosclerotic
lesions in HHcy ApoE−/− mice. Previous studies have
revealed that ER stress is a cross-point that links cellular
processes with numerous risk factors that exist in all stages of
atherosclerosis, and is believed to have a critical role in
endothelial dysfunction (22,23),
activation of inflammatory reactions (24) and foam cell formation (25). Activation of ER stress is
associated with the severity and clinical complications of
atherosclerosis in humans. In a previous study, directional
coronary atherectomy specimens demonstrated that ruptured plaques
exhibited a markedly increased expression of the ER chaperones
GRP78 and CCAAT-enhancer-binding protein homologous protein (CHOP)
(26). These findings further
supported the hypothesis that vulnerability of artery plaques in
HHcy mice may be partially attributed to ER stress activation.
Therapeutic interventions that reduce ER stress may
be considered promising strategies to reduce Hcy-induced vascular
injury and consequent plaque instability. In the present study,
administration of atorvastatin to ApoE−/− treated with
methionine resulted in a decreased number and size of necrotic
cores, and increased collagen content in the plaques, as compared
with the plaques in the methionine group. Downregulation of TNF-α
and MMP-9 in the lesions of the atorvastatin-treated mice further
substantiated these findings. Atorvastatin is widely used in the
treatment and prevention of atherosclerosis. Atorvastatin has
previously been shown to downregulate GRP78, caspase-12 and CHOP
expression in myocardial cells (27), and attenuate myocardial ischemia
reperfusion injury via inhibiting ER stress-related apoptosis
(28). Atorvastatin may function
as a pharmacological inhibitor of ER stress. In the present study,
treatment with atorvastatin significantly suppressed ER stress
activation and reduced plaque vulnerability, thus indicating that
atorvastatin may maintain ER homeostasis and prevent HHcy-induced
atherosclerotic lesion progression. However, atorvastatin did not
reduce plasma Hcy levels in ApoE−/− mice, thus
indicating that the plaque stabilizing effects of atorvastatin
against Hcy injury may not depend on the lowering of Hcy levels.
Inhibition of ER stress may serve as an important role in the
plaque stabilizing effects of atorvastatin.
The present study also demonstrated that
atorvastatin could suppress Hcy-induced ER stress activation in
macrophages. Macrophage infiltration plays a crucial role
throughout the entire process of atherogenesis and plaque rupture.
Cytokines secreted by macrophages, including MMPs and TNF-α, can
attenuate plaque stability and prompt rupture of atherosclerotic
plaques. In the present study, treatment with Hcy increased MMP-9
and TNF-α expression in murine macrophages; however, Hcy-induced
MMP-9 and TNF-α mRNA expression was markedly attenuated by
atorvastatin. Thapsigargin attenuated the protective effects of
atorvastatin against Hcy-induced ER stress and MMP-9 and TNF-α
production, thus suggesting that ER stress pathways have
predominant roles in Hcy-induced inflammation in macrophages.
Atorvastatin may inhibit the Hcy-induced inflammatory response via
suppressing ER stress in macrophages.
In conclusion, the results of the present study
provide a novel insight into the protective effects of atorvastatin
against Hcy-induced vascular injury. Hcy markedly promoted the
development of atherosclerotic plaques and plaque instability.
Atorvastatin was used to antagonize Hcy-induced injury in HHcy
ApoE−/− mice and in macrophages, and it was shown to
target ER molecules and inhibit the development of atherosclerotic
lesions. Atorvastatin may therefore attenuate the progression of
atherosclerotic lesions and plaque vulnerability by regulating ER
stress activation, which may provide a novel interpretation of its
pleiotropic effects. However, further studies are required to fully
understand the relationship between atorvastatin and ER stress
activation in the treatment of atherosclerosis.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81300220).
References
|
1
|
Silvestre-Roig C, de Winther MP, Weber C,
Daemen MJ, Lutgens E and Soehnlein O: Atherosclerotic plaque
destabilization: Mechanisms, models, and therapeutic strategies.
Circ Res. 114:214–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hopkins PN: Molecular biology of
atherosclerosis. Physiol Rev. 93:1317–1542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gautier EL, Huby T, Witztum JL, Ouzilleau
B, Miller ER, Saint-Charles F, Aucouturier P, Chapman MJ and Lesnik
P: Macrophage apoptosis exerts divergent effects on atherogenesis
as a function of lesion stage. Circulation. 119:1795–1804. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao S, Miao C, Tian H, Sang H, Yang N,
Jiao P, Han J, Zong C and Qin S: Endoplasmic reticulum stress
promotes macrophage-derived foam cell formation by up-regulating
cluster of differentiation 36 (CD36) expression. J Biol Chem.
289:4032–4042. 2014. View Article : Google Scholar :
|
|
6
|
Gao J, Ishigaki Y, Yamada T, Kondo K,
Yamaguchi S, Imai J, Uno K, Hasegawa Y, Sawada S, Ishihara H, et
al: Involvement of endoplasmic stress protein C/EBP homologous
protein in arteriosclerosis acceleration with augmented biological
stress responses. Circulation. 124:830–839. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McAlpine CS, Bowes AJ, Khan MI, Shi Y and
Werstuck GH: Endoplasmic reticulum stress and glycogen synthase
kinase-3β activation in apolipoprotein E-deficient mouse models of
accelerated atherosclerosis. Arterioscler Thromb Vasc Biol.
32:82–91. 2012. View Article : Google Scholar
|
|
8
|
Zhou J and Austin RC: Contributions of
hyperhomocysteinemia to atherosclerosis: Causal relationship and
potential mechanisms. Biofactors. 35:120–129. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qiu G and Hill JS: Atorvastatin inhibits
ABCA1 expression and cholesterol efflux in THP-1 macrophages by an
LXR-dependent pathway. J Cardiovasc Pharmacol. 51:388–395. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Llaverias G, Noé V, Peñuelas S,
Vázquez-Carrera M, Sánchez RM, Laguna JC, Ciudad CJ and Alegret M:
Atorvastatin reduces CD68, FABP4, and HBP expression in
oxLDL-treated human macrophages. Biochem Biophys Res Commun.
318:265–274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gómez-Hernández A, Sánchez-Galán E,
Martín-Ventura JL, Vidal C, Blanco-Colio LM, Ortego M, Vega M,
Serrano J, Ortega L, Hernández G, et al: Atorvastatin reduces the
expression of prostaglandin E2 receptors in human carotid
atherosclerotic plaques and monocytic cells: Potential implications
for plaque stabilization. J Cardiovasc Pharmacol. 47:60–69. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jia F, Wu C, Chen Z and Lu G:
AMP-activated protein kinase inhibits homocysteine-induced
dysfunction and apoptosis in endothelial progenitor cells.
Cardiovasc Drugs Ther. 25:21–29. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bao XM, Wu CF and Lu GP: Atorvastatin
attenuates homocysteine-induced apoptosis in human umbilical vein
endothelial cells via inhibiting NADPH oxidase-related oxidative
stress-triggered p38MAPK signaling. Acta Pharmacol Sin.
30:1392–1398. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Institutes of Health; The National Academies
Press, Washington, DC: 2011
|
|
15
|
Jacobsen DW, Gatautis VJ, Green R,
Robinson K, Savon SR, Secic M, Ji J, Otto JM and Taylor LM Jr:
Rapid HPLC determination of total homocysteine and other thiols in
serum and plasma: Sex differences and correlation with cobalamin
and folate concentrations in healthy subjects. Clin Chem.
40:873–881. 1994.PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
17
|
Shah PK: Molecular mechanisms of plaque
instability. Curr Opin Lipidol. 18:492–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cavieres V, Valdes K, Moreno B,
Moore-Carrasco R and Gonzalez DR: Vascular hypercontractility and
endothelial dysfunction before development of atherosclerosis in
moderate dyslipidemia: Role for nitric oxide and interleukin-6. Am
J Cardiovasc Dis. 4:114–122. 2014.PubMed/NCBI
|
|
19
|
McLaren JE, Michael DR, Ashlin TG and
Ramji DP: Cytokines, macrophage lipid metabolism and foam cells:
Implications for cardiovascular disease therapy. Prog Lipid Res.
50:331–347. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vizzardi E, Cavazzana I, Sciatti E,
Bonadei I, D'Aloia A, Tincani A, Franceschini F and Metra M:
Evaluation of ascending aorta wall in rheumatoid arthritis by
tissue and strain Doppler imaging during anti-tumor necrosis
factor-α therapy. Clin Cardiol. 37:738–743. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim HS and Lip GY: Circulating matrix
metalloproteinase-9 levels in atherosclerotic vascular disease: A
possible measurement of systemic or specific disease
pathophysiology? J Intern Med. 263:620–622. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galán M, Kassan M, Kadowitz PJ, Trebak M,
Belmadani S and Matrougui K: Mechanism of endoplasmic reticulum
stress-induced vascular endothelial dysfunction. Biochim Biophys
Acta. 1843:1063–1075. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Civelek M, Manduchi E, Riley RJ, Stoeckert
CJ Jr and Davies PF: Chronic endoplasmic reticulum stress activates
unfolded protein response in arterial endothelium in regions of
susceptibility to atherosclerosis. Circ Res. 105:453–461. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou AX and Tabas I: The UPR in
atherosclerosis. Semin Immunopathol. 35:321–332. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oh J, Riek AE, Weng S, Petty M, Kim D,
Colonna M, Cella M and Bernal-Mizrachi C: Endoplasmic reticulum
stress controls M2 macrophage differentiation and foam cell
formation. J Biol Chem. 287:11629–11641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Myoishi M, Hao H, Minamino T, Watanabe K,
Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H,
Gabbiani G, et al: Increased endoplasmic reticulum stress in
atherosclerotic plaques associated with acute coronary syndrome.
Circulation. 116:1226–1233. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song XJ, Yang CY, Liu B, Wei Q, Korkor MT,
Liu JY and Yang P: Atorvastatin inhibits myocardial cell apoptosis
in a rat model with post-myocardial infarction heart failure by
downregulating ER stress response. Int J Med Sci. 8:564–572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia JG, Xu FF, Qu Y, Song DG, Shen H and
Liu XH: Atorvastatin post-conditioning attenuates myocardial
ischemia reperfusion injury via inhibiting endoplasmic reticulum
stress-related apoptosis. Shock. 42:365–371. 2014. View Article : Google Scholar : PubMed/NCBI
|