Introduction
Understanding the complete set of proteins targeted
by bacterial toxin components, such as the anthrax lethal factor
(LF), is important for the understanding of its mode of action in
order to generate therapeutic agents or biomarkers. The ingestion
of raw food contaminated with Bacillus anthracis spores
causes gastrointestinal (GI) anthrax disease which is divided in
three clinical phases: I, fainting accompanied by fever; II,
abdominal pain with vomiting; and III, intensified abdominal pain
accompanied with bleeding (1).
Reported cases have indicated that lesions further down the GI
tract, in the mid-jejunum, terminal ilium or cecum, result in
single or multiple ulcerations and edema (2). In addition, a study in a mouse model
for GI anthrax have suggested that the Peyer's patches is the
specific site for B. anthracis growth following gastric
inoculation (3). However, the
mechanisms associated with this pathogenic bacteria in the enteric
system are not well understood due to a low number of clinical
cases, resulting in mortality rates of 20–60% (4). Furthermore, recent cases from India
and Iran, the latter being fatal, highlight the importance of
understanding the specific manifestation of GI anthrax and the
administration of early treatments (5,6).
Therefore, it is important to understand the mode of action of this
pathogenic bacteria, in particular its toxin's components.
One of the components of anthrax toxin is the LF, a
zinc-dependent metalloproteinase whose entrance to the cell is
through a protective antigen (PA)-mediated endocytosis (7). The LF (90 kDa) is composed of four
domains: Domain I binds to the translocon PA; domain II recognizes
the substrate; domain III is a duplication of a structural fragment
of domain II; and domain IV contains the catalytic center (8). Once in the cell cytosol, the LF
cleaves the N-terminal of mitogen-activated protein kinase kinase
(MAPKK), resulting in the inhibition of the MAPK pathway (9). The MAPKKs are involved in a number of
important cellular pathways including cell proliferation,
embryogenesis (10) and
angiogenesis (11). The disruption
of these proteins promotes macrophage apoptosis due to the
inhibition of p38 MAPK pathways, suggesting the requirement for p38
to allow the transcription of genes involved in the inhibition of
apoptosis (12). Furthermore, as
pathogens survive and replicate in macrophages, these cells undergo
pyroptosis, releasing their cytosolic contents into the
extracellular space (13). A
previous study suggested the requirement of the direct proteolytic
activity of LF on the MAPKKs for the antiproliferative and
pro-apoptotic effects of the toxin at the intestinal epithelium
(14). Despite the efforts to
identify the LF targets in human cells, the mechanisms that cause
macrophage death and the impact that it has on the innate immune
response are not well understood (15).
Levinsohn et al (16) demonstrated an LF-mediated direct
proteolytic cleavage at the N-terminal of NOD-like receptor protein
1 (NLRP1). This reaction yields an inflammasome response in the
NLRP1B BMAJ mouse macrophage cell line. The inflammasomes are
considered as the multimeric protein complexes that occur in
response to danger signals within the cytoplasm and provide a
scaffold for the activation of caspase-1 (17). Considering this, if an additional
substrate undergoes LF-mediated cleavage, which results in
apoptosis, then a novel LF-interacting partners may be suggested to
promote this biological process in GI anthrax. Therefore, it is
important to better understand GI anthrax at the molecular level to
enable the generation of novel therapeutic agents (18,19).
Furthermore, identifying new anthrax LF interactions may aid in
elucidating the complete pathways of the disease. The current study
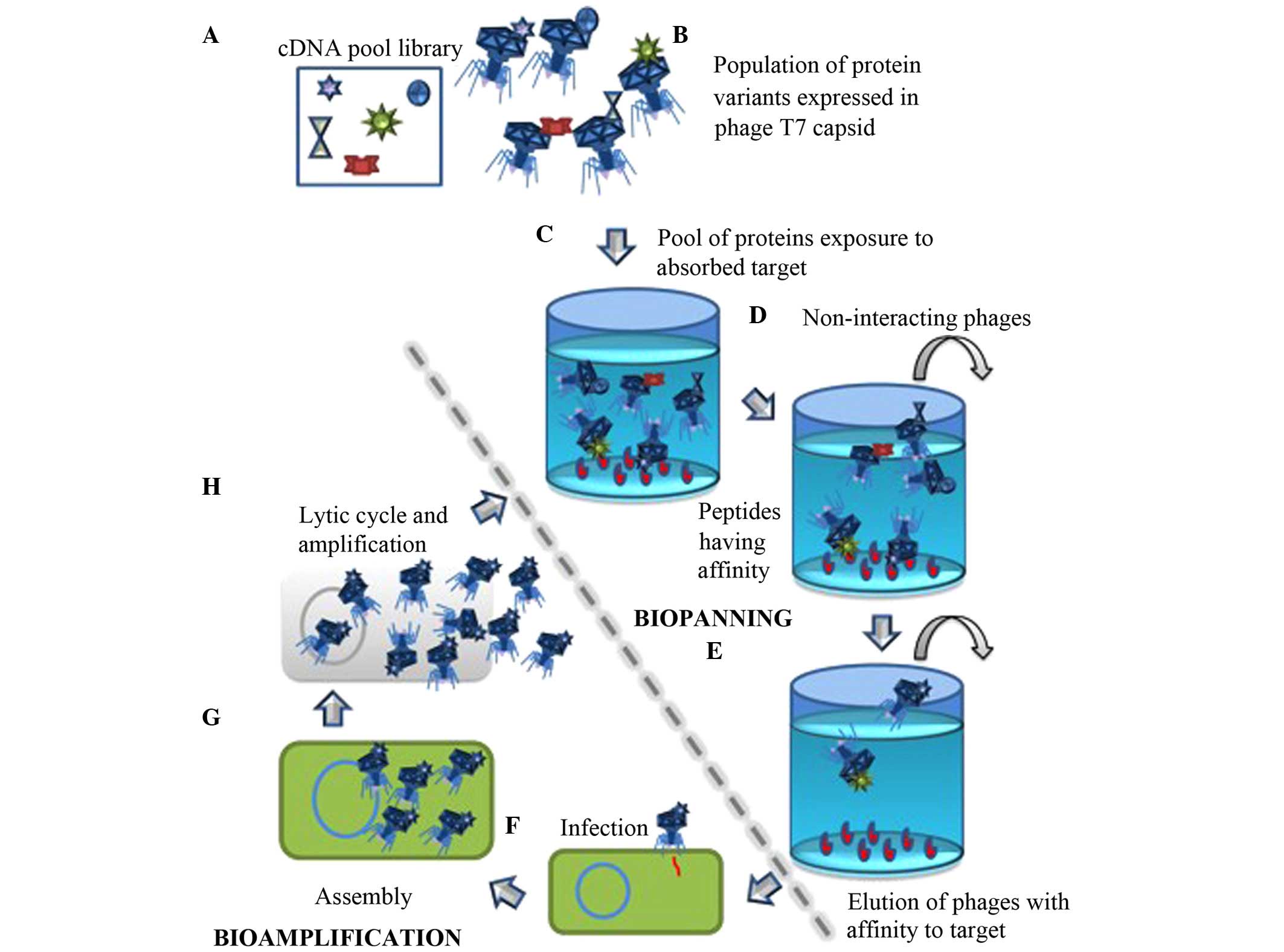
takes advantages of the combinatorial high throughput screening
that may be performed using T7 phage display (PD), in order to
select and identify proteins in human stomach tissue that may serve
as a target to interact with LF in vitro. In addition, these
observations may aid in the generation of new therapeutic agents or
biosensors for the disease at the GI level.
Materials and methods
The system and methods used were based on the
Novagen T7Select™ libraries procedures. Pre-made human stomach T7PD
cDNA libraries (Novagen; EMD Millipore, Billerica, MA, USA), at
1.4×1010 PFU/ml, were used to perform the experiments.
Two types of lethal factor were used as 'baits': Wild type (LF-WT),
which has metalloproteinase activity due to a zinc dependent
catalytic center on the fourth domain; and a site directed mutant
in the LF active site (LF-MT) E687A, which lacks cleavage activity
(20). The toxin component was
purified and provided by Dr. Stephen Leppla, Laboratory of
Parasitic Diseases at National Institutes of Allergy and Infectious
Disease (Bethesda, MD, USA). The host strain used for T7PD
screenings was an Escherichia coli Rosetta 5615 (R5615)
(Novagen; EMD Millipore). These bacterial cells carry a plasmid
with an ampicillin resistant gene that supplies an additional 10A
capsid protein, whose expression is controlled by a lacUV5
promoter. The complete procedure was performed four times for each
type of LF at four separated events of biopanning cycles. A
biopanning cycle consisted of three consecutive rounds of
biopanning, which was performed in the four independent
experiments.
Adsorption
LF-WT and LF-MT were diluted with Tris buffered
saline (TBS; Tris-HCl 10 mM, pH 8, 150 mM NaCl) to 4.5 μg/ml
and transferred to sterile 96-well polystyrene microplates (Greiner
Bio-One International GmbH, Kremsmünster, Austria) to start the LF
coating. The microplate was covered with parafilm and incubated
overnight at 4°C. The microplate wells with the adsorbed target
were washed three times with 300 μl TBS to remove unbound
protein. A solution of 5% casein was prepared as a blocking agent
and 200 μl was added to each well and incubated overnight at
4°C. Finally, the microplate wells were washed five times with 200
μl TBS to remove the excess blocking agent. The washed
microplates were stored with 100 μl TBS until use.
Biopanning
Aliquots of 100 μl containing
~1.4×107 clones of the stomach T7PD cDNA library were
applied to the coated wells to initiate the protein-protein
interactions. The microplates were incubated for 1 h at room
temperature. To remove phages expressing non-interacting peptides,
five stringent washes were performed using 200 μl TBS with
0.2% Tween 20 (TBST). Phage particles with specific binding
properties to the target were eluted using 200 μl 1% sodium
dodecyl sulfate (SDS). The bioamplification of the LF interacting
partners was performed by infection with a previously induced R5615
with isopropyl-β-D-thiogalacto pyranoside (Sigma-Aldrich, St.
Louis, MO, USA) at 1 mM. Subsequently, the purification of the
phage particles was performed by centrifuging 1,500 μl of
the lysed culture at 8,000 × g for 10 min at room temperature,
following which the supernatant was transferred to a clean tube.
The biopanning product (supernatant) was used to perform the second
selection round by adding aliquots of 100 μl containing
~1×107 clones to each well. A third biopanning round was
conducted using aliquots of 100 μl containing
~1×109 clones from the product of the second round, in
order to promote specificity and reduce random interactions
(Fig. 1).
Overlay assay and polymerase chain
reaction (PCR)
A series of dilutions (from
102–1010) of the phage lysates were performed
in order to quantify the number of amplified phages following the
third round of biopanning. Selected dilutions
(106–107) were mixed with a fresh culture of
R5615 (OD600 0.5) on top agar [Luria Bertani (LB; Difco
Laboratories, Inc., Detroit, MI, USA) broth 0.7% agar]. The mix was
transferred to a petri dish, containing bottom agar (LB agar with
50 μg/ml ampicillin). The plates were incubated at 37°C
until plaques were observed. Following phage quantification,
plaques were extracted and transferred to a clean tube with fresh
induced R5615 culture. Once the phage particles were isolated,
their DNA was extracted. The amplification of the cloned cDNA was
performed by PCR using GoTaq Green Master Mix (Promega Corporation,
Madison, WI, USA), following the manufacturer's instructions. The
amplification reaction was performed as follows: 10 pmol of each PD
cloning site specific primer T7Select forward 5′-GGA GCT GTC GTA
TTC CAGTC-3′ and T7Select reverse 5′-GCT GAT ACC ACC CTT CAA G-3′
(Novagen; EMD Millipore). A total of 1–2 μl of phage lysate
was used as the template DNA. All the PCR reactions were performed
with a total volume of 25 μl.
The thermal cycler (T100; Bio-Rad Laboratories,
Inc., Hercules, CA, USA) conditions consisted of an initial
denaturation step at 94°C for 3 min, followed by 30 cycles of 94°C
for 30 sec, 51°C annealing for 30 sec, 72°C extension for 30 sec,
and a final extension at 72°C for 6 min. The PCR control reactions
were prepared to compare the experimental results. PCR control
reactions consisted on a known and already amplified PD clone DNA
sample (positive control) and a reaction tube without DNA sample
(negative control). A 1.8% agarose gel (Denville Scientific, Inc.,
South Plainfield, NJ, USA) was run, using a horizontal
electrophoresis unit to confirm the presence of amplicons. For this
step, 1X Tris-acetate-EDTA was used as running buffer, and the gel
was analyzed using a Gel Documentation system (Bio-Rad
Laboratories, Inc.). The amplicons were sent to MCLAB (San
Francisco, CA, USA) for sequencing. Sequences were refined using
BioEdit 7.2.5 (Ibis Biosciences, Carlsbad, CA, USA). The DNA was
translated to amino acid sequences using the bioinformatics web
tool Expasy (http://web.expasy.org/translate/; SIB Swiss Institute
of Bioinformatics, Lausanne, Switzerland). The DNA and protein
sequences were analyzed using the National Center for Biotechnology
Information BLASTN/P for human species, with
this program able to compare sequence homologies (21).
Specificity test and minimum
concentration for interaction (MCI)
The LFs (WT & MT) were immobilized on 96-well
microplates at 22.5 μg/ml. All the biopannings were
performed using 5% casein as blocking agent. If the specificity
test revealed interaction of the T7PD candidates with casein, the
blocking agent was then changed to 3% bovine serum albumin, and the
specificity test repeated. Individual phages expressing selected
peptides were exposed to experimental wells and empty wells that
contained only blocking agents. Following five washes with TBST,
and an elution using 1% SDS, the product of interactions (~400
μl) was inoculated on 5 ml cultures of R5615. The cultures
were incubated at 37°C for 4 h or until lysis was detected. For the
MCI assay, the LFs (WT & MT) at 1, 2 and 3 μg/ml were
adsorbed onto microplate wells. To test the MCI, an individual
candidate that showed affinity to both types of LF was used for
this test. The candidate was exposed to the LFs following the
biopanning protocols. The elution product from the wells was
inoculated on R5615 5 ml cultures, which were then incubated at
37°C until lysis was detected. For both assays the OD600
readings were measured using the Eppendorf UV biophotometer
(Eppendorf, Hamburg, Germany). The OD readings were utilized to
identify the lysis of the bacteria vs. the control (data not
shown).
In silico analysis of LF-interacting
partners
Multiple sequence alignments and the hydrophobicity
of peptides that showed putative affinity to LF in vitro
were determined using the Centre for Integrative Bioinformatics
PRALINE web tools (http://www.ibi.vu.nl/programs/pralinewww/) (22). Default parameters were used for
this analysis. The full-length reference sequences for peptides
with conserved amino acids used for comparisons were obtained from
Uniprot databases.
Results
Selection and DNA isolation
Following four different rounds of selection,
through the three rounds of biopannings a total of 192 interacting
phages were isolated and amplified. A total of 124 putative
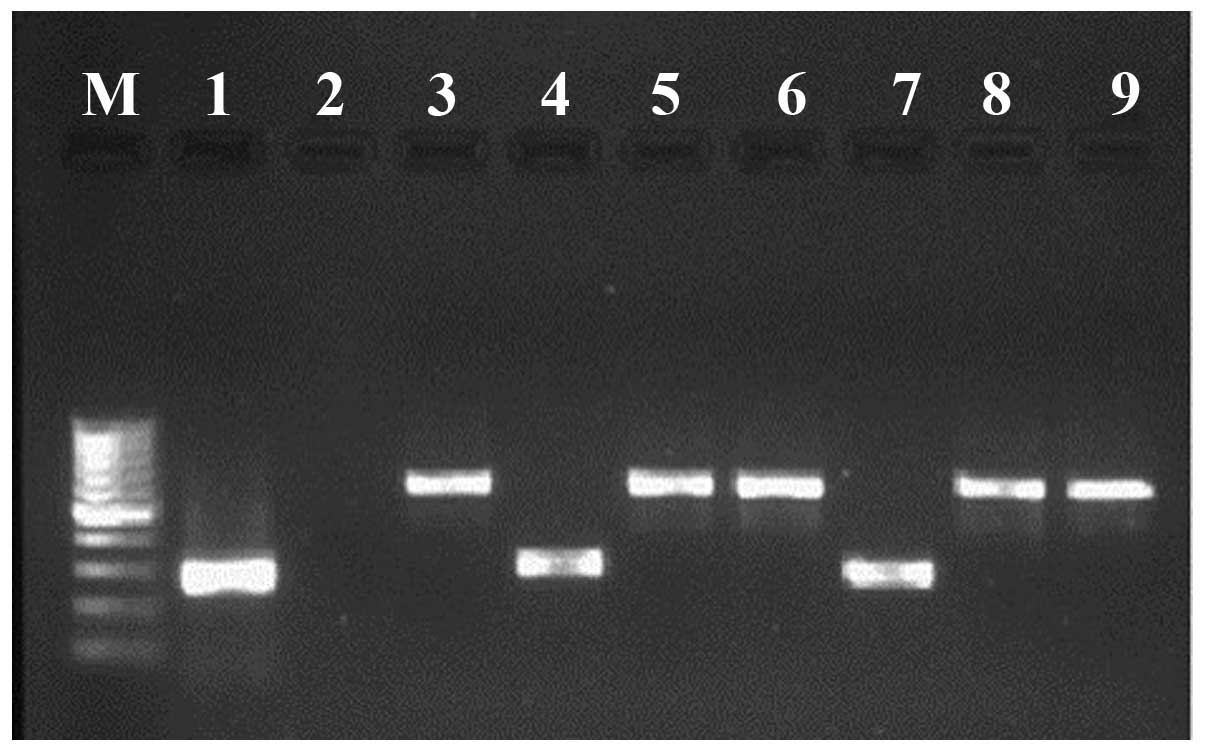
LF-interacting clones were amplified by PCR and verified in a 1.8%
agarose gel. The total number of identified clones for LF-WT was
54. In addition, a total of 70 clones were identified as LF-MT
interacting partners. Amplicon size varied from 300–1,000 base
pairs in the majority of cases for both types of LF (Fig. 2).
Identification of candidates
Following the sequencing of the 124 clones, a total
of 84 were selected for the present study, and after the in
silico analysis, 33 different known human proteins were
identified. From these proteins, 60% demonstrated interaction with
LF-MT, and 36% with LF-WT. Furthermore, 9% of the identified
proteins were obtained when either type of LF was used as bait. In
general, the LF-MT interacting partners were predominantly
associated with the peptidase A1, lipase gastric, and kruppel
C2H2-type zinc-finger protein (ZNF) families. By contrast, proteins
identified for LF-WT belonged predominantly to the peptidase A1
family. In addition, proteins associated with DNA binding, antigen
recognition and the cell cycle were identified. The proteins that
interacted with both types of LF were pepsin A, gastric
triacylglycerol lipase and a ribosomal protein.
The distribution of the isolated clones following
the biopanning was ~12% related to the peptidase A1 family, 7%
Kruppel and 6% to the abhydrolase family. The remaining clones had
lower frequencies per family. Furthermore, the in silico
analysis revealed conserved domains, including abhydrolase 1,
aspartyl proteases, xylanase inhibitors and BRICHOS. These domains
correspond to the proteins gastric lipase (GL), pepsin A,
gastricsin and gastrokine-1, respectively. The remaining proteins
did not show putative conserved domains or motifs. The size of the
three largest peptides consisted of 170 amino acids (aa)
(gastricsin), 156aa (pepsin A) and 122aa (A kinase anchor protein)
residues. The smallest peptide (5aa) identified in silico,
shared similarity with Ras-related protein Rab-34.
Specificity test and MCI
Following the specificity test a total of 10
proteins (Table I) were observed
to interact with a minimum of one type of LF. Of these, two were
specific to LF-MT, four to LF-WT and three showed affinity to both
types of targets tested. The 10th protein (Gastric
lipase) demonstrated an inconclusive affinity to LF due to its
ability to interact with multiple baits and all the blocking agents
used. The GL candidate interaction remains poorly understood due to
its affinity to the blocking agents. In Fig. 3A, an example of the lysis detection
for this test is presented. As the pepsin A3 pre-protein (46aa in
length) showed affinity to both types of LF in vitro, this
candidate was used to determine the MCI. For both targets used the
required concentration of LF to allow interactions was detected at
1 μg/ml (Fig. 3B).
 | Table ICandidates that showed putative
affinity to LF following the T7 phage display specificity test. |
Table I
Candidates that showed putative
affinity to LF following the T7 phage display specificity test.
| Peptide
similarity | 'Bait' | Peptide
sequence | Type of blocking
agent |
|---|
| Pepsin A3
preprotein | LF-MT & WT |
ACISGFQGMNLPTES
GELWILGDVFIRQYFT
VFDRANNQVGLAPVA | NL B |
| Pepsin A
preprotein | LF-MT |
ASSYYTGSLNWVPVTVEGH
WQITVDSITM NGEAIACAE
GCQAIVDTGTSLLTGPTSPIA
NIQSDIGASENSDGDMV
VSCSAISSLPDIVFTINGVQYPV
PPSAYILQ SEGSCISGFQGMNL
PTESGELWILGDVFIR QY
FTVFDRANNQVGLAPVA | +C
NL C/T |
| Gastric
triacylglycerol lipase | ND |
ANVTAMNVPIAVWNGGKDP
LADPQDVGLLLPKLPNL
IYHKEIPFYNHLDFIWAMDA
PQEVYNDIVSMISEDKK | +C/T
+B |
| Interleukin
enhancer-binding factor 2 | LF-WT |
AVTPSEKAYEKPPEKKE
GEEEEENTEEPPQGE
EEESMETQE | NL C |
| Copper transporting
ATPase | LF-WT |
AREQGQDLHTP
LGRTVRRIRYI | NL C |
| Probable
phospholipid ATPase | LF-WT | ALLTYWENSLC | NL C |
| Cytochrome c
oxidase | LF-WT |
AELGQPGNLLGNDHI
YNVIVTAHAFVIIF
FIVIPIIIGGFGN | NL C |
| Death ligand signal
enhancer | LF-MT |
ALEFKPSTASSTGCRSGQW | NL C |
| GDNF-inducible zinc
finger protein 1 | LF-MT & WT |
LNLVIVQVSSGPGVSSAPA | NL C |
| Homo sapiens
chromosome 11 open reading frame 96 | LF-MT & WT |
AECWRECEWVCAG
GHGGAVCKIGVANHRT
RAWSGYPPPTQRGRASPH
TLTAEFALGRVKKA | NL C |
Multiple sequence alignment and
hydrophobicity scale
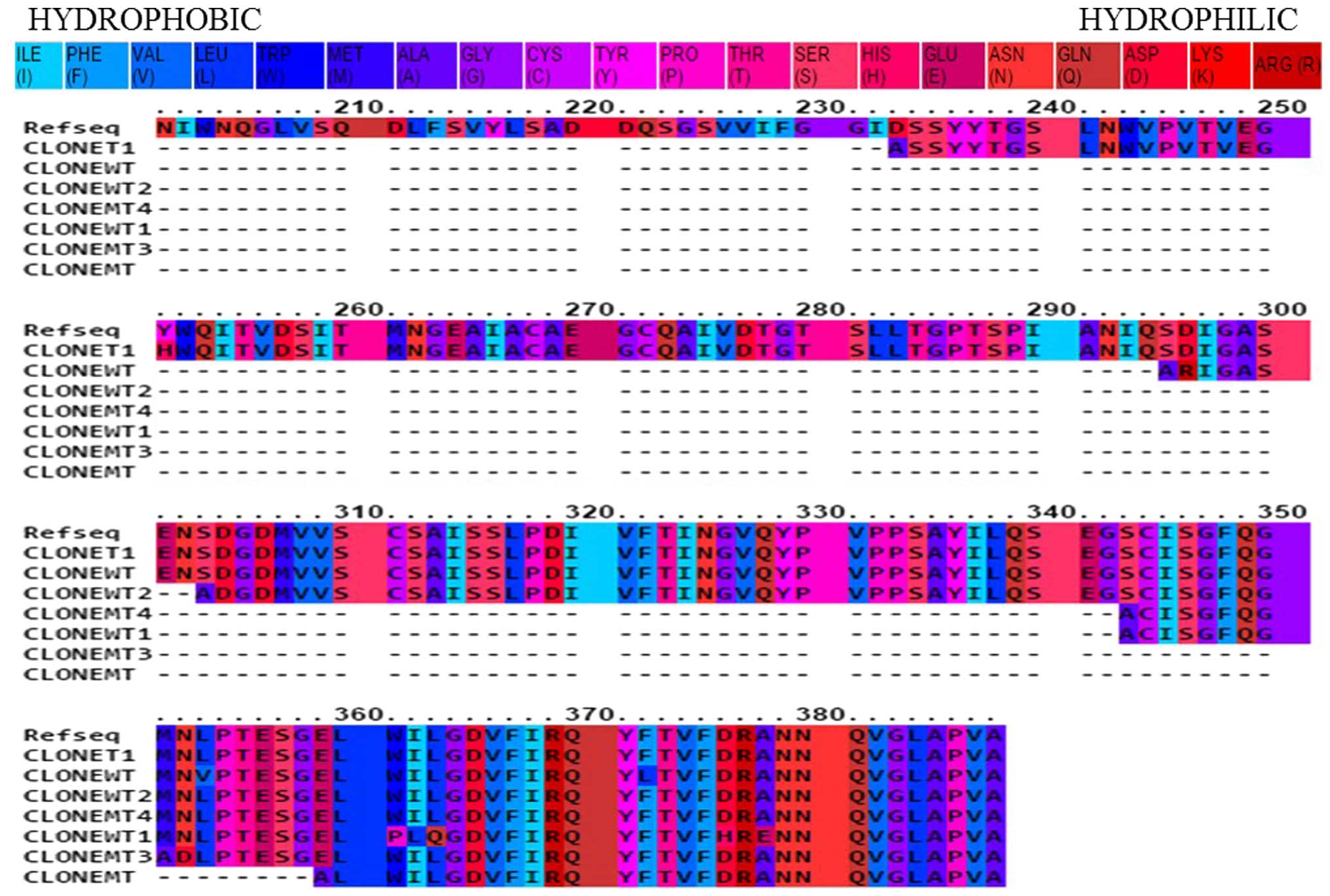
From all the putative LF-interacting partners, the
peptide sequences corresponding to pepsin A exhibited regions in
common following PRALINE alignment (Fig. 4). These clones were identified in
different biopanning events, and the peptides varied in size. The
detected peptides belongs to the pepsin A (EC:3.4.23.1).
The identity coverage in the putative pepsin PD
clones compared with the reference sequence protein range from
15–20%. The hydrophobicity analysis using PRALINE indicated that
the remaining identified peptides consist mostly of hydrophilic
residues (data not shown). However, several hydrophobic regions
were detected.
Discussion
Currently, the identified targets that undergo
direct cleavage by LF belong to protein families such as NLRPs and
MAPKKs (23,24). However, the mechanisms involved in
GI anthrax infection are not well known (25), meaning that the toxin components
targets remain to be identified. In the current study, a number of
proteins are suggested to be putative LF targets through the T7PD
technique. The data suggest a more diverse group of proteins have
affinity to the LF-MT than the LF-WT. This may be due to the
mutated catalytic center in LF-MT. Having no metalloproteinase
activity enables the selection and identification of interacting
proteins with the protease domain, however, avoiding the cleavage
of the protein and the loss of valuable data following the washing
steps. Following the in silico analysis, the same putative
interacting partners for LF-MT and LF-WT were identified, for
example, GL and pepsin A preprotein. Therefore, proteins such as GL
and pepsin A preprotein may be interacting with domains II and III
of LF as they were present in both types of LF tested. As reported
by Pannifer et al (8),
these domains are involved in the recognition of substrate by the
metalloproteinase. For this reason, any interactions between the
substrate recognition domains and human stomach proteins were able
to be detected by PD. However, isolation from PD using LF-WT as
bait was possible due to multiple interactions with a non-catalytic
region of LF (e.g. domains II and III). However, despite GL being a
constant candidate isolated through the biopanning cycles, the
interactions of this candidate with LF were not determined. This
candidate showed promiscuous interaction properties, being
considered as a possible non-specific LF interacting peptide.
The putative conserved domains obtained (Fig. 4) may represent a step towards
understanding GI anthrax. The detection at independent events,
following all biopanning cycles, of proteins having affinity for
LF-WT and LF-MT, such as the gastric triacylglycerol lipase and
pepsin A, represent the most valuable candidates. This data
suggests that both sequences may have interactions with the LF at
least in vitro. For pepsin A, a member of the peptidase A1
protein family, the minimum conserved sequence for being a
LF-interacting partner isolated by PD was of 30 residues that
matches with the aspartyl protease domain. Several members from
different peptidase families have been proposed as anthrax LF
inhibitors, resulting in the survival of macrophage cells due to an
interference of LF protease activity (26). The pepsins are well known to have a
preference for hydrophobic ligands, and some of these may serve as
inhibitors, such as pepstatin (27). Considering this, the interactions
between the hydrophobic residues presented in Fig. 3 and the hydrophobic amino acids
from domain III proposed by Pannifer et al (8) may explain and support our data. The
largest peptide identified in the present study was gastricsin,
which belongs to the peptidase A1 family. This contained domains
that belong to aspartyl protease and xylanase inhibitors, and did
not have considerable conserved regions with pepsin A, suggesting
that the protease domain may be recognizing LF. As a result,
peptides from this protein family may be tested in order to
identify potential as LF inhibitors.
A previous study established an association between
the MAPKK stress signaling pathways and time, as an explanation of
the alteration of these proteins in in vivo models, however,
not in vitro (28). The
same study suggested a difference in the regulation of these
proteins during stress conditions. For this reason, the duration of
interactions may have served an important role in the identified
interactions, explaining the absence of these proteins in the
current study. An alternative explanation may be that due to the
presence of low abundance mRNA for this protein in the stomach,
compared with other tissues (29).
However, stomach MAPKK's levels are comparable to other tissues,
which could be due to high stability of these proteins. Considering
that human stomach cDNA libraries are generated using mRNAs, this
may explain the non-isolation and identification of the MAPKKs. In
the current study, proteins from different families were identified
as putative LF-binding partners, however, not at the high frequency
as the previously mentioned ones. These proteins were confirmed
through a specificity test, consisting of exposing individual
candidates to LF and to the blocking agent individually.
The interleukin enhancer-binding factor 2 peptide
consisted predominantly of hydrophilic residues. The group of
interleukin enhancers, such as interleukin 2 (IL-2), have the
property of binding to antigen receptors (30). Previous studies suggested IL-2
inhibition due to the LF-mediated disruption of the MAPKK pathway
(31). Therefore, the present
study proposes a possible alternate route for the inhibition of
IL-2 as an immune response during anthrax disease. Two additional
isolated peptides were part of the P-type ATPases and showed
putative affinity to LF. P-type ATPases are considered as anion,
cation and lipid pumps located in cell membranes; these enzymes
generate electrochemical gradients using metabolic energy and
additionally mediate cell signaling pathways (32). Due to their ability to maintain
homeostasis through the transport of different ions such as
Ca2+, the lack of activity from these enzymes can result
in neurological disorders (33).
Accordingly, the disruption of these ion pumps may result in an
electrochemical imbalance within the cell, and potentially promote
the circulatory shock stage in patients with GI anthrax. A previous
study of cardiomyocytes infected with the anthrax lethal toxin
indicated disruption of their function and of the intracellular
calcium transport; however, the underlying mechanism is not clear
(28).
An additional LF-interacting partner isolated
through T7PD was cytochrome c oxidase (CcO).
CcO is a metalloprotein that maintains the proton gradient
and synthesis of ATP, and in addition activates oxygen as the
terminal acceptor of the transport electron chain (34). A previous study postulated that
CcO and additional proteins involved in energy pathways were
inactivated in macrophages via an s-nitrosylation process, driven
by B. anthracis-derived nitric oxide, resulting in cell
death (35). While the net charge
of the T7PD isolate similar to CcO is neutral, it is possible that
the interaction with LF is due to a hydrophobic region present in
the peptide. These results suggest a possible inhibition of
CcO, followed by its release, through the proteolytic
activity of LF in GI anthrax, resulting in a reduction in the
efficiency of cellular respiration. The identified death ligand
enhancer (DELE) is known to be involved in apoptosis by activating
tumor necrosis factor (TNF)-α and TNF-related apoptosis-inducing
ligand; however, this process is due to the binding of death
associated protein (DAP3) to DELE (36). The same study additionally
suggested that the disruption of DELE reduced the rate of apoptosis
in cells; therefore the in vitro interaction between DELE
and LF is unclear. This may be due to LF altering the conformation
of DELE, increasing its affinity for DAP3, followed by the rest of
the apoptosis signaling pathways.
Glial cell-derived neurotrophic factor (GDNF) zinc
finger protein was suggested as a transcriptional repressor of
HOXA10 gene, which is associated with morphogenesis
(37). The ZNFs are considered as
residues of cysteine and histidine, whose center binds zinc and
provides structural stability to the rest of the protein in order
to allow interactions with DNA (38). These protein motifs are known to be
involved in the transcriptional process. The amino acid sequences
for these motifs varied in length from 20–22aa and were identified
as the mutant-interacting partners. Despite the identification of
two types of ZNFs, including the GDNF, further studies are required
in order to understand the origin of these motifs.
The mechanisms of GI anthrax are poorly understood
due to the deficit of in vivo models of the disease
(39). Burgos (40) suggested that the use of T7PD to
determine LF-binding proteins from different human tissues revealed
the presence of proteins including cytochrome c oxidase,
ribosomal proteins and ATPase. This supports the data of the
current study, and indicates the identity of the profile of
proteins involved in GI anthrax, and aids in the understanding of
how this pathogenic bacteria evades the immune system, breaks down
epithelial barriers and its dissemination out from the GI
system.
Through T7PD, the present study provided a profile
of potential proteins from the human stomach that binds LF. Further
studies both in vitro and in vivo are necessary in
order to confirm and understand the complete set of events that
promote tissue death, and the identity of proteins cleaved by the
metalloproteinase LF, resulting in apoptosis. The generation of
in vivo models will serve to determine which of the reported
proteins are cleaved by the anthrax LF. Additionally, considering
the constant in vitro interactions in the present study,
these highly conserved peptides may be synthesized and tested as
inhibitors in in vivo assays. In conclusion, the use of T7PD
in the present study is a platform from which to begin unraveling
GI anthrax at the molecular level, to elucidate the disease
pathways and generate novel therapeutic agents.
Acknowledgments
The authors would like to thank Dr Stephen Leppla
for providing the purified LF, and for his comments and suggestions
on the manuscript. The current study was supported by a Enhancing
Biomedical Sciences and Biomedical Engineering in Science and
Technology (RISE2BEST) grant (grant no. NIH-R25GM088023), from the
National Institute of General Medical Sciences. The authors would
also like to thank Miss Lorein Moya for her assistance with the
specificity test analysis. The present study was previously
presented at the 115th Annual Meeting of the American Society for
Microbiology in New Orleans, Louisiana, USA.
References
|
1
|
Akbulut A, Akbulut H, Özgüler M, İnci N
and Yalçın Ş:: Gastrointestinal anthrax: A case and review of
literature. Adv Infect Dis. 2:67–71. 2012. View Article : Google Scholar
|
|
2
|
Sirisanthana T and Brown AE: Anthrax of
the gastrointestinal tract. Emerg Infect Dis. 8:649–651. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Glomski IJ, Piris-Gimenez A, Huerre M,
Mock M and Goossens PL: Primary involvement of pharynx and peyer's
patch in inhalational and intestinal anthrax. PLoS Pathog.
3:e762007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tonry JH, Popov SG, Narayanan A, Kashanchi
F, Hakami RM, Carpenter C, Bailey C and Chung MC: In vivo murine
and in vitro M-like cell models of gastrointestinal anthrax.
Microbes Infect. 15:37–44. 2013. View Article : Google Scholar
|
|
5
|
Iqbal N, Basheer A, Ramesh AN, Vimal J,
Mookkappan S, Kanungo R, Anandhalakshmi and Princess I:
Gastrointestinal anthrax in coastal south India: A critical alert
on a fatal masquerader. JMM Case Rep. 2:e0.0000132015. View Article : Google Scholar
|
|
6
|
Hashemi SA, Azimian A, Nojumi S, Garivani
T, Safamanesh S and Ghafouri M: A case of fatal gastrointestinal
anthrax in north eastern Iran. Case Rep Infect Dis.
2015:8758292015.PubMed/NCBI
|
|
7
|
Bouzianas DG: Medical countermeasures to
protect humans from anthrax bioterrorism. Trends Microbiol.
17:522–528. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pannifer AD, Wong TY, Schwarzenbacher R,
Renatus M, Petosa C, Bienkowska J, Lacy DB, Collier RJ, Park S,
Leppla SH, et al: Crystal structure of the anthrax lethal factor.
Nature. 414:229–233. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Duesbery NS, Webb CP, Leppla SH, Gordon
VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K,
Paull KD and Vande Woude GF: Proteolytic inactivation of
MAP-kinase-kinase by anthrax lethal factor. Science. 280:734–737.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pearson G, Robinson F, Beers Gibson T, Xu
BE, Karandikar M, Berman K and Cobb MH: Mitogen-activated protein
(MAP) kinase pathways: Regulation and physiological functions.
Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
11
|
Yang J, Boerm M, McCarty M, Bucana C,
Fidler IJ, Zhuang Y and Su B: Mekk3 is essential for early
embryonic cardiovascular development. Nat Genet. 24:309–313. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park JM, Greten FR, Li ZW and Karin M:
Macrophage apoptosis by anthrax lethal factor through p38 MAP
kinase inhibition. Science. 297:2048–2051. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun C, Fang H, Xie T, Auth RD, Patel N,
Murray PR, Snoy PJ and Frucht DM: Anthrax lethal toxin disrupts
intestinal barrier function and causes systemic infections with
enteric bacteria. PloS One. 7:e335832012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu S, Moayeri M and Leppla SH: Anthrax
lethal and edema toxins in anthrax pathogenesis. Trends Microbiol.
22:317–325. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levinsohn JL, Newman ZL, Hellmich KA,
Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH and Moayeri M:
Anthrax lethal factor cleavage of Nlrp1 is required for activation
of the inflammasome. PLoS Pathog. 8:e10026382012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moayeri M, Sastalla I and Leppla SH:
Anthrax and the inflammasome. Microbes Infect. 14:392–400. 2012.
View Article : Google Scholar :
|
|
18
|
Lightfoot YL, Yang T, Sahay B, Zadeh M,
Cheng SX, Wang GP, Owen JL and Mohamadzadeh M: Colonic immune
suppression, barrier dysfunction and dysbiosis by gastrointestinal
Bacillus anthracis infection. PloS One. 9:e1005322014. View Article : Google Scholar
|
|
19
|
Baldari CT, Tonello F, Paccani SR and
Montecucco C: Anthrax toxins: A paradigm of bacterial immune
suppression. Trends Immunol. 27:434–440. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Klimpel KR, Arora N and Leppla SH: Anthrax
toxin lethal factor contains a zinc metalloprotease consensus
sequence which is required for lethal toxin activity. Mol
Microbiol. 13:1093–1100. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Altschul SF, Gish W, Miller W, Myers EW
and Lipman DJ: Basic local alignment search tool. J Mol Biol.
215:403–410. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pirovano W, Feenstra KA and Heringa J:
PRALINETM: A strategy for improved multiple alignment of
transmembrane proteins. Bioinformatics. 24:492–497. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hellmich KA, Levinsohn JL, Fattah R,
Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH and Moayeri M:
Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive
and toxin-resistant macrophages. PLoS One. 7:e497412012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vitale G, Pellizzari R, Recchi C,
Napolitani G, Mock M and Montecucco C: Anthrax lethal factor
cleaves the N-terminus of MAPKKs and induces tyrosine/threonine
phosphorylation of MAPKs in cultured macrophages. Biochem Biophys
Res Commun. 248:706–711. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bishop BL, Lodolce JP, Kolodziej LE, Boone
DL and Tang WJ: The role of anthrolysin O in gut epithelial barrier
disruption during Bacillus anthracis infection. Biochem Biophys Res
Commun. 394:254–259. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Menard A, Papini E, Mock M and Montecucco
C: The cytotoxic activity of Bacillus anthracis lethal factor is
inhibited by leukotriene A4 hydrolase and metallopeptidase
inhibitors. Biochem J. 320:687–691. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kuzmič P, Sun CQ, Zhao ZC and Rich DH:
Long range electrostatic effects in pepsin catalysis. Tetrahedron.
47:2519–2534. 1991. View Article : Google Scholar
|
|
28
|
Kandadi MR, Hua Y, Ma H, Li Q, Kuo SR,
Frankel AE and Ren J: Anthrax lethal toxin suppresses murine
cardiomyocyte contractile function and intracellular Ca2+ handling
via a NADPH oxidase-dependent mechanism. PloS One. 5:e133352010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Uhlen M, Oksvold P, Fagerberg L, Lundberg
E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S,
et al: Towards a knowledge-based human protein atlas. Nat
Biotechnol. 28:1248–1250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Durand DB, Shaw JP, Bush MR, Replogle RE,
Belagaje R and Crabtree GR: Characterization of antigen receptor
response elements within the interleukin-2 enhancer. Mol Cell Biol.
8:1715–1724. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fang H, Cordoba-Rodriguez R, Lankford CS
and Frucht DM: Anthrax lethal toxin blocks MAPK kinase-dependent
IL-2 production in CD4+ T cells. J Immunol. 174:4966–4971. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Palmgren MG and Nissen P: P-type ATPases.
Annu Rev Biophys. 40:243–266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Veen S, Sørensen DM, Holemans T, Holen
HW, Palmgren MG and Vangheluwe P: Cellular function and
pathological role of ATP13A2 and related P-type transport ATPases
in Parkinson's disease and other neurological disorders. Front Mol
Neurosci. 7:482014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brunori M, Antonini G, Malatesta F, Sarti
P and Wilson MT: Cytochrome-c oxidase. Subunit structure and proton
pumping. Eur J Biochem. 169:1–8. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chung MC, Narayanan A, Popova TG,
Kashanchi F, Bailey CL and Popov SG: Bacillus anthracis derived
nitric oxide induces protein S-nitrosylation contributing to
macrophage death. Biochem Biophys Res Commun. 430:125–130. 2013.
View Article : Google Scholar
|
|
36
|
Harada T, Iwai A and Miyazaki T:
Identification of DELE, a novel DAP3-binding protein which is
crucial for death receptor-mediated apoptosis induction. Apoptosis.
15:1247–1255. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morinaga T, Enomoto A, Shimono Y, Hirose
F, Fukuda N, Dambara A, Jijiwa M, Kawai K, Hashimoto K, Ichihara M,
et al: GDNF-inducible zinc finger protein 1 is a sequence-specific
transcriptional repressor that binds to the HOXA10 gene regulatory
region. Nucleic Acids Res. 33:4191–4201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brown RS, Sander C and Argos P: The
primary structure of transcription factor TFIIIA has 12 consecutive
repeats. FEBS Lett. 186:271–274. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xie T, Sun C, Uslu K, Auth RD, Fang H,
Ouyang W and Frucht DM: A new murine model for gastrointestinal
anthrax infection. PloS One. 8:e669432013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Burgos R: Isolation of interacting
peptides to Bacillus anthracis lethal toxin (LF) by T7 phage
display (unpublished Master's dissertation). University of Puerto
Rico; 2010
|