Introduction
Resident hepatic stellate cell (HSC) activation has
been described as a central event in the development of hepatic
fibrosis; therefore, activated HSCs are considered a major target
for anti-fibrotic therapy (1). It
has been reported that transforming growth factor-β 1 (TGF-β1)/Smad
signaling is a pivotal pathway in hepatic fibrogenesis, and
suppression of the TGF-β1/Smad signaling pathway may inhibit
collagen production and eventually alleviate hepatic fibrosis
(2–5). Despite recent progress in hepatic
fibrosis research, the mechanism underlying hepatic fibrogenesis
remains to be fully elucidated, and certain issues in the
approaches to anti-fibrotic treatment still need to be
addressed.
The ras-related C3 botulinum toxin substrate 1
(Rac1) signaling pathway has been reported to regulate various
biological processes, including cell proliferation, apoptosis,
redox signaling, and gene transcription (6–8).
Previous studies have shown that the overexpression of Rac1 is a
novel pathway involved in hepatic fibrogenesis (9,10).
In keratinocytes, Rac1 has been reported to regulate
TGF-β1-mediated epithelial cell plasticity and matrix
metalloproteinase (MMP)9 production (11). In addition, Rac1 may regulate the
expression of Smad2/3 in pancreatic carcinoma cells; the
suppression of Rac1 weakened the transcription and phosphorylation
of Smad2 but enhanced the expression of Smad3 (12). Our previous study (13) indicated that S-adenosylmethionine
(SAM) reduces the expression of Rac1, Smad3 and Smad4 in activated
HSCs; however, the association between Rac1 and Smads in activated
HSCs has yet to be revealed.
In the present study, LX-2 cells, a cell line
derived from activated human HSCs (14–18),
were used to demonstrate that SAM suppresses the expression of
Smad3/4 via methylating the Rac1 promoter in activated HSCs.
Materials and methods
Materials and chemicals
LX-2 cells were purchased from BioHermes Bio &
Medical Technology Co., Inc. (Meishan, China). Dulbecco's modified
Eagle's medium (DMEM) was purchased from Wisent Inc.
(Saint-Jean-Baptiste, QC, Canada). Fetal bovine serum (FBS) was
obtained from Biological Industries (Beit Haemek, Israel), and SAM
was from Abbott Laboratories (Lake Bluff, IL, USA). DC Protein
Assay Reagent was supplied by Bio-Rad Laboratories, Inc.,
(Hercules, CA, USA). Cell Counting kit-8 (CCK-8) was purchased from
Dōjindo Laboratories (Kumamoto, Japan), and Matrigel Basement
Membrane Matrix and Cell Culture Inserts were obtained from BD
Biosciences (San Jose, CA, USA). The QuantiTech Reverse
Transcription and QuantiFast SYBR Green PCR kits for reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) were
obtained from Qiagen (Hilden, Germany). The PrimeScript RT Master
Mix for methylation-specific PCR (MSP) was purchased from Takara
Bio, Inc., (Otsu, Japan). Transwell chambers were from EMD
Millipore (Billerica, MA, USA). The primary monoclonal antibodies,
mouse anti-human Rac1 (cat. no ab33186) and mouse anti-human
Smad3/4 (cat. no. ab75512/ab130242), polyclonal rabbit anti-human
β-actin (cat. no. ab8227), the polyclonal horseradish-peroxidase
conjugated goat anti-mouse IgG (cat. no. ab6789) and goat
anti-rabbit IgG (cat. no. ab6721) secondary antibodies were all
purchased from Abcam (Cambridge, UK.)
Cell culture
LX-2 cells were maintained in DMEM (high glucose)
medium supplemented with 10% FBS and 100 U/ml
penicillin-streptomycin solution (Biological Industries) in an
incubator containing 5% CO2. Cells were divided into
three treatment groups and incubated with 0 mM, 4 mM and 6 mM of
SAM for 24 h.
Cell proliferation assay
LX-2 cells were recovered from the culture flask
using 0.25% trypsin and 0.1% ethylenediaminetetraacetic acid, and
were then seeded onto 96-well microplates at a density of
1×104 cells/well. The cell viability was evaluated with
CCK-8, according to the manufacturer's protocol, and was expressed
as relative cell viability, using the following formula: Percentage
of cell viability (%) = OD of treated sample/OD of control sample)
× 100%. OD indicates optical density. The experiment was performed
in triplicate.
Plasmid transfection
LX-2 cells were transfected with plasmids encoding
Rac1 protein or an empty expression vector (Shanghai GeneChem Co.,
Ltd., Shanghai, China) using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. The Rac1 overexpression
plasmid was fluorescently-labeled. The untransfected,
vector-transfected and Rac1 plasmid-transfected cells were all
harvested following incubation for 48 h. Transfection efficiency
was evaluated by RT-qPCR, western blotting and inverted fluorescent
microscopy using an Axio Scope A1 microscope (Carl Zeiss AG,
Oberkochen, Germany).
Transwell migration and invasion
assays
Transwell migration and invasion assays were
performed using the 8-mm pore size Transwell system. For the cell
invasion assay the chambers were coated with Matrigel, according to
the manufacturer's protocol. Briefly, the chambers set on 24-well
cluster plates were covered with 20 µl Matrigel and were
incubated for 30 min at 37°C, whereas non-coated chambers were used
for the cell migration assay. LX-2 cells post-transfection and
control cells were resuspended in DMEM containing 0.5% FBS, and
were seeded at a density of 2×104 cells/well on the
upper chamber of the Transwell. Complete medium (0.5 ml) was added
to the lower chamber. Following a 24 h incubation, the inner
surface of the upper chamber was gently scrubbed using a cotton
bud. The cells were fixed in methanol and stained with crystal
violet. Cells that traversed the Transwell membrane were counted,
and images corresponding to the entire membrane surface were
captured using an Olympus inverted microscope equipped with a
charge-coupled device camera (CKX41-F32FL; Olympus Corp., Tokyo,
Japan). Five evenly spaced fields of cells were counted in each
well. The assay was performed in triplicate. A group of cells was
also treated with 50 µM NSC23766 (Tocris Bioscience,
Bristol, UK) for 48 h, a Rac1-specific inhibitor.
Western blot analysis
Proteins were extracted in lysis buffer [30 mM Tris,
pH 7.5, 150 mM sodium chloride, 1 mM phenylmethylsulfonyl fluoride,
1 mM sodium orthovanadate, 1% Nonidet P-40, 10% glycerol, and
phosphatase and protease inhibitor cocktail (Roche Diagnostics,
GmbH, Mannheim, Germany)]. Total proteins (30 µg) were then
separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and electrophoretically transferred onto
polyvinylidene fluoride membranes (0.45 µl; EMD Millipore).
They were quantified using a DC Protein Assay. The membranes were
blocked with 5% non-fat milk and Tris-buffered saline with Tween 20
for 1 at room temperature. Next, they were probed with primary
antibodies (Rac1, 1:1,000; Smad3, 1:5,000; Smad4, 1:1,000; β-actin,
1:1,000) overnight at 4°C, and were then incubated with the
secondary antibodies (goat anti-mouse IgG, 1:2,000; goat
anti-rabbit IgG, 1:2,000) for 1 h at room temperature. The blots
were visualized using Immobilon Western Chemiluminescent HRP
Substrate (EMD Millipore). Images of the western blotting products
were captured and analyzed using Quantity One V4.31 (Bio-Rad
Laboratories, Inc.).
RT-qPCR
Total RNA was extracted from the cells with TRIzol
(Invitrogen; Thermo Fisher Scientific, Inc.) and quantified using a
spectrophotometer (NanoDrop 2000; Thermo Fisher Scientific, Inc.).
The samples were treated with gDNA Wipeout Buffer from the
QuantiFast SYBR kit. RT was performed with 1 µg RNA in a
total reaction volume of 20 µl containing 200 U Moloney
murine leukemia virus reverse transcriptase, according to the
manufacturer's protocol, using the following protocol: 42°C for 15
min and 95°C for 3 min. The PCR primers (β-actin, forward
5′-AGCGAGCATCCCCCAAAGTT, reverse 5′-GGGCACGAAGGCTCATCATT; Rac1,
forward 5′-CCCTATCCTATCCGCAAACA, reverse 5′-CGCACCTCAGGATACCACTT;
and α-smooth muscle actin (SMA), forward 5′-ACTGGGACGACGACATGGAAAAG
and reverse 5′-TAGATGGGGACATTGTGGGT) were designed and validated by
Sangon Biotech Co., Ltd. (Shanghai, China). qPCR was performed at
95°C for 5 min, followed by 40 cycles of amplification at 95°C for
10 sec and 60°C for 30 sec on an Applied Biosystems 7500 Real-Time
PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.).
The fluorescent signals were collected during the extension phase,
quantification cycle (Cq) values of the sample were calculated, and
transcript levels were analyzed using the 2−ΔΔCq method
(19). The results were normalized
to β-actin and cDNA was prepared 30 times.
MSP
Genomic DNA (gDNA; 1 µg) isolated from LX-2
cells was extracted when the cells were treated with lysis buffer
and incubated with proteinase K for 10 min, next cells were treated
with bisulfite using the EZ DNA Methylation kit (Zymo Research
Corp., Irvine, CA, USA), according to the manufacturer's protocol.
The specific primers (methylated DNA: 164 bp PCR product, forward
5′-TTAATTAAAGTGTTGGGATGATAGAC, reverse
5′-AAATCTCTTAAACCTAAAAAACGAT; and unmethylated DNA: 164 bp PCR
product, forward 5′-AATTAAAGTGTTGGGATGATAGATGT and reverse
5′-AAATCTCTTAAACCTAAAAAACAAT) were designed and synthesized by
Sangon Biotech Co., Ltd. Normal lymphocytes collected from healthy
human blood (Dr Xuejia Lu, Affiliated Drum Tower Hospital of
Nanjing University Medical School, Nanjing, China) were used as the
negative control, whereas normal lymphocytes treated with
bisulfite-converted gDNA were used as the positive control. The
modified DNA was immediately used for MSP. The PCR was carried out
using PrimeScript RT Master Mix (Takara Bio, Inc.) under the
following conditions: 98°C for 10 min, 53°C for 30 min, 8 cycles of
53°C for 6 min and 37°C for 30 min. The methylated and unmethylated
DNA was distinguished after running on a 3% agarose gel and was
stained with GelRed DNA-binding dye (Biotium, Inc., Hayward, CA,
USA).
Immunofluorescence (IF)
LX-2 cells were seeded onto 24-well plates
containing a sterile glass slide at a density of 2×104
cells/well. Following incubation in high-glucose DMEM medium
supplemented with 10% FBS and antibiotics at 37°C in a humidified
incubator containing 5% CO2 for 24 h, the cells coated
on the glass slide were washed three times with cold
phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde
for 20 min at room temperature. Following blocking with 1% non-fat
milk for 2 h, the cells were incubated with primary antibodies
(Smad3 and Smad4) at a dilution of 1:250 overnight at 4°C.
Subsequently, the slides were washed three times with 0.5%
PBS-Tween 20 and incubated with fluorescein
isothiocyanate-conjugated goat anti-mouse IgG H&L Alexa Fluor
488 secondary antibody (cat. no. ab150117; Abcam) at a dilution of
1:1,000 at 37°C for 2 h. Eventually, the slides were washed and
examined using the inverted fluorescence microscope. Five random
high power field images were taken in each group.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
(SPSS Inc., Chicago, IL, USA). Results are presented as the mean ±
standard deviation of at least three independent experiments. The
significant differences among the groups were evaluated by a
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
SAM suppresses the expression of Smad3/4
in LX-2 cells
To determine the mechanism underlying SAM-induced
reversal of LX-2 HSC activation, the protein expression levels of
Smad3/4 were examined in cells treated with various concentrations
of SAM for 24 h. As shown in Fig.
1, the protein expression of Smad3 and Smad4 in the SAM-treated
groups (4 and 6 mM SAM) was significantly decreased compared with
in the control group (P<0.05), as determined by western
blotting. This result indicates an inhibitory effect of SAM on the
expression of Smad3/4.
Rac1 enhances the proliferation,
migration and invasion of LX-2 cells
Following transfection of the cells with plasmids
encoding Rac1 protein or an empty expression vector for 48 h, the
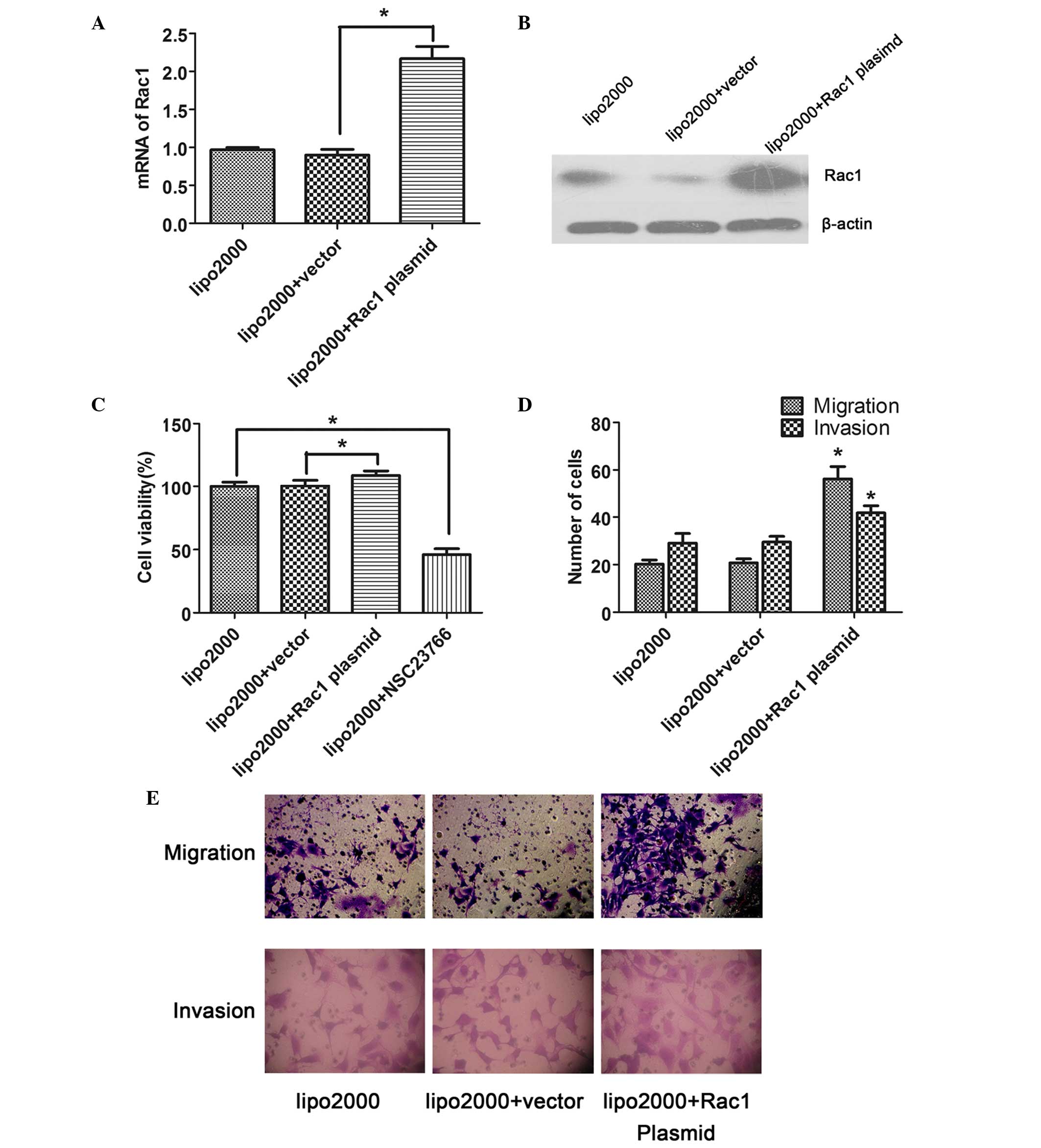
expression of Rac1 in the LX-2 cells was examined. The mRNA
expression levels of Rac1 in the Rac1 plasmid-transfected cells
were significantly increased compared with in the
vector-transfected cells, as determined by RT-qPCR (P<0.05;
Fig. 2A). Similarly, the protein
expression levels of Rac1 were also increased in response to Rac1
plasmid transfection (Fig. 2B),
confirming that the transfection was successful.
To investigate the role of Rac1 in the proliferation
of LX-2 cells, CCK-8 assay was performed. The results showed that
the overexpression of Rac1 significantly facilitated the
proliferation of LX-2 cells compared with the vector-transfected
cells (P<0.05); however, proliferation was reduced when the
activity of Rac1 was suppressed by a specific inhibitor, NSC23766
(Fig. 2C).
Transwell assay was performed in order to determine
the effects of Rac1 on the migration and invasion of the
transfected LX-2 cells. The results indicated that the number of
migrated or invaded cells was significantly increased in the Rac1
plasmid-transfected group compared with in the vector-transfected
group (P<0.05; Fig. 2D and
E).
Rac1 promotes the expression of Smad3/4
in LX-2 cells
Compared with the vector-transfected cells, Rac1
plasmid-transfected LX-2 cells exhibited a marked increase in
Smad3/4 protein expression (Fig. 3A
and B). Smad3/4 expression detected by IF was consistent with
the results of the western blot analysis (Fig. 3C). These findings indicate that
Rac1 may act as part of an upstream signaling pathway that promotes
the expression of Smad3/4 in LX-2 cells. In addition, the
expression levels of α-SMA were detected by RT-qPCR. The mRNA
expression levels of α-SMA were significantly increased in the Rac1
plasmid-transfected cells compared with in the vector-transfected
cells (Fig. 3D), thus indicating
that the overexpression of Rac1 facilitates the activation of
HSCs.
SAM increases methylation of the Rac1
promoter
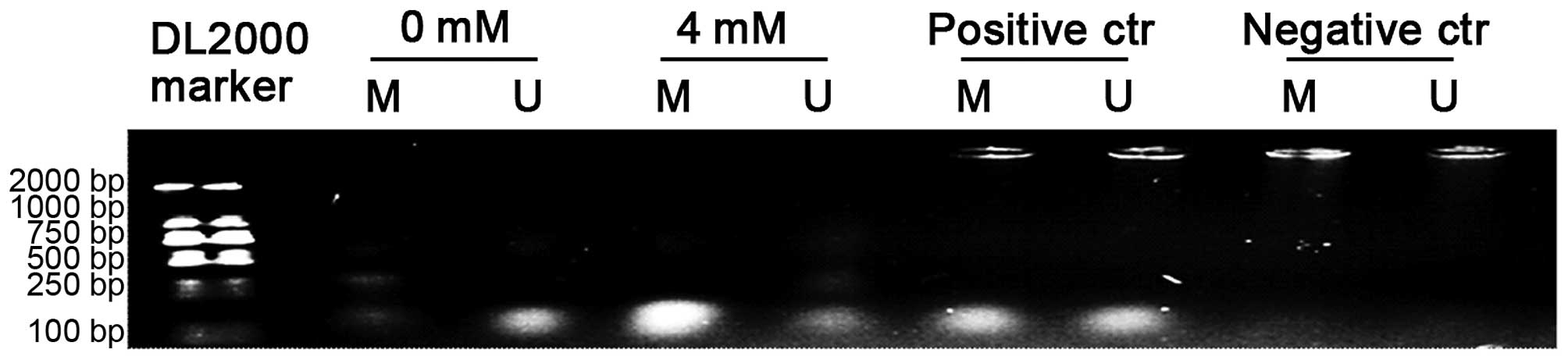
gDNA was extracted from LX-2 cells treated with 4 mM
SAM for 24 h, and from untreated control cells for MSP. As shown in
Fig. 4, a strong signal was
observed in the bisulite-converted lymphocyte DNA (positive
control) amplified with both methylated and unmethylated
allele-specific MSP primers, which were not effective at amplifying
the negative control. A markedly increased methylation status was
shown in the SAM-treated cells compared with the untreated ones,
which indicated that the Rac1 promoter could be methylated by SAM
in LX-2 cells, explaining the inhibitory effect of SAM on Rac1
expression.
Discussion
Rac1, which is a member of the Rho family of
GTPases, is an important intracellular transducer. Rac1 is known to
induce cytoskeletal rearrangement, and is required for cell
migration, proliferation and gene transcription. Previous studies
have reported that Rac1 regulates the TGF-β1/Smad signaling pathway
(11,20). In addition, Rac1 may regulate the
expression of Smad2/3 in pancreatic carcinoma cells, facilitating
cell proliferation and mobility, whereas the suppression of Rac1
weakens the transcription and phosphorylation of Smad2 but enhances
that of Smad3 (12). In
keratinocytes, Rac1 has been reported to regulate TGF-β1-mediated
epithelial cell plasticity and MMP9 production (11); however, the interaction between
Rac1 and Smad4 has not yet been detected. Rac1 has also been shown
to facilitate the synthesis of collagen, as mediated by TGF-β1
(21). Our previous study
(13) demonstrated that SAM
inhibited the activated phenotype of HSCs, probably via Rac1, and
that SAM attenuated the expression of Smad3/4; however, the
association between Rac1 and Smads in activated HSCs is currently
unknown.
The present study focused on the regulating effect
of Rac1 on the expression of Smad3/4 in activated HSCs, and on
further demonstrating the anti-fibrotic mechanism of SAM. To
validate the effects of Rac1, plasmids encoding Rac1 protein or an
empty expression vector were transfected into LX-2 cells. The
results demonstrated that the expression levels of Smad3 and Smad4
were markedly increased by overexpression of Rac1, which had not
previously been demonstrated. Conversely, the expression levels of
Smad3 and Smad4 were decreased when the expression of Rac1 was
downregulated by NSC23766. Furthermore, following our previous
study, the mechanism underlying the inhibitory effects of SAM on
Rac1 expression was explored. SAM, which is an intermediate product
of L-methionine metabolism, is a precursor of glutathione and an
endogenous methyl donor. SAM regulates the expression of target
genes through altering the methylation status; therefore, the
present study examined the methylation status of the Rac1 promoter.
The results indicated that the Rac1 promoter was highly methylated
by SAM.
Smad2/3 form a hetero-oligomeric complex with Smad4,
which translocates into the nucleus of HSCs where it induces target
gene transcription and mediates hepatic fibrosis (1). It has previously been reported that
partly suppressing the TGF-β1/Smad signaling pathway could delay or
reverse hepatic fibrosis (22,23);
however, a complete block of the TGF-β1/Smad pathway also increases
the risk of tumorigenesis. Therefore, targeting selected Smads in
TGF-β1/Smad could comprise a potential treatment for fibrosis
(24).
The present study demonstrated that Rac1 may have a
promising role in the regulation of Smad3/4 expression, thus
revealing a novel mechanism for the reversal of liver fibrosis. The
present findings suggested that the expression of Smad3/4 in
activated HSCs could be enhanced by the overexpression of Rac1,
whereas SAM suppressed the expression of Smad3/4, probably via
methylation of the Rac1 promoter, which would result in subsequent
downregulation. The present study provided a molecular basis for a
potential application of SAM and Rac1 in the treatment of hepatic
fibrosis.
Acknowledgments
The present study was supported by the Natural
Science Foundation of Jiangsu Province (grant no. BK2011094) and
the Fundamental Research Funds for the Central Universities (grant
no. 20620140710).
References
|
1
|
Lee UE and Friedman SL: Mechanisms of
hepatic fibrogenesis. Best Pract Res Clin Gastroenterol.
25:195–206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bian EB, Huang C, Wang H, Chen XX, Zhang
L, Lv XW and Li J: Repression of Smad7 mediated by DNMT1 determines
hepatic stellate cell activation and liver fibrosis in rats.
Toxicol Lett. 224:175–185. 2014. View Article : Google Scholar
|
|
3
|
He Y, Huang C, Sun X, Long XR, Lv XW and
Li J: MicroRNA-146a modulates TGF-beta1-induced hepatic stellate
cell proliferation by targeting SMAD4. Cell Signal. 24:1923–1930.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y, Wang Z, Wang J, Lam W, Kwong S, Li
F, Friedman SL, Zhou S, Ren Q, Xu Z, et al: A histone deacetylase
inhibitor, largazole, decreases liver fibrosis and angiogenesis by
inhibiting transforming growth factor-β and vascular endothelial
growth factor signalling. Liver Int. 33:504–515. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Szuster-Ciesielska A, Mizerska-Dudka M,
Daniluk J and Kandefer-Szerszeń M: Butein inhibits ethanol-induced
activation of liver stellate cells through TGF-β, NFκB, p38, and
JNK signaling pathways and inhibition of oxidative stress. J
Gastroenterol. 48:222–237. 2013. View Article : Google Scholar :
|
|
6
|
Bosco EE, Mulloy JC and Zheng Y: Rac1
GTPase: A “Racˮ of all trades. Cell Mol Life Sci. 66:370–374. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nagase M and Fujita T: Role of
Rac1-mineralocorticoid-receptor signalling in renal and cardiac
disease. Nat Rev Nephrol. 9:86–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elnakish MT, Hassanain HH, Janssen PM,
Angelos MG and Khan M: Emerging role of oxidative stress in
metabolic syndrome and cardiovascular diseases: Important role of
Rac/NADPH oxidase. J Pathol. 231:290–300. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Muhanna N, Doron S, Wald O, Horani A, Eid
A, Pappo O, Friedman SL and Safadi R: Activation of hepatic
stellate cells after phagocytosis of lymphocytes: A novel pathway
of fibrogenesis. Hepatology. 48:963–977. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bopp A, Wartlick F, Henninger C, Kaina B
and Fritz G: Rac1 modulates acute and subacute genotoxin-induced
hepatic stress responses, fibrosis and liver aging. Cell Death Dis.
4:e5582013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santibáñez JF, Kocić J, Fabra A, Cano A
and Quintanilla M: Rac1 modulates TGF-beta1-mediated epithelial
cell plasticity and MMP9 production in transformed keratinocytes.
FEBS Lett. 584:2305–2310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ungefroren H, Groth S, Sebens S, Lehnert
H, Gieseler F and Fändrich F: Differential roles of Smad2 and Smad3
in the regulation of TGF-β1-mediated growth inhibition and cell
migration in pancreatic ductal adenocarcinoma cells: Control by
Rac1. Mol Cancer. 10:672011. View Article : Google Scholar
|
|
13
|
Zhang F, Zhuge YZ, Li YJ and Gu JX:
S-adenosylmethionine inhibits the activated phenotype of human
hepatic stellate cells via Rac1 and matrix metalloproteinases. Int
Immunopharmacol. 19:193–200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang C, Zeisberg M, Mosterman B, Sudhakar
A, Yerramalla U, Holthaus K, Xu L, Eng F, Afdhal N and Kalluri R:
Liver fibrosis: Insights into migration of hepatic stellate cells
in response to extracellular matrix and growth factors.
Gastroenterology. 124:147–159. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu L, Hui AY, Albanis E, Arthur MJ,
O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL and Eng FJ: Human
hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis
of hepatic fibrosis. Gut. 54:142–151. 2005. View Article : Google Scholar
|
|
16
|
Hendrickson H, Chatterjee S, Cao S,
Morales Ruiz M, Sessa WC and Shah V: Influence of caveolin on
constitutively activated recombinant eNOS: Insights into eNOS
dysfunction in BDL rat liver. Am J Physiol Gastrointest Liver
Physiol. 285:G652–G660. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Werneburg NW, Yoon JH, Higuchi H and Gores
GJ: Bile acids activate EGF receptor via a TGF-alpha-dependent
mechanism in human cholangiocyte cell lines. Am J Physiol
Gastrointest Liver Physiol. 285:G31–G36. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Taimr P, Higuchi H, Kocova E, Rippe RA,
Friedman S and Gores GJ: Activated stellate cells express the TRAIL
receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis.
Hepatology. 37:87–95. 2003. View Article : Google Scholar
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFbeta-mediated fibroblastic transdifferentiation and cell
migration. J Cell Sci. 115:3193–3206. 2002.PubMed/NCBI
|
|
21
|
Hubchak SC, Sparks EE, Hayashida T and
Schnaper HW: Rac1 promotes TGF-beta-stimulated mesangial cell type
I collagen expression through a PI3K/Akt-dependent mechanism. Am J
Physiol Renal Physiol. 297:F1316–F1323. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tang LX, He RH, Yang G, Tan JJ, Zhou L,
Meng XM, Huang XR and Lan HY: Asiatic acid inhibits liver fibrosis
by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One.
7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Karaa A, Thompson KJ, McKillop IH, Clemens
MG and Schrum LW: S-adenosyl-L-methionine attenuates oxidative
stress and hepatic stellate cell activation in an
ethanol-LPS-induced fibrotic rat model. Shock. 30:197–205.
2008.PubMed/NCBI
|
|
24
|
Inagaki Y and Okazaki I: Emerging insights
into Transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|