Introduction
Atherosclerosis is a complicated vascular disease,
and is generally recognized as a chronic inflammatory disease
(1–3). It is characterized by infiltration of
various inflammatory cells, including T cells, mast cells and
macrophages, into atherosclerotic plaques at all stages of
development of atherosclerosis (4). Atherosclerosis is initiated by
subendothelial retention of apolipoprotein B-containing
lipoproteins followed by adhesion and penetration of inflammatory
cells from the blood into the arterial intima (5). By secreting cytokines and growth
factors, these inflammatory cells, together with resident vascular
wall cells, can promote the migration and proliferation of vascular
smooth muscle cells (VSMCs), which participate in the formation of
neointima and finally plaque formation (6–8).
In addition to intimal inflammation, numerous
previous studies have focused on the correlation between
adventitial inflammation and atherosclerosis. Schwartz and Mitchell
(9) were the first to report that
cellular infiltration of the adventitia is associated with
atheromatous plaques in 1962. T lymphocytes and B lymphocytes,
together with a small proportion of macrophages, have been observed
in areas of adventitial inflammation (10,11).
Furthermore, a previous study demonstrated that the degree of
adventitial inflammation is positively correlated with the severity
of atherosclerotic lesion (11).
Although a number of studies support the concept that adventitial
inflammation is derived from the spread of intimal inflammation
(12–14), a recent study showed that
adventitial inflammation may be responsible for the accumulation of
macrophages in the intima and involved in atherosclerotic lesion
development as an important early event (6,15,16).
Shimokawa et al (17)
suggested that in vivo chronic treatment of the coronary
adventitia with interleukin (IL)-1β can induce intimal thickening,
which is the earliest pathological change in atherosclerosis.
Subsequently, the authors further found that treatment of the
coronary adven-titia with other key inflammatory cytokines [tumor
necrosis factor (TNF)-α and IL-1α] also induces
arteriosclerosis-like changes in the coronary intima (18). These studies strongly indicate that
adventitial inflammation can trigger the development of
atherosclerosis; however, the mechanism involved in this process
has not been clearly defined.
As mentioned, inflammation-induced VSMC
proliferation and migration from the media to intima are key early
events in lesion development (19). VSMCs respond to specific
extracellular stimuli secreted by inflammatory cells or vascular
wall cells to induce signal transduction to the nucleus. This
activates the expression of a series of genes controlling VSMC
proliferation and migration, thus promoting VSMC migration from the
media to the intima as well as VSMC proliferation. Previous studies
have shown that the Janus kinase/signal transducer and activator of
transcription (JAK/STAT) signaling pathway is important role in
atherosclerotic lesion development (20–22).
Notably, it has been verified that the JAK2/STAT3 signaling pathway
is critical in VSMC proliferation and migration (23–25).
Tyrosine phosphorylation occurs sequentially on JAK2 and STAT3,
which in turn activates downstream target gene expression
influencing VSMC proliferation and migration (26–30).
In the current study, the adventitial inflammatory process was
initiated by an wrapping agar suspension containing interleukin
(IL)-1β around the adventitia to determine the role of the
JAK2/STAT3 signaling pathway in mediating adventitial
inflammation-induced vascular proliferation and lesion
formation.
Materials and methods
Animals
A total of 15, ten-week old, male Sprague-Dawley
(SD) rats (weight, 180–200 g) were obtained from the Vital River
Laboratories, Co., Ltd. (Beijing, China). All rats were housed at
22°C with a 12:12-h light-dark cycle and free access to standard
rodent chow and tap water in a rodent facility. Animal experiments
were approved by the Animal Research Committee of the 101st
Hospital of PLA (Wuxi, China). All procedures were performed in
accordance with the guidelines of the Animal Research Committee of
the 101st Hospital.
Preparation of IL-1β sustained-release
agar suspension
A 100-mg slurry of CNBr-activated Sepharose
4B (45–165 µm diameter; Sigma-Aldrich, St. Louis, MO, USA)
was washed with 1 mM HCl by vortexing and centrifugation (328 × g
for 5 min) four times. The slurry was then washed three times with
a solution containing 0.5 M NaCl and 1 M NaHCO3,
followed by centrifugation (328 × g for 5 min). Then, 100 µg
IL-1β (Beijing Sino Biological Technology Co., Ltd., Beijing,
China) was added to the slurry precipitation and mixed thoroughly.
This mixture was incubated at room temperature for 1 h followed by
incubation at 4°C overnight. The following day, the sample was
centrifuged briefly for 5 min at 328 × g and the liquid was
discarded. The remaining solid phase was suspended in 1 M Tris-HCl
to form a slurry, and the mixture was incubated at room temperature
for 1 h, it was then centrifuged and the liquid was removed. Saline
was added and the beads were washed 3 times and resuspended in 2 ml
saline. The final concentration of IL-1β was 50 mg/ml.
Tyrosine kinase (JAK2) inhibitor AG490
sustained-release gel preparation
P-F127 (0.3 g; Sigma-Aldrich) was dissolved in 1 ml
phosphate-buffered saline (PBS), and then 30 µg AG490
(Sigma-Aldrich) was added. This mixture was vortexed and incubated
at 4°C overnight. The final concentration of AG490 was 100
µM.
Construction of the adventitial
inflammation-induced intimal proliferation rat model
Fifty SD male rats were randomly divided into an
experimental group and a control group (n=25). Prior to surgery,
rats were anesthetized by intraperitoneal injection of 2% sodium
pentobarbital (50 mg/kg body weight; Sigma-Aldrich). The animals
were supine-fixed on a surgical board. Following removing the neck
hair using a razor and routinely disinfecting the skin using
iodophor disinfection solution, a 3-cm longitudinal incision was
made along the midline to expose 2 to 3 cm of the left common
carotid artery. Gauze (2×0.5 cm; Medicom Inc., Ltd., Shanghai,
China) was placed beneath the artery. For the experimental group,
50 µl agar suspensions with IL-1β was placed on the gauze,
and for the control group, 50 µl of the agar suspension
without IL-1β was placed on the gauze. The gauze was wrapped around
the blood vessel using surgical cord (Medicom Inc., Ltd.) and
sutures were used to close the incision. Sodium penicillin (100
kIU; Shandong Lukang Pharmaceutical Co., Ltd., Jining, China) was
injected intra-muscularly once every three days to prevent
infection. The animals were given free access to standard rodent
chow and tap water, and five animals from each group were
sacrificed at 2 h, 8 h, 24 h, 48 h and 1 week post-surgery. Target
blood vessels were excised (~4 mm) and fixed in 4%
paraformalde-hyde (Beyotime Institute of Biotechnology, Shanghai,
China) and the remaining tissue was frozen in liquid nitrogen. For
AG490 treatment, both sides of the carotid artery of SD male rats
(n=15) were surgically separated, and an experimental side and a
control side were assigned. For experimental sides, 150 µl
AG490 sustained release P-F127 gel was applied, and for control
sides, 150 µl gel with blank solution was applied.
Subsequently, 50 µl agar suspension with IL-1β was applied
to both sides, and surgery was completed as described above. Five
animals were randomly selected for sacrifice at 8 h, 48 h, and 1
week post-surgery, and samples were obtained as described
above.
Histopathological examination
Conventional methods were used to fix and dehydrate
the vascular tissue, and embed the tissues in paraffin (Chuandong
Chemical Co., Ltd., Chongqing, China). The paraffin-embedded
tissues were cut into 4-µm slices, and sections were stained
using hematoxylin and eosin (H&E; Beyotime Institute of
Biotechnology). Changes in blood vessel layers were observed using
a microscope (BX51; Olympus, Tokyo, Japan). Image-Pro Plus 6.0
software (Media Cybernetics, Rockville, MD, USA) was used to
measure layer areas and calculate the intima/media ratio (IMR).
Western blot analysis
Proteins were extracted from the frozen vascular
tissues by homogenization using E.Z.N.A DNA/RNA/Protein Isolation
kit (Omega Bio-Tek, Inc., Norcross, GA, USA) and quantified using
the bicinchoninic acid assay (Beyotime Institute of Biotechnology).
In total, 15 µg (to detect total JAK2 and STAT3 proteins) or
30 µg (to detect phosphorylated proteins) protein were
resolved using 8% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to polyvinylidene fluoride
membranes (Merck Millipore, Billerica, MA, USA). Membranes were
blocked with 5% bovine serum albumin (Sigma-Aldrich) for 1 h at
room temperature, and then incubated with rabbit polyclonal
anti-rat STAT3 antibody (1:400; cat. no. sc-7179; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), rabbit polyclonal
anti-rat JAK2 antibody (1:1,000, cat. no. ab39636; Epitomics,
Burlingame, CA, USA), rabbit polyclonal anti-rat phosphorylated
JAK2 (p-JAK2) antibody (1:1,000; cat. no. ab68268; Epitomics), and
rabbit anti-rat phosphorylated STAT3 (p-STAT3) antibodies (1:1,000;
cat. no. ab76315; Epitomics) overnight at 4°C overnight. Rabbit
anti-glyceraldehyde-3-phosphate dehydrogenase antibody (1:1,000;
cat. no. sc-25778; Santa Cruz Biotechnology, Inc.) was used as a
loading control for normalization. Membranes were washed and
incubated with goat anti-rabbit horseradish peroxidase-conjugated
secondary antibody (1:5,000; cat. no. zb-2301; Beijing Zhongshan
Jinqiao Biotechnology Co., Ltd., Beijing, China) at 37°C for 1 h.
Chemiluminescence was detected using an enhanced chemiluminescence
reagent, and Quantity One software (version 4.6.2; Bio-Rad
Laboratories, Inc., Hercules, CA, USA) was used to measure band
intensity.
Immunohistochemical staining
Deparaffinized and rehydrated tissue sections were
placed in a microwavable vessel with antigen retrieval buffer
(citric acid-sodium citrate buffer, pH 6.0) and exposed to
microwaves for 15 min in a microwave oven at 750 W. Using an
Envision immunohistochemical staining kit (Dako, Carpinteria, CA,
USA), paraffin sections were subsequently stained with rabbit
anti-rat α-actin polyclonal antibody (1:100; cat. no. sc-53142;
Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-rat
proliferating cell nuclear antigen (PCNA; 1:100; cat. no. sc-7907;
Santa Cruz Biotechnology, Inc.), rabbit polyclonal anti-rat
embryonic smooth muscle myosin heavy chain (SM-emb) antibody
(1:100; cat. no. sc-65734; Santa Cruz Biotechnology, Inc.), rabbit
polyclonal anti-rat p-JAK2 (1:100; cat. no. kg11151; Nanjing KeyGen
Biotech. Co., Ltd., Nanjing, China), rabbit polyclonal anti-rat
p-STAT3 (1:100; cat. no. kg11045-1; Nanjing KeyGen Biotech. Co.,
Ltd.) to determine the corresponding proteins in tissues.
Statistical analysis
SPSS17.0 software (SPSS Software, Inc., Chicago, IL,
USA) was used to perform statistical analysis. Experimental data
were collected from multiple animals under each experimental
condition (n=5) and are presented as the mean ± standard deviation.
One-way analysis of variance was conducted to compare means of
three or more groups. The Student-Newman-Keuls (SNK) method was
used for pairwise comparisons. A t-test was used to compare
vascular protein intensity from the left and right sides of the
same rat. P<0.05, was considered to indicate a statistically
significant difference.
Results
Adventitial administration of IL-1β
induces smooth muscle cell proliferation, migration, and neointimal
formation
The model of adventitial inflammation induced by
treatment with IL-1β has been well documented in previous study
(17). In the present study, the
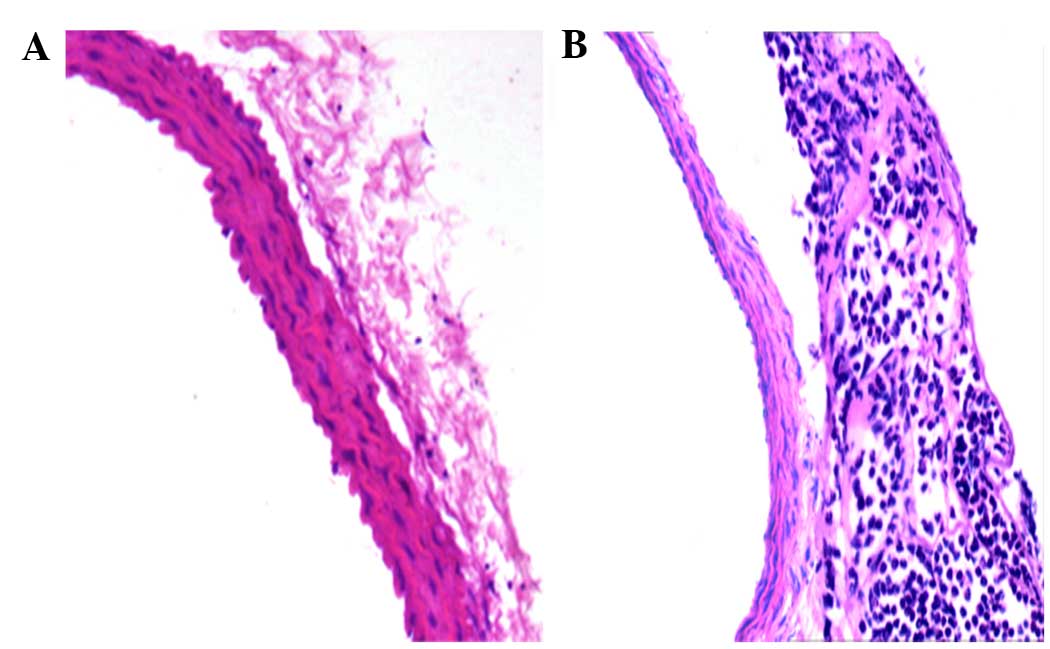
results of H&E staining demonstrated leucocyte, including
numerous neutrophils, infiltration into the vascular adventitia 2 h
after surgery (Fig. 1), which
indicated that adventitial inflammation was triggered and the model
was successfully established.
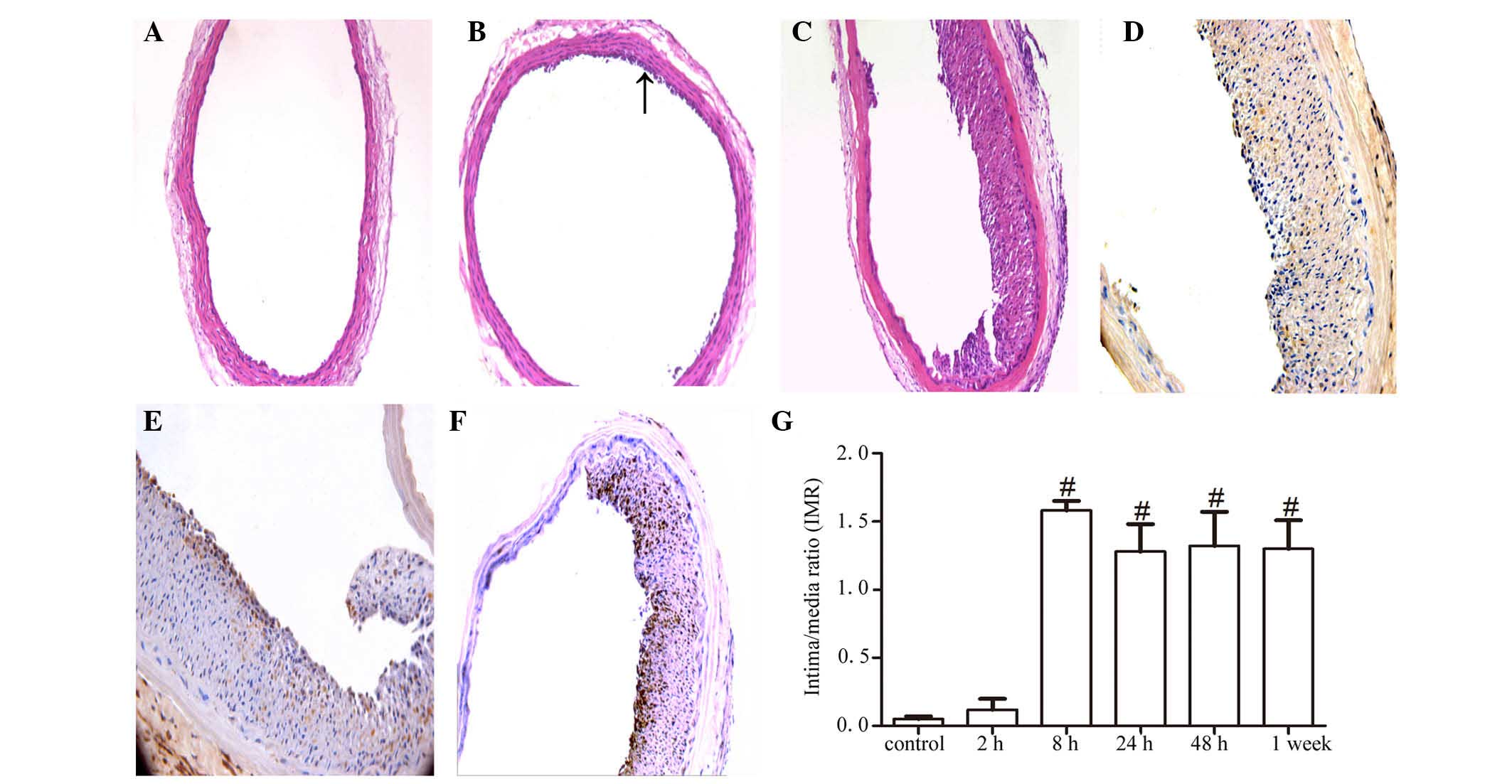
At all the time points, the vascular morphology of
the control group was normal and no intimal proliferation responses
were observed (Fig. 2A). After
surgery (2 h), cell proliferation and migration towards the lumen
were visible in the experimental group (Fig. 2B). At 8 h, intimal proliferation
and luminal narrowing of the blood vessels was observed, which was
shown by H&E staining (Fig.
2C) and immunohistochemical staining with anti-rat PCNA
antibody (Fig. 2D), respectively.
Further immunohistochemical staining with the anti-rat α-actin
antibody (Fig. 2E) and anti-rat
SM-emb antibody (Fig. 2F) showed
positive staining in the majority of neointimal cells, indicating
that the cells that has proliferated and migrated towards the lumen
were VSMCs. Statistical analysis of IMR results from different time
points revealed significant differences in intima proliferation
over time (P<0.05). As early as 8 h post-surgery, the IMR of
mice in the experimental group was significantly higher than that
in the control group (P<0.05; Fig.
2G).
Adventitial administration of IL-1β
activates the JAK2/STAT3 pathway in vascular tissue
After the adventitial inflammatory response was
induced by treatment with IL-1β, protein expression of members of
the JAK2/STAT3 pathway were evaluated using western blotting. As
shown in Fig. 3A and B, the
vascular expression of total JAK2 and STAT3 was comparable between
the experimental group and the control group at all the examined
time points. However, treatment with IL-1β resulted in a gradual
and progressive increase in vascular p-JAK2 which peaked at 8 h
post-treatment, and then gradually decreased to normal levels.
Similarly, after IL-1β treatment, the p-STAT3 level increased at 2
h post-treatment, peaked at 24 h post-treatment, was maintained
until 48 h post-treatment, and then gradually decreased.
Immunohistochemical staining revealed that p-JAK2 and p-STAT3 were
expressed in VSMCs, particularly in proliferating cells and
migrating cells within the lumen and near the neointima (Fig. 3C and D).
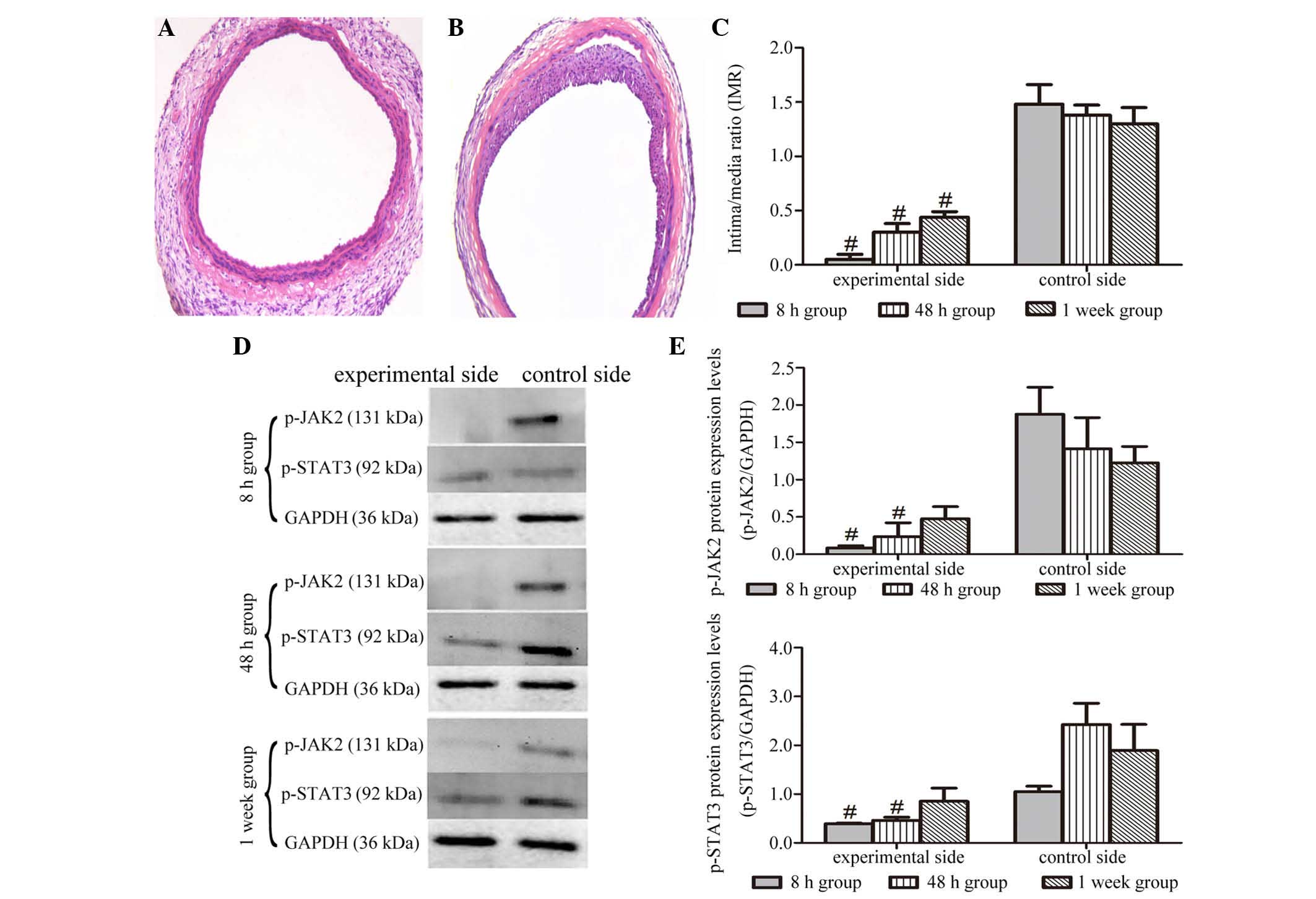
AG490 administration inhibits
IL-1β-induced intimal proliferation and phosphorylation of JAK2 and
STAT3
Compared with the control group, VSMC proliferation
and migration, and intimal proliferation in the experimental group
administered the JAK2 inhibitor (AG490) were significantly
suppressed (Fig. 4A and B). IMR
measurements and statistical analysis showed that the arterial IMR
of rats in the AG490-treated group was significantly decreased
compared that in the group without AG490 treatment at all the
examined time points (Fig.
4C).
The effect of AG490 administration on the
IL-1β-increased expression of p-JAK2 and p-STAT3 was also
investigated by western blot analysis. As shown in Fig. 4D and E, The IL-1β-increased
expression of p-JAK2 was significantly inhibited by AG490
administration at the 8 and 48 h time points. One week after
treatment, although the expression of p-JAK2 in the artery of
experimental side was lower than that of control side, there was no
significant difference between the two values. Similarly, the
increase of p-STAT3 induced by IL-1β was also suppressed by AG490
treatment at 8 and 48 h time points, and no significant difference
in the expression of p-STAT3 was observed between the two groups at
1 week.
Discussion
Studies have demonstrated that artery adventitial
inflammation is involved in the initiation and development of
atherosclerosis. Chemical cues, such as endotoxin,
lipopolysaccharide and interleukins, have been used to induce
adventitial inflammation (31). In
the present study, an adventitial inflammation model was
successfully created by wrapping IL-1β around the rat carotid. In
this model, adventitial inflammation induced VSMC proliferation and
migration as well as intimal proliferation. In addition, the
expression of p-JAK2 and p-STAT3 increased following IL-1β
treatment. Furthermore, an inhibitor of JAK2/STAT3 pathway, AG490,
suppressed IL-1β-induced intimal proliferation and phosphorylation
of JAK2 and STAT3.
Neointimal formation, which is considered to be a
marker for early atherosclerosis, is the main pathological process
of atherosclerosis (32,33). It has been reported that the
excessive proliferation and migration of VSMCs from the arterial
media into the intima under the stimulation of cytokines and growth
factors participates in neointimal formation during the development
of atherosclerosis (6–8) or restenosis after percutaneous
transluminal coronary angioplasty (34). In the current study, the results
showed that adventitial treatment with IL-1β resulted in neointimal
formation. Furthermore, immunohistochemical staining with antibody
against rat α-actin and SM-emb, markers of VSMCs, indicated that
VSMCs had migrated into the neointima, and were the main cellular
component of the neointima, confirming their role in neointima
formation. These results suggested that the initial step in
atherosclerosis had been triggered by adventitial inflammation
induced by treatment with IL-1β, which was consistent with the
results of a previous study (17).
The JAK2/STAT3 pathway can be triggered when a
growth factor or cytokine binds to its receptor which then
phosphorylates JAK2. The phosphorylated JAK2 subsequently
phosphorylates STAT3, which results in the dimerization and
translocation of STAT3 to the nucleus. Finally, STAT3 in the
nucleus binds to the promoter region of its target genes to
regulate the biological behavior of cells (35). The JAK2/STAT3 pathway is involved
in numerous critical functions in normal and malignant cells, such
as differentiation, proliferation, survival and angiogenesis
(36,37). During the development of
atherosclerosis, activation of the JAK/STAT pathway, including the
JAK2/STAT3 pathway have been observed in atherosclerotic lesions
(38,39). Moreover, the JAK2/STAT3 pathway
mediates the proliferation and migration of VSMCs (25). In the current study, expression of
JAK2 and STAT3 was analyzed to determine the time course of
induction by IL-1β. Immunohistochemistry revealed that following
stimulation, the phosphorylation of JAK2 and STAT3 occurred
primarily in proliferating and migrating VSMCs, indicating that
VSMCs migrate and proliferate following autophosphorylation rather
than indirect activation via other cells. The results of western
blot analysis demonstrated that following IL-1β administration,
JAK2 phosphorylation occurred at 2 h and peaked at 8 h; and STAT3
phosphorylation was initiated at 2 h and peaked at 24 h, remaining
elevated for up to a week. These results indicated that the
JAK2/STAT3 pathway was activated at the early stages of IL-1β
treatment. In a rat carotid artery balloon injury experiment, JAK2
and STAT3 tyrosine phosphorylation levels peaked after 1 week, and
VSMC proliferation and migration appeared 96 h post-surgery
(40). In the present study,
neointimal changes were observed as early as 8 h post-surgery, and
phosphorylation levels of the two proteins peaked relatively
earlier, indicating that JAK2 and STAT3 phosphorylation levels are
correlated with VSMC proliferation and migration.
Numerous studies have shown that AG490 is a JAK2
inhibitor, which competes for binding with tyrosine kinases, thus
inhibiting JAK2 phosphorylation. In the present study, 8 and 48 h
post-AG490 treatment, p-JAK2 levels were lower than the
detection range of western blot analysis. Although minimal levels
of p-JAK2 could be detected 1 week post-AG490 treatment,
this may have resulted from the reduction in AG490 effectiveness
due to dose elimination by degradation. It can be inferred from
this experiment that AG490 administration inhibits the
IL-1β-induced activation of the JAK2/STAT3 pathway. Notably,
IL-1β-induced VSMC proliferation and migration and neointimal
formation was also significantly suppressed following AG490
administration, which suggested that VSMC proliferation and
migration, and neointimal formation induced by IL-1β is dependent
on the JAK2/STAT3 signal transduction pathway. This result was
similar to that of several previous studies that demonstrated that
AG490 effectively inhibits VSMC proliferation and migration by
inhibiting JAK2 phosphorylation (41–43).
However, the downstream molecules of the JAK2/STAT3 signaling
pathway regulating the final intimal proliferation process remain
unclear. A previous study demonstrated that metalloproteinases
(MMPs) regulate the migration, proliferation, and death of VSMCs
(44). Furthermore, members of
MMPs family have been reported to be regulated by STAT3 (45,46).
These results imply that MMPs may act downstream of the JAK2/STAT3
signaling pathway and therefore functionally regulate the final
intimal proliferation process induced by IL-1β. This hypothesis
requires further investigation.
In this study, IL-1β administration to the vascular
adventitia was used as a model of adventitial inflammation, rat
carotid artery VSMC proliferation, migration, and neointima
formation were induced, which confirmed the role of adventitial
inflammation in the development of atherosclerosis and was
consistent with the results of a previous study (17), however the mechanisms underlying
this remain unclear. The present study found that the JAK2/STAT3
signaling pathway was activated by adventitial inflammation, and
administration of AG490 inhibited JAK2/STAT3 signaling accompanied
by decreases in VSMC proliferation, migration, and neointima
formation. These results indicate that the JAK2/STAT3 pathway is
important in neointimal formation induced by adventitial
inflammation and may be a potential therapeutic target for the
clinical treatment of arterial atherosclerosis and restenosis
following vascular injury. Notably, an oral JAK1/JAK2 inhibitor,
ruxolitinib, has been approved to use in the treatment of
myelofibrosis by the FDA (47),
which indicates that agents blocking the JAK2/STAT3 signaling
pathway are available in the clinic besides being feasible in
theory.
Numerous major inflammatory cytokines have been
demonstrated to induce arteriosclerosis-like changes in the
coronary intima in various studies (17,18).
The current study, however, only investigated the mechanisms
involved in IL-1β-induced intimal proliferation. Further studies
are required to explore whether the JAK2/STAT3 signaling pathway
universally mediates intimal proliferation induced by these
inflammatory cytokines.
References
|
1
|
Clarke MC, Talib S, Figg NL and Bennett
MR: Vascular smooth muscle cell apoptosis induces
interleukin-1-directed inflammation: Effects of
hyperlipidemia-mediated inhibition of phagocytosis. Circ Res.
106:363–372. 2010. View Article : Google Scholar
|
|
2
|
Kleemann R, Zadelaar S and Kooistra T:
Cytokines and atherosclerosis: A comprehensive review of studies in
mice. Cardiovasc Res. 79:360–376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sprague AH and Khalil RA: Inflammatory
cytokines in vascular dysfunction and vascular disease. Biochem
Pharmacol. 78:539–552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soehnlein O and Weber C: Myeloid cells in
atherosclerosis: Initiators and decision shapers. Semin
Immunopathol. 31:35–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Subbotin VM: Neovascularization of
coronary tunica intima (DIT) is the cause of coronary
atherosclerosis. Lipoproteins invade coronary intima via
neovascularization from adventitial vasa vasorum, but not from the
arterial lumen: A hypothesis. Theor Biol Med Model. 9:112012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim S and Park S: Role of vascular smooth
muscle cell in the inflammation of atherosclerosis. BMB Rep.
47:1–7. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vogt F, Zernecke A, Beckner M, Krott N,
Bosserhoff AK, Hoffmann R, Zandvoort MA, Jahnke T, Kelm M, Weber C
and Blindt R: Blockade of angio-associated migratory cell protein
inhibits smooth muscle cell migration and neointima formation in
accelerated atherosclerosis. J Am Coll Cardiol. 52:302–311. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schwartz CJ and Mitchell JR: Cellular
infiltration of the human arterial adventitia associated with
atheromatous plaques. Circulation. 26:73–78. 1962. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watanabe M, Sangawa A, Sasaki Y, Yamashita
M, Tanaka-Shintani M, Shintaku M and Ishikawa Y: Distribution of
inflammatory cells in adventitia changed with advancing
atherosclerosis of human coronary artery. J Atheroscler Thromb.
14:325–331. 2007. View
Article : Google Scholar
|
|
11
|
Parums D and Mitchinson MJ: Demonstration
of immunoglobulin in the neighbourhood of advanced atherosclerotic
plaques. Atherosclerosis. 38:211–216. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao L, Moos MP, Gräbner R, Pédrono F, Fan
J, Kaiser B, John N, Schmidt S, Spanbroek R, Lötzer K, et al: The
5-lipoxygenase pathway promotes pathogenesis of
hyperlipidemia-dependent aortic aneurysm. Nat Med. 10:966–973.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Moos MP, John N, Gräbner R, Nossmann S,
Günther B, Vollandt R, Funk CD, Kaiser B and Habenicht AJ: The
lamina adventitia is the major site of immune cell accumulation in
standard chow-fed apolipoprotein E-deficient mice. Arterioscler
Thromb Vasc Biol. 25:2386–2391. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Galkina E, Kadl A, Sanders J, Varughese D,
Sarembock IJ and Ley K: Lymphocyte recruitment into the aortic wall
before and during development of atherosclerosis is partially
L-selectin dependent. J Exp Med. 203:1273–1282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rayner K, Van Eersel S, Groot PH and Reape
TJ: Localisation of mRNA for JE/MCP-1 and its receptor CCR2 in
atherosclerotic lesions of the ApoE knockout mouse. J Vasc Res.
37:93–102. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kortelainen ML and Porvari K: Adventitial
macrophage and lymphocyte accumulation accompanying early stages of
human coronary atherogenesis. Cardiovasc Pathol. 23:193–197. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shimokawa H, Ito A, Fukumoto Y, Kadokami
T, Nakaike R, Sakata M, Takayanagi T, Egashira K and Takeshita A:
Chronic treatment with interleukin-1 beta induces coronary intimal
lesions and vasospastic responses in pigs in vivo. The role of
platelet-derived growth factor. J Clin Invest. 97:769–776. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fukumoto Y, Shimokawa H, Ito A, Kadokami
T, Yonemitsu Y, Aikawa M, Owada MK, Egashira K, Sueishi K, Nagai R,
et al: Inflammatory cytokines cause coronary arteriosclerosis-like
changes and alterations in the smooth-muscle phenotypes in pigs. J
Cardiovasc Pharmacol. 29:222–231. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tedgui A and Mallat Z: Cytokines in
atherosclerosis: Pathogenic and regulatory pathways. Physiol Rev.
86:515–581. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liao M, Xu J, Clair AJ, Ehrman B, Graham
LM and Eagleton MJ: Local and systemic alterations in signal
transducers and activators of transcription (STAT) associated with
human abdominal aortic aneurysms. J Surg Res. 176:321–328. 2012.
View Article : Google Scholar
|
|
21
|
Mo ZC, Xiao J, Liu XH, Hu YW, Li XX, Yi
GH, Wang Z, Tang YL, Liao DF and Tang CK: AOPPs inhibits
cholesterol efflux by down-regulating ABCA1 expression in a
JAK/STAT signaling pathway-dependent manner. J Atheroscler Thromb.
18:796–807. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beyazit Y, Sayilir A, Suvak B and Torun S:
The missing link between obesity and hepatocellular carcinoma:
IL-6-mediated STAT-3 activation as a key player in
hepatocarcinogenesis. Digestion. 84:185–186. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Manea A, Tanase LI, Raicu M and Simionescu
M: Jak/STAT signaling pathway regulates nox1 and nox4-based NADPH
oxidase in human aortic smooth muscle cells. Arterioscler Thromb
Vasc Biol. 30:105–112. 2010. View Article : Google Scholar
|
|
24
|
Ortiz-Munoz G, Martin-Ventura JL,
Hernandez-Vargas P, Mallavia B, Lopez-Parra V, Lopez-Franco O,
Muñoz-Garcia B, Fernandez-Vizarra P, Ortega L, Egido J and
Gomez-Guerrero C: Suppressors of cytokine signaling modulate
JAK/STAT-mediated cell responses during atherosclerosis.
Arterioscler Thromb Vasc Biol. 29:525–531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song B, Jin H, Yu X, Zhang Z, Yu H, Ye J,
Xu Y, Zhou T, Oudit GY, Ye JY, et al: Angiotensin-converting enzyme
2 attenuates oxidative stress and VSMC proliferation via the
JAK2/STAT3/SOCS3 and profiling-1/MAPK signaling pathways. Regul
Pept. 185:44–51. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jinno T, Iwai M, Li Z, Li JM, Liu HW, Cui
TX, Rakugi H, Ogihara T and Horiuchi M: Calcium channel blocker
azelnidipine enhances vascular protective effects of AT1 receptor
blocker olmesartan. Hypertension. 43:263–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Banes AK, Shaw SM, Tawfik A, Patel BP,
Ogbi S, Fulton D and Marrero MB: Activation of the JAK/STAT pathway
in vascular smooth muscle by serotonin. Am J Physiol Cell Physiol.
288:C805–C812. 2005. View Article : Google Scholar
|
|
28
|
Sun J, Zheng J, Ling KH, Zhao K, Xie Z, Li
B, Wang T, Zhu Z, Patel AN, Min W, et al: Preventing intimal
thickening of vein grafts in vein artery bypass using STAT-3 siRNA.
J Transl Med. 10:22012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dronadula N, Liu Z, Wang C, Cao H and Rao
GN: STAT-3-dependent cytosolic phospholipase A2 expression is
required for thrombin-induced vascular smooth muscle cell motility.
J Biol Chem. 280:3112–3120. 2005. View Article : Google Scholar
|
|
30
|
Watanabe S, Mu W, Kahn A, Jing N, Li JH,
Lan HY, Nakagawa T, Ohashi R and Johnson RJ: Role of JAK/STAT
pathway in IL-6-induced activation of vascular smooth muscle cells.
Am J Nephrol. 24:387–392. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimokawa H, Morishige K, Miyata K,
Kandabashi T, Eto Y, Ikegaki I, Asano T, Kaibuchi K and Takeshita
A: Long-term inhibition of Rho-kinase induces a regression of
arteriosclerotic coronary lesions in a porcine model in vivo.
Cardiovasc Res. 51:169–177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zeller I, Knoflach M, Seubert A,
Kreutmayer SB, Stelzmüller ME, Wallnoefer E, Blunder S, Frotschnig
S, Messner B, Willeit J, et al: Lead contributes to arterial
intimal hyperplasia through nuclear factor erythroid 2-related
factor-mediated endothelial interleukin 8 synthesis and subsequent
invasion of smooth muscle cells. Arterioscler Thromb Vasc Biol.
30:1733–1740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Sukhova GK, Sun JS, Xu WH, Libby P
and Shi GP: Lysosomal cysteine proteases in atherosclerosis.
Arterioscler Thromb Vasc Biol. 24:1359–1366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pauletto P, Sartore S and Pessina AC:
Smooth-muscle-cell proliferation and differentiation in neointima
formation and vascular restenosis. Clin Sci (Lond). 87:467–479.
1994. View Article : Google Scholar
|
|
35
|
Raible DJ, Frey LC and Brooks-Kayal AR:
Effects of JAK2-STAT3 signaling after cerebral insults. JAKSTAT.
3:e295102014.PubMed/NCBI
|
|
36
|
Levy DE and Darnell JE Jr: Stats:
Transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sansone P and Bromberg J: Targeting the
interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol.
30:1005–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Seki Y, Kai H, Shibata R, Nagata T,
Yasukawa H, Yoshimura A and Imaizumi T: Role of the JAK/STAT
pathway in rat carotid artery remodeling after vascular injury.
Circ Res. 87:12–18. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Recinos A III, LeJeune WS, Sun H, Lee CY,
Tieu BC, Lu M, Hou T, Boldogh I, Tilton RG and Brasier AR:
Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in
aortic adventitia of LDL receptor-deficient mice. Atherosclerosis.
194:125–133. 2007. View Article : Google Scholar
|
|
40
|
Shibata R, Kai H, Seki Y, Kato S, Wada Y,
Hanakawa Y, Hashimoto K, Yoshimura A and Imaizumi T: Inhibition of
STAT3 prevents neointima formation by inhibiting proliferation and
promoting apoptosis of neointimal smooth muscle cells. Hum Gene
Ther. 14:601–610. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Neria F, Caramelo C, Peinado H,
González-Pacheco FR, Deudero JJ, de Solis AJ, Fernández-Sánchez R,
Peñate S, Cano A and Castilla MA: Mechanisms of endothelial cell
protection by blockade of the JAK2 pathway. Am J Physiol Cell
Physiol. 292:C1123–C1131. 2007. View Article : Google Scholar
|
|
42
|
Neeli I, Liu Z, Dronadula N, Ma ZA and Rao
GN: An essential role of the Jak-2/STAT-3/cytosolic phospholipase
A(2) axis in platelet-derived growth factor BB-induced vascular
smooth muscle cell motility. J Biol Chem. 279:46122–46128. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sasaguri T, Teruya H, Ishida A, Abumiya T
and Ogata J: Linkage between alpha(1) adrenergic receptor and the
Jak/STAT signaling pathway in vascular smooth muscle cells. Biochem
Biophys Res Commun. 268:25–30. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Newby AC: Matrix metalloproteinases
regulate migration, proliferation and death of vascular smooth
muscle cells by degrading matrix and non-matrix substrates.
Cardiovasc Res. 69:614–624. 2006. View Article : Google Scholar
|
|
45
|
Zhang L, Li Y, Liu Y, Wang X, Chen M, Xing
Y and Zhu D: STAT3-mediated MMP-2 expression is required for
15-HETE-induced vascular adventitial fibroblast migration. J
Steroid Biochem Mol Biol. 149C:106–117. 2015. View Article : Google Scholar
|
|
46
|
Jo DH and Kim JH, Cho CS, Cho YL, Jun HO,
Yu YS, Min JK and Kim JH: STAT3 inhibition suppresses proliferation
of retinoblastoma through down-regulation of positive feedback loop
of STAT3/miR-17-92 clusters. Oncotarget. 5:11513–11525. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mesa RA, Verstovsek S, Gupta V,
Mascarenhas JO, Atallah E, Burn T, Sun W, Sandor V and Gotlib J:
Effects of ruxolitinib treatment on metabolic and nutritional
parameters in patients with myelofibrosis from COMFORT-I. Clin
Lymphoma Myeloma Leuk. 15:214.e1–221.e1. 2015. View Article : Google Scholar
|