Introduction
Acyl-coenzyme A: cholesterol acyltransferase 1
(ACAT1) is an intracellular enzyme which catalyzes the formation of
cholesteryl esters from cholesterol and long-chain fatty
acyl-coenzyme A (1). In adult
human tissues, ACAT1 is found in a number of different tissues and
cell types, including hepatocytes and Kupffer cells, adrenal
glands, neurons and macrophages, and accounts for >80% of the
total ACAT enzyme activity measured in vitro (2–4). In
addition to storing cholesterol intracellularly, ACAT1 has
important physiological roles, including in hepatic lipoprotein
assembly, dietary cholesterol absorption and steroidogenesis.
Cholesterol and its metabolites have been implicated in the
pathogenesis of multiple human diseases, including atherosclerosis,
cancer, neurodegenerative diseases and diabetes. ACAT1 is important
in the formation of macrophage-derived foam cells in
atherosclerotic lesions (1). In
addition, ACAT1 expression was indicated to serve as a potential
prognostic marker in prostate cancer, specifically in
differentiating indolent and aggressive forms of cancer (5). ACAT1 knockdown gene therapy was shown
to decrease amyloid-β in a mouse model of Alzheimer's disease
(6). In addition, ACAT1 represents
a novel biomarker in adipose tissue associated with type 2 diabetes
in obese individuals (7). Thus,
understanding how cells maintain the homeostasis of cholesterol and
its metabolites is an important area of research.
The human ACAT1 gene contains two promoters (P1 and
P7), each located on a different chromosome (1 and 7) (8). Northern blot analyses have revealed
the presence of four ACAT1 mRNAs (7.0-, 4.3-, 3.6- and 2.8-knt),
present in almost all human tissues and cells examined (8). These messengers share the same coding
sequence. The 2.8- and 3.6-knt messengers, comprising more than
70–80% of the total ACAT1 mRNAs, are produced from the P1 promoter
(8). In the present study, a
luciferase reporter vector containing human ACAT1 gene P1 promoter,
and eight different 5′-deletion constructs of the P1 promoter was
constructed to analyze the transcriptional function of the human
ACAT1 gene P1 promoter using a luciferase reporter gene transient
expression system. The present study may provide an experimental
basis for investigating the regulatory mechanisms of the ACAT1 gene
P1 promoter during various biological processes associated with
cholesterol homeostasis, in particular the development of
atherosclerosis.
Materials and methods
Materials
RPMI-1640, Dulbecco's modified Eagle's medium, and
fetal calf serum were obtained from Gibco, Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). T4 DNA ligase, pGL3 luciferase
reporter vectors, pRL-TK vector and a Dual-luciferase reporter
assay system were purchased from Promega Corporation (Madison, WI,
USA). Pyrobest DNA polymerase, DNA Marker DL2000, λ-EcoT14 I digest
DNA Marker and all restriction enzymes were purchased from Takara
Biotechnology Co., Ltd. (Dalian, China). dNTPs were obtained from
MBI Fermentas, Thermo Fisher Scientific, Inc. The plasmid
purification mini kit was purchased from Watson Biotechnologies,
Inc. (Shanghai, China). Primers were synthesized by Invitrogen,
Thermo Fisher Scientific, Inc. The Gel Extraction kit was obtained
from Axygen Biosciences (Hangzhou, China). DNA sequences were
analyzed using an ABI 3730 automated DNA sequence analyzer by
Invitrogen, Thermo Fisher Scientific, Inc. All other reagents were
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
THP-1 cells were purchased from the Institute of
Biochemistry and Cell Biology, Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences (Shanghai, China). HepG2,
HEK293 and Hela cells were obtained from Institute of Biochemistry
and Cell Biology (Shanghai Institutes for Biological Sciences;
Chinese Academy of Sciences, Shanghai, China). HepG2, HEK293 and
Hela cells were maintained in Dulbecco's modified Eagle's medium
(DMEM) and THP-1 cells in RPMI-1640 medium. Culture medium was
supplemented with 10% heat-inactivated fetal bovine serum (FBS), 50
U/ml penicillin (Gibco; Thermo Fisher Scientific, Inc.), and 50
µg/ml streptomycin (Gibco; Thermo Fisher Scientific, Inc.),
and cells were maintained in a humidified atmosphere of 5%
CO2 and 95% air at 37°C.
Construction of luciferase reporter
vectors
The previously described cloning vector pMD19-T-P1
constructed in our laboratory (9)
containing the −603/+65 region of human ACAT1 gene P1 promoter was
used in this study. In the current study, the ACAT1 gene P1
promoter was isolated from the pMD19-T-P1 vector following
digestion with KpnI/XhoI, and the fragment was
subcloned into the multiple cloning sites of the Firefly luciferase
reporter gene vector pGL3-Enhancer to obtain P1E-1.
Transcription Element Search System (www.cbil.upenn.edu/cgi-bin/tess/tess)
was used to predict transcription factor binding sites in the DNA
sequence of the human ACAT1 P1 promoter. According to the analysis
of biological information, the P1E-1 plasmid was used to generate
deletions of the ACAT1 gene P1 promoter with varying 5′ ends and an
identical 3′ end at +65 by polymerase chain reaction (PCR). The
primer sequences, presented in Table
I, were designed according to the human ACAT1 gene P1 promoter
GenBank sequence (www.ncbi.nlm.nih.gov/genbank; GenBank accession no.
AF143319), all included a 6-bp KpnI/XhoI linker. All
PCR reactions were performed in a total volume of 50 µl
containing 1X Pyrobest buffer II, 1.5 mmol/l MgCl2, 200
µmol/l deoxynucleotides, 200 nmol/l each primer, 2 units
Pyrobest DNA polymerase and 0.5 µg DNA. Cycling conditions
were as follows: 23 Cycles of pre-denaturation at 94°C for 5 min,
denaturation at 94°C for 30 sec, annealing at 52°C for 30 sec, and
extension at 72°C for 1 min (Bio-Rad T100 PCR Thermal Cycler;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). The PCR products
were then digested with KpnI and XhoI. The digested
PCR products were analysed using agarose gel electrophoresis and
then retrieved using a gel extraction kit (Axygen Biosciences). The
pGL3 Enhancer plasmid was also digested by double digestion and the
large fragment was retrieved. During a ligation reaction, the
recycled PCR products and empty pGL3 Enhancer vector were incubated
overnight with T4 DNA ligase at 4°C, transformed into E.
coli DH5α (Takara Biotechnology Co., Ltd.) and screened. The
plasmid was extracted and identified by PCR amplification,
restriction enzyme digestion mapping and DNA sequencing. The
5′-deletion constructs of the P1 promoter represent bases −547 to
+65 bp (P1E-2), −498 to +65 bp (P1E-3), −428 to +65 bp (P1E-4),
−363 to +65 bp (P1E-5), −324 to +65 bp (P1E-6), −256 to +65 bp
(P1E-7), −188 to +65 bp (P1E-8) and −125 to +65 bp (P1E-9). These
constructs were then identified by restriction enzyme digestion
mapping and DNA sequencing. Sequences were determined on an ABI
3730 automated DNA sequence analyzer by Invitrogen (Thermo Fisher
Scientific, Inc.). The plasmids were purified by the Wizard
purification plasmid DNA purification system (Promega Corporation,
Madison, WI, USA) to transfect cells.
 | Table ISequences of the primers used in the
amplification of 5′-deletion fragments of human ACAT1 gene P1
promoter. |
Table I
Sequences of the primers used in the
amplification of 5′-deletion fragments of human ACAT1 gene P1
promoter.
| Name | Sequence (5′–3′) | Position (bp) |
|---|
| P1E-2 |
| Sense |
5′-ATGATCAggtaccCAGGTTTTTTCCCCTTATC-3′ | −547 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-3 |
| Sense |
5′-ATGATCAggtaccTTCAAACGGTAAGGAATC-3′ | −498 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-4 |
| Sense |
5′-ATGATCAggtaccCTGGCTAGTTCTACG-3′ | −428 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-5 |
| Sense |
5′-ATGATCAggtaccGGCTTCTTCCAGTCG-3′ | −363 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-6 |
| Sense |
5′-ATGATCAggtaccGCTCCATGCTACCACGC-3′ | −324 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-7 |
| Sense |
5′-ATGATCAggtaccACATTCTACTGCTGGGGTG-3′ | −256 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-8 |
| Sense |
5′-ATGATCAggtaccAGCTTCCTTGGCAAGGTTGCC-3′ | −188 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
| P1E-9 |
| Sense |
5′-ATGATCAggtaccGGGAAGGGGCGGGGAGG-3′ | −125 to +65 |
| Antisense | 5′-GATCActcgagGTCGACTCCGGGAAGCTCTCC-3′ | |
Transient transfection
A series of luciferase reporter constructs
containing the ACAT1 gene P1 promoter were transfected into THP-1
cells using the Diethylaminoethyl dextran (DEAE-dextran) method
(10). Briefly, after washing
twice with phosphate-buffered saline (PBS), 1×106 cells
were transfected with 0.5 µg ACAT1 promoter/Luc plasmids and
10 ng Renilla Luciferase Reporter Vector (pRL-TK) as
internal control in 200 µl STBE (25 mmol/l Tris-HCl, pH 7.4;
5 mmol/l KCl, 0.7 mmol/l CaCl2, 137 mmol/l NaCl, 0.6
mmol/l Na2HPO4 and 0.5 mmol/l
MgCl2) containing 60 µg DEAE-dextran. The cells
were incubated for 20 min at 37°C, washed once with RPMI-1640
without FBS, resuspended in 500 µl fresh RPMI-1640 with 10%
FBS, and plated in a 24-well plate. After 48 h, the cells were
harvested. The series of luciferase reporter constructs containing
the ACAT1 gene P1 promoter were separately transfected into HepG2,
HEK-293 and HeLa cells using Lipofectamine 2000 liposome
(Invitrogen, Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Briefly, the day prior to
transfection, cells reached 80–90% confluence. Then, the cells were
transfected with the plasmid DNA and Lipofectamine 2000 at a ratio
of 0.8 µg DNA:2 µl lipid per well in serum free
Opti-minimal essential medium (Gibco; Thermo Fisher Scientific,
Inc.) at 37°C for 4 h. Cells were then grown for 48 h in DMEM
containing FBS. All the cells were transfected with pGL3-Enhancer
and pGL3-Control, which served as negative and positive controls,
respectively. Each transfection reaction was performed in
triplicate.
Luciferase assay
The transfected cells were washed twice with cold
phosphate-buffered saline (PBS) and lysed in 100 µl passive
lysis buffer (Promega Corporation, Madison, WI, USA) and
centrifuged at 12,000 × g for 30 sec at 4°C after placing at room
temperature for 15 min. Then, 20 µl of the cell lysate was
mixed with 100 µl Luciferase Assay Reagent II (Promega
Corporation) for luciferase activity measurement in a Lumat LB9507
luminometer (EG&G Bertold, Freiburg, Germany), 100 µl
Stop & Glo Reagent (Promega Corporation) was then added for
luciferase activity measurement. For the luciferase activity assay,
the Dual-luciferase reporter assay system (Promega Corporation) was
used. Results were obtained from three different transfection
experiments after normalization to the internal control, thymidine
kinase activity. Experimental variations are indicated as the mean
± standard deviation.
Statistical analysis
Results are presented are the mean ± standard
deviation from at least three independent experiments performed in
duplicate. Statistical analysis was performed using one-way
analysis of variance and Tukey's post-hoc test with SPSS software
(version 12.0; SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Biological information analysis of human
ACAT1 gene P1 promoter
The ACAT1 gene P1 promoter is contiguous with the
coding sequence and spans from −603 to +65 of the ACAT1 genomic
sequence (9). Using sequence
analysis (Transcription Element Search System), neither a typical
TATA box nor a typical CAAT box was found. However, four copies of
a typical GC box (Sp1) were found in this region. In the P1
promoter sequence, other potential binding sites for various
transcription factors existed, including CCAAT enhancer binding
protein α (C/EBPα), serum response factor (SRF), hepatocyte nuclear
factor-3 (HNF-3), winged helix protein, c-Myb, GATA binding factor,
nuclear factor-1 (NF-1), cyclic AMP-response element-binding
protein and nuclear factor-κB (NF-κB). Fig. 1 shows the biological information
analysis of the human ACAT1 gene P1 promoter DNA sequence.
Identification of luciferase reporter
vectors
The ACAT1 gene P1 promoter and a series of
5′-deletion fragments were amplified by PCR with their recombinant
constructs as templates. The electrophoresis of these PCR products
showed nine different fragments in 0.9% agarose gel (Fig. 2). The recombinant constructs of P1
promoter and its deletions were identified by restriction enzyme
digestion mapping. When these recombinant constructs were digested
with KpnI and XhoI, two fragments were generated,
5,037 bp pGL3-Enhancer vector and 685 bp ACAT1 gene P1 promoter in
length, or 611, 562, 492, 427, 388, 320, 252 and 189 bp 5′-deletion
fragments in length (Fig. 3). DNA
sequencing was conducted using an ABI 3730 automated DNA sequence
analyzer. The sequencing results were consistent with GenBank
database. Figure 4 showed the
partial sequence of the human ACAT1 gene P1 promoter.
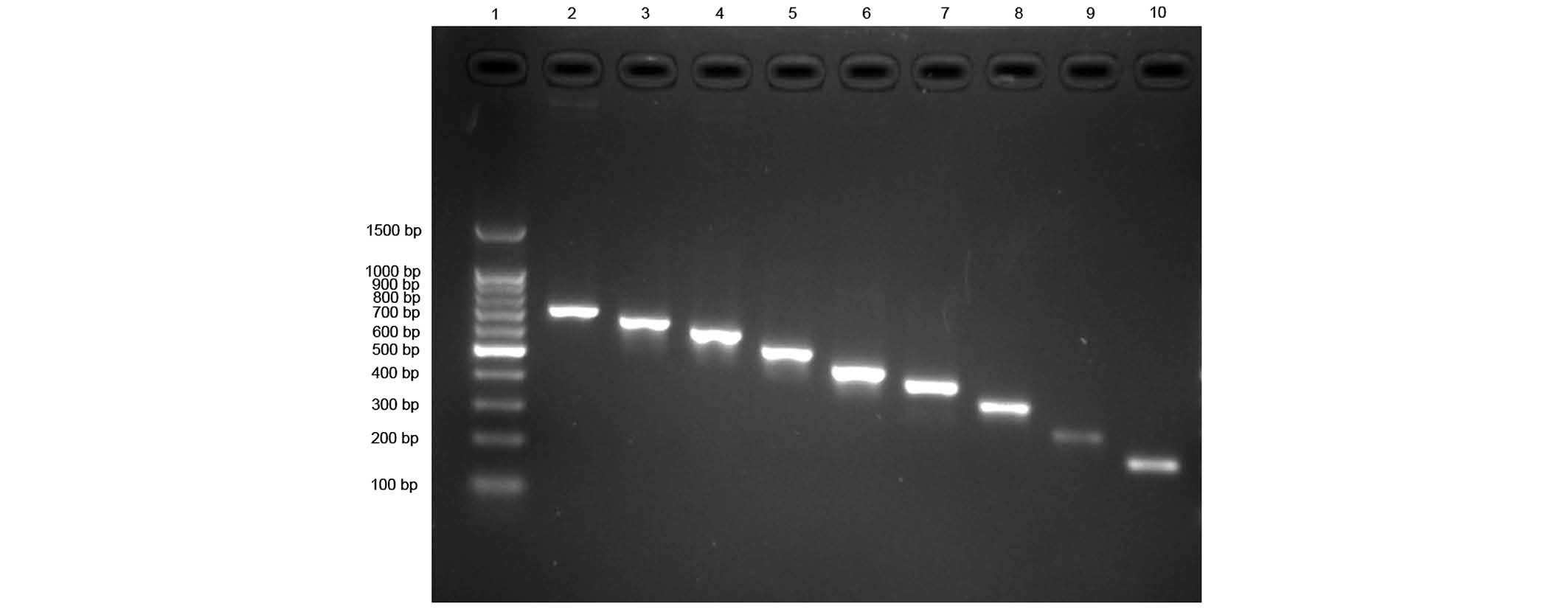 | Figure 2Fragments of the ACAT1 gene P1
promoter and its deletions amplified by PCR with the recombinant
plasmid as template. Lane 1, 100 bp DNA Ladder; lane 2, PCR product
of P1E-1 (with plasmid pGL3E-P1 DNA template, 685 bp); lane 3, PCR
product of P1E-2 (611 bp); lane 4, PCR product of P1E-3 (562 bp);
lane 5, PCR product of P1E-4 (492 bp); lane 6, PCR product of P1E-5
(472 bp); lane 7, PCR product of P1E-6 (388 bp); lane 8, PCR
product of P1E-7 (320 bp); lane 9, PCR product of P1E-8 (252
bp);and lane 10, PCR product of P1E-9 (189 bp). PCR, polymerase
chain reaction; ACAT1, acyl coenzyme A: Cholesterol acyltransferase
1. |
 | Figure 3Identification of the recombinant
plasmids of ACAT1 gene P1 promoter and its deletions by enzyme
digestion. Lane 1, 100 bp DNA Ladder; lane 2, P1E-1 (pGL3E-P1, 685
bp); lane 3, P1E-2 (611 bp); lane 4, P1E-3 (562 bp); lane 5, P1E-4
(492 bp); lane 6, P1E-5 (472 bp); lane 7, P1E-6 (388 bp); lane 8,
P1E-7 (320 bp); lane 9, P1E-8 (252 bp); lane 10, P1E-9 (189 bp);
and lane 11, 1 kbp DNA Ladder Marker. All the recombinants were
digested with KpnI/XhoI restriction enzymes. ACAT1,
acyl coenzyme A: Cholesterol acyltransferase 1. |
Transcriptional activity of human ACAT1
gene P1 promoter and its deletions in diverse cell lines
The recombinant constructs of the P1 promoter and
its deletions, including P1E-1, P1E-2, P1E-3, P1E-4, P1E-5, P1E-6,
P1E-7, P1E-8 and P1E-9 were transfected in THP-1, HepG2, HEK-293
and HeLa cells, respectively. The transcriptional activities were
detected by a luciferase activity assay analysis method.
pGL3-Enhancer and pGL3-Control served as negative and positive
controls respectively. The results (Fig. 5) demonstrated that the activity of
the ACAT1 gene P1 promoter with and without deletions in THP-1
cells was increased compared with that in HepG2, HEK293 and HeLa
cells. Low activity was detected in HEK293 and HeLa cells
transfected with ACAT1 gene P1 promoter and its deletions. As shown
in Fig. 5, the luciferase activity
of P1E-9 was greater than that of the other deletions when
transfected into THP-1, HepG2, HEK293 and HeLa cells. Moreover, the
activity of P1E-9 was highest when it was transfected into THP-1
cells, which was 97.7 fold higher than the pGL3-Enhancer. The
maximal transcriptional activity was located within the 189-base
pair region from −125 to +65, which suggested that the core
sequence of ACAT1 gene P1 promoter was located in this region.
Multiple GC boxes, which are likely to be Spl binding sites are
present within this region (Fig.
1). The luciferase activities of P1E-1 were notably higher than
those of P1E-2 in THP-1, HepG2, HEK-293 and HeLa cells, which
suggested that the region from the −603 to −547 may contain
positive cis-acting regulatory element(s). The luciferase
activity of P1E-2, P1E-3, P1E-4 and P1E-5 were markedly lower than
that of P1E-6, P1E-7, P1E-8 and P1E-9 in THP-1, HepG2, HEK 293 and
HeLa cells. Thus, it was speculated that there were the
transcription repressors in the sequence from −547 to −324. Further
studies are required to investigate the activating or repressing
function of these regions.
 | Figure 5Effects of deletions on the
transcription activities of the ACAT1 gene P1 promoter in different
cell lines. (A) Schematic presentation of the ACAT1 gene P1
promoter region and its eight truncated forms. (B) Relative
luciferase activities in cells transfected with each truncated
reporter construct. The transfected cells were determined by
luciferase after 48 h transfection. In each set of experiments,
luciferase activity was normalized to pRL-TK plasmid. The bars were
presented as the mean ± standard deviation of three independent
transfection experiments. *P<0.05 vs. P1E-1, P1E-2,
P1E-3, P1E-4, P1E-5, P1E-6, P1E-7 or P1E-8 in THP-1 cells.
#P <0.05 vs. P1E-1, P1E-2, P1E-3, P1E-4, P1E-5,
P1E-6, P1E-7 or P1E-8 in HepG2 cells. %P<0.05 and
&P<0.05 vs. P1E-1, P1E-2, P1E-3, P1E-4, P1E-5,
P1E-6, P1E-7 or P1E-8 in HEK293 and Hela cells, respectively.
ACAT1, acyl coenzyme A: Cholesterol acyltransferase 1. |
Discussion
Cholesterol is essential to the growth and viability
of cells. In mammalian cells, the homeostasis of free cholesterol
and cholesteryl ester is strictly controlled. ACAT1 is a key enzyme
in the regulation of the metabolic homeostasis of cholesterol and
cholesteryl ester. The main mode of sterol-specific regulation of
ACAT1 has been identified at the post-translational level,
involving allosteric regulation by its substrate cholesterol
(11). ACAT1, however, could also
be regulated at the transcriptional level. Yang et al
(12) showed that a glucocorticoid
response element in the human ACAT1 P1 promoter responsible for the
dexamethasone-induced elevation of ACAT1 gene expression could be
functionally bound to the glucocorticoid receptor. In human THP-1
monocytic cells, the combination of interferon-γ and
all-trans retinoic acid enhanced ACAT1 P1 promoter activity
in a synergistic manner (10).
Previous studies showed that insulin could upregulate ACAT1
expression and enzyme activity in human monocyte-derived
macrophages at the transcriptional level by activating the P1
promoter (9). Other factors
associated with atherosclerosis upregulate ACAT1 gene expression in
human monocyte-macrophages, such as 1, 25-dihydroxyvitamin D
(3) or 9-cis-retinoic acid,
transforming growth factor-β1, serotonin, salusin-β and leptin
(13–17). However, incretins, hydrogen
sulfide, adiponectin and Ghrelin downregulated ACAT1 expression to
suppress the development of atherosclerosis (18–21).
However, the molecular mechanism of non-sterol-specific mediated
ACAT1 regulation remains largely unknown.
To investigate the regulatory mechanisms of the
ACAT1 gene in diverse cell lines during the disorder of cholesterol
metabolism, a series of deletions of the P1 promoter were generated
with varying 5′ ends and an identical 3′ end at +65 according to
the analysis of biological information, and analyzed the functional
regions of ACAT1 gene P1 promoter. A series of 5′-deletion
luciferase reporter gene constructs of the P1 promoter were
successfully constructed. Then the transcriptional activities of P1
promoter and its deletions, P1E-1, P1E-2, P1E-3, P1E-4, P1E-5,
P1E-6, P1E-7, P1E-8 and P1E-9, were detected after transient
transfection into THP-1, HepG2, HEK293 and Hela cells,
respectively. The results showed that the activities of the ACAT1
gene P1 promoter and its deletions in THP-1 cells were higher than
that in HepG2, HEK293 and HeLa cells. There were low activities
observed when each reporter vector was transfected into HEK293 and
HeLa cells respectively. It was hypothesized that different
deletions of the ACAT1 gene P1 promoter have different effects on
transcription activities, and that the activity varies with cell
type. Smith et al (22)
demonstrated quantitatively that ACAT1 is the predominant
transcript isoform in human peripheral blood mononuclear cells and
liver, which is consistent with the present results. Thus, the
ACAT1 gene P1 promoter may drive gene expression with cell type
specificity.
In this study, the P1 promoter characterization of
the ACAT1 gene was conducted and THP-1, HepG2, HEK 293 and HeLa
cell lines were selected for deletion analysis. Results from the
deletion analysis of ACAT1 gene P1 promoter indicated that there
were several potential regions containing positive or negative
cis-acting regulatory element(s). The results suggested that
the region from −603 to −547 may contain positive cis-acting
regulatory element(s). Bioinformatics analysis of the region from
−603 to −547 found three cis-acting regulatory elements: C/EBPα,
SNF and HNF-3 binding sites. The results of deletion analysis
suggested that there were transcription repressors in the region
from −547 to −324. Bioinformatics analysis of the region from −547
to −324 found several cis-acting regulatory elements,
including winged helix protein, c-Myb, NF-κB, GATA binding factor,
NF-1, Sp1 binding sites. The activity of P1E-9 was notably higher
than those of the other deletions in THP-1, HepG2, HEK293 and HeLa
cells. Moreover, the activity of P1E-9 was highest in THP-1 cells,
which was 97.7 multiple of pGL3-Enhancer. Thus, it was presumed
that the core sequence of the ACAT1 gene P1 promoter was located in
the region from −125 to +65. There was neither a typical TATA box
nor a typical CAAT box, however, four copies of a typical GC box
(Sp1) were found in this region, which was important for the
transcriptional activity of the ACAT1 gene P1 promoter. The results
suggested that the activities of the potential positive or negative
elements of the P1 promotor in different cell lines determined the
differences in ACAT1 gene expression. Further studies on the
effects of site-directed mutagenesis are required for these
potential transcription factor binding sites of the ACAT1 gene P1
promoter in different cell lines.
In conclusion, luciferase reporter vectors
containing human ACAT1 gene P1 promoter with and without deletions
were successfully constructed, and the transcriptional activities
in diverse cell lines and the transcriptional activities of
different regions of P1 promoter were determined. Deletion analysis
revealed the core sequence of the ACAT1 gene P1 promoter and
potential positive or negative cis-acting regulatory
element(s). These finding may explain why the ACAT1 gene expression
varied between cell types specificity and provided an experimental
basis for investigating the regulatory mechanisms of the ACAT1 gene
P1 promoter in various biological processes associated with
cholesterol homeostasis, particularly the development of
atherosclerosis. Further investigation is required to determine the
association between cis-acting elements and the interacting
trans-acting factors within the ACAT1 gene P1 promoter regulatory
region. The results may contribute to the development of novel
treatment strategies and/or the determination of additional
therapeutic targets for the prevention and treatment of diseases
associated with cholesterol metabolism imbalance.
Acknowledgments
This study was supported by the National Natural
Scientific Foundation of China (grant no. 30471921 to Dr Bei Cheng
and grant no. 30700767 to Dr Wei Zhai), and the Natural Scientific
Foundation of Hubei province of China (grant nos. 2013CFB073 and
2013CFB088).
References
|
1
|
Tabas I: The stimulation of the
cholesterol esterification pathway by atherogenic lipoproteins in
macrophages. Curr Opin Lipidol. 6:260–268. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang CC, Sakashita N, Ornvold K, Lee O,
Chang ET, Dong R, Lin S, Lee CY, Strom SC, Kashyap R, et al:
Immunological quantitation and localization of ACAT-1 and ACAT-2 in
human liver and small intestine. J Biol Chem. 275:28083–28092.
2000.PubMed/NCBI
|
|
3
|
Lee O, Chang CC, Lee W and Chang TY:
Immunodepletion experiments suggest that acyl-coenzyme A:
Cholesterol acyl-transferase-1 (ACAT-1) protein plays a major
catalytic role in adult human liver, adrenal gland, macrophages,
and kidney, but not in intestines. J Lipid Res. 39:1722–1727.
1998.PubMed/NCBI
|
|
4
|
Sakashita N, Miyazaki A, Takeya M,
Horiuchi S, Chang CC, Chang TY and Takahashi K: Localization of
human acyl-coenzyme A: Cholesterol acyltransferase-1 (ACAT-1) in
macrophages and in various tissues. Am J Pathol. 156:227–236. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saraon P, Trudel D, Kron K,
Dmitromanolakis A, Trachtenberg J, Bapat B, van der Kwast T, Jarvi
KA and Diamandis EP: Evaluation and prognostic significance of
ACAT1 as a marker of prostate cancer progression. Prostate.
74:372–380. 2014. View Article : Google Scholar
|
|
6
|
Murphy SR, Chang CC, Dogbevia G, Bryleva
EY, Bowen Z, Hasan MT and Chang TY: Acat1 knockdown gene therapy
decreases amyloid-β in a mouse model of Alzheimer's disease. Mol
Ther. 21:1497–1506. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dharuri H, 't Hoen PA, van Klinken JB,
Henneman P, Laros JF, Lips MA, El Bouazzaoui F, van Ommen GJ,
Janssen I, van Ramshorst B, et al: Downregulation of the acetyl-CoA
metabolic network in adipose tissue of obese diabetic individuals
and recovery after weight loss. Diabetologia. 57:2384–2392. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li BL, Li XL, Duan ZJ, Lee O, Lin S, Ma
ZM, Chang CC, Yang XY, Park JP, Mohandas TK, et al: Human acyl-coA:
Cholesterol acyltransferase-1 (ACAT-1) gene organization and
evidence that the 4.3-Kilobase ACAT-1 mRNA is produced from two
different chromosomes. J Biol Chem. 274:11060–11071. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ge J, Zhai W, Cheng B, He P, Qi B, Lu H,
Zeng Y and Chen X: Insulin induces human acyl-coenzyme A:
Cholesterol acyl-transferase 1 gene expression via MAP kinases and
CCAAT/enhancer-binding protein α. J Cell Biochem. 114:2188–2198.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang JB, Duan ZJ, Yao W, Lee O, Yang L,
Yang XY, Sun X, Chang CC, Chang TY and Li BL: Synergistic
transcriptional activation of human Acyl-coenzyme A: Cholesterol
acyltransterase-1 gene by interferon-gamma and all-trans-retinoic
acid THP-1 cells. J Biol Chem. 276:20989–20998. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chang TY, Chang CC and Cheng D:
Acyl-coenzyme A: Cholesterol acyltransferase. Annu Rev Biochem.
66:613–638. 1997. View Article : Google Scholar
|
|
12
|
Yang L, Yang JB, Chen J, Yu GY, Zhou P,
Lei L, Wang ZZ, Cy Chang C, Yang XY, Chang TY and Li BL:
Enhancement of human ACAT1 gene expression to promote the
macrophage-derived foam cell formation by dexamethasone. Cell Res.
14:315–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maung K, Miyazaki A, Nomiyama H, Chang CC,
Chang TY and Horiuchi S: Induction of acyl-coenzyme A: Cholesterol
acyltransferase-1 by 1,25-dihydroxyvitamin D(3) or 9-cis-retinoic
acid in undifferentiated THP-1 cells. J Lipid Res. 42:181–187.
2001.PubMed/NCBI
|
|
14
|
Lei L, Xiong Y, Chen J, Yang JB, Wang Y,
Yang XY, Chang CC, Song BL, Chang TY and Li BL: TNF-alpha
stimulates the ACAT1 expression in differentiating monocytes to
promote the CE-laden cell formation. J Lipid Res. 50:1057–1067.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hori M, Miyazaki A, Tamagawa H, Satoh M,
Furukawa K, Hakamata H, Sasaki Y and Horiuchi S: Up-regulation of
acyl-coenzyme A: Cholesterol acyltransferase-1 by transforming
growth factor-beta1 during differentiation of human monocytes into
macrophages. Biochem Biophys Res Commun. 320:501–505. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Suguro T, Watanabe T, Kanome T, Kodate S,
Hirano T, Miyazaki A and Adachi M: Serotonin acts as an
up-regulator of acyl-coenzyme A: Cholesterol acyltransferase-1 in
human monocyte-macrophages. Atherosclerosis. 186:275–281. 2009.
View Article : Google Scholar
|
|
17
|
Hongo S, Watanabe T, Arita S, Kanome T,
Kageyama H, Shioda S and Miyazaki A: Leptin modulates ACAT1
expression and cholesterol efflux from human macrophages. Am J
Physiol Endocrinol Metab. 297:E474–E482. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nagashima M, Watanabe T, Terasaki M,
Tomoyasu M, Nohtomi K, Kim-Kaneyama J, Miyazaki A and Hirano T:
Native incretins prevent the development of atherosclerotic lesions
in apolipoprotein E knockout mice. Diabetologia. 54:2649–2459.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao ZZ, Wang Z, Li GH, Wang R, Tan JM,
Cao X, Suo R and Jiang ZS: Hydrogen sulfide inhibits
macrophage-derived foam cell formation. Exp Biol Med (Maywood).
236:169–176. 2011. View Article : Google Scholar
|
|
20
|
Furukawa K, Hori M, Ouchi N, Kihara S,
Funahashi T, Matsuzawa Y, Miyazaki A, Nakayama H and Horiuchi S:
Adiponectin down-regulates acyl-coenzyme A: Cholesterol
acyltransferase-1 in cultured human monocyte-derived macrophages.
Biochem Biophys Res Commun. 317:831–836. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wan JJ, Cheng B, Wang YF, Mei CL, Liu W,
Ke L and He P: Ghrelin down-regulates ACAT-1 in THP-1 derived foam
cells via growth hormone secretagogue receptor-dependent pathway.
Zhonghua Xin Xue Guan Bing Za Zhi. 37:1030–1034. 2009.In
Chinese.
|
|
22
|
Smith JL, Rangaraj K, Simpson R, Maclean
DJ, Nathanson LK, Stuart KA, Scott SP, Ramm GA and de Jersey J:
Quantitative analysis of the expression of ACAT genes in human
tissues by real-time PCR. J Lipid Res. 45:686–696. 2004. View Article : Google Scholar : PubMed/NCBI
|