Introduction
Previous studies have demonstrated that apoptosis is
an important process in injured to the lung epithelium in paraquat
(PQ) poisoning (1,2). The mechanisms of apoptosis are highly
complex and involve oxidative/nitrative stress impairment to the
function of mitochondria with the reduction of the mitochondrial
membrane potential (ΔΨ), resulting in an increase of superoxide
formation and reduced production of adenosine 5′-triphosphate (ATP)
(3,4). Previous studies suggest that
adenosine 5′-monophosphate-activated protein kinase (AMPK) is one
of the most important molecules in the stimulation of ATP
production and protection against oxidative/nitrative stress in
mitochondria (5,6). However, whether dysfunction of the
mitochondria and reduction of ATP via inactivation of AMPK serve
crucial roles in the PQ-induced apoptosis of alveolar epithelial
type II cells remains to be fully elucidated.
Adiponectin (APN), an adipocytokine predominantly
secreted by adipocytes and found at high levels in the plasma, has
been reported to exhibit anti-insulin resistance and
anti-inflammatory and anti-apoptotic properties (7–9).
Circulating APN is present as trimeric, hexameric or oligomeric
complexes of monomers, and cleavage to produce the C-terminal
globular domain, gAd, has been proposed as an important regulatory
step in the action of APN. This is due to the fact that this
C-terminal fragment has been reported to mediate potent
physiological effects (10,11).
Previous studies have suggested that the potential role of gAd
against apoptosis is mediated via the activation of AMPK (12–14).
APN-knockout mice exhibit inhibition of AMPK, a reduction in the
levels of ATP, an increase of mitochondrial swelling and ROS/RNS
production, resulting in augmented apoptosis, while administration
of gAd reversed these effects. The protective effects of gAd have
been reported to be reversed by administration of the AMPK
inhibitor (compound C) (15). It
remains unclear whether APN may protect alveolar type II cells from
PQ poisoning via activation of AMPK and increasing the production
of ATP in mitochondria.
Therefore, the aims of the current study were: i) To
determine whether mitochondrial function is impaired and ATP is
reduced in PQ-poisoned alveolar type II cells via inactivation of
AMPK; ii) to determine the effect of gAd against PQ-induced
apoptosis and in alveolar type II cells; and if so, iii) whether
gAd mediates protective effects via activation of AMPK and
increasing production of ATP.
Materials and methods
Materials
All reagents were purchased from Sigma-Aldrich (St.
Louis, MO, USA), unless otherwise specified. RPMI 1640 medium was
obtained from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). Penicillin/streptomycin was obtained from Wisent, Inc.
(Saint-Bruno, QC, Canada). PQ was purchased from Tokyo Chemical
Industry Co., Ltd. (Tokyo, Japan). The globular domain of APN (gAd)
was purchased from Beijing Adipobiotech, Inc. (Beijing, China).
Compound C, an AMPK Inhibitor, was obtained from Merck Millipore
(Darmstadt, Germany). The Annexin V-Fluorescein Isothiocyanate
(FITC) Apoptosis Detection kit I was from Nanjing KeyGen Biotech
Co., Ltd. (Nanjing, China). JC-1 and dihydroethidium (DHE) were
purchased from Molecular Probes; Thermo Fisher Scientific, Inc. The
Nitrate/Nitrite Colorimetric Assay kit was from Nanjing Jiancheng
Biochemical Reagent Co. (Nanjing, China). The ATP Bioluminescent
Somatic Cell assay kit was from Sigma-Aldrich.
Cell cultures and treatments
The human lung adenocarcinoma cell line A549
(Shanghai Institute of Biochemistry and Cell Biology, Shanghai,
China) was grown in a monolayer culture in RPMI 1640 medium
supplemented with 10% fetal bovine serum, 1% glutamine, 1% (v/v)
streptomycin/penicillin. The culture medium was refreshed two to
three times per week and was subcultured upon reaching 80%
confluence. The cells were then seeded onto flat-bottom six-well or
96-well plates with growth medium at 37°C in a humidified
atmosphere at 5% CO2, followed by administration of PQ
at 300 µM for 24 h for the PQ, PA and PCA groups.
Subsequently, gAd (2.5 µg/ml) was added and the cells were
cultured for another 24 h, in order to investigate the role of gAd
in PQ-induced cytotoxicity. PCA group cells were also treated with
compound C (1 µM) for 30 min prior to the addition of PQ.
The optimal doses and time points for gAd, PQ and compound C were
determined through preliminary experiments (data not shown).
Measurement of cell viability
The 3-(4,
5-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
was conducted as previously described (16). Briefly, following PQ challenge,
culture media was refreshed with media containing MTT reagent (5
mg/ml) and cells were incubated under standard conditions for an
additional 4 h. The culture media was then carefully aspirated and
100 µl dimethylsulfoxide was added per well to solubilize
the formazan crystals. Following agitation, absorbance was measured
spectrophotometrically at a wavelength of 490 nm using a Benchmark
Plus Microplate Spectrophotometer (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Viabilities of the challenged cells were
expressed relative to control cells.
Measurement of necrosis and
apoptosis
Apoptotic and necrotic cell death were measured
using flow cytometry with the Annexin V-FITC Apoptosis Detection
kit I (17). At 24 h after
administration of gAd, cells were trypsinized (Thermo Fisher
Scientific, Inc.) without ethylenediaminetetraacetic acid and
labelled with the fluorochromes in the absence of light for 10 min
at room temperature. Fluorescence was measured by flow cytometric
analysis using a FACSCalibur flow cytometer (BD Biosciences, San
Jose, CA, USA) to monitor green fluorescence (525 nm band-pass
filter) for Annexin V-fluorescein conjugate and red fluorescence
(575 nm band-pass filter) for PI respectively. The Annexin
V-FITC-/PI-cell population was regarded as normal, while Annexin
V-FITC+/PI-cells were taken as an indicator of early apoptosis,
Annexin V-FITC+/PI+ as late apoptosis and Annexin V-FITC-/PI+ as
necrosis. The data were analyzed using CellQuest software (BD
Biosciences). A minimum of 30,000 gated events were acquired per
sample.
Measurement of superoxide production
Production of superoxides was evaluated
intracellularly using the superoxide-sensitive dye DHE (18). DHE is oxidized by superoxides to a
novel product, which binds to DNA enhancing intracellular
fluorescence. Culture medium was aspirated and cells were incubated
with DHE (5 µM final concentration) for 10 min at 37°C in
the dark. Following two rinses in phosphate-buffered saline, cells
were photographed using an Axiostart 50 (Zeiss, Oberkochen,
Germany) microscope equipped with a Canon PowerShot G5
epifluorescence attachment (Canon, Inc., Tokyo, Japan). Between
five and six photographs were captured from each well. The
fluorescence intensity values for each photo were determined using
Adobe Photoshop software (version 4; Adobe Systems, Inc., San Jose,
CA, USA).
Measurement of nitrites
Nitrites were measured in the culture supernatants
by a colorimetric assay using a procedure based on the Greiss
reaction with sodium nitrite as the standard (19). Culture medium was collected and
stored at −80°C until analysis with the Nitrate/Nitrite
Colorimetric Assay kit, according to the manufacturer's
instructions.
Measurement of ΔΨ
The membrane potential assay was conducted as
previously described (20). The
relative ΔΨ was assessed using a laser scanning confocal microscope
(SP8; Leica Microsystems GmbH, Wetzlar, Germany) analysis of cells
stained with JC-1. Cells were incubated with 10 µg/ml JC-1
for 30 min at 37°C. Subsequent to applying the dye, the cells were
scanned with the confocal microscope using a 10x objective lens.
Fluorescence was excited by the 488-nm line of an argon laser and
the 543-nm line of a helium/neon laser. The red emission of the dye
is due to a potential-dependent aggregation in the mitochondria,
reflecting high ΔΨ. Green fluorescence indicates the monomeric form
of JC-1, appearing in the cytosol following mitochondrial membrane
depolarization, indicating low ΔΨ. The ratio of red/green indicated
the mitochondrial membrane depolarization states of different
cells.
Measurement of ATP
The amount of ATP was measured in the cultured cells
using a luminometer (Bio Orbit 1251 Luminometer; Bio-Orbit, Turku,
Finland) as previously described (21). Subsequent to exposure to different
experimental conditions, cells were collected to react with
different working solutions of from the ATP Bioluminescent Somatic
Cell assay kit, according to the manufacturer's instructions. All
reactions for a given sample were monitored simultaneously and
calibrated with the addition of an ATP standard from the kit.
Statistics
Data are presented as the mean ± standard error
(n=5) and were analyzed for statistical significance by one- or
two-way analysis of variance with were least significant difference
or Student-Newman-Keuls post hoc analyses. Statistical analysis and
the significance of the data were determined using SPSS software,
version 12.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
gAd reduced PQ-mediated cytotoxicity
As presented in Fig.
1, PQ significantly reduced the cell viability of A549 cells,
while gAd alleviated the PQ-induced cytotoxicity without any
effects on the cells alone. However, pretreatment with compound C
significantly reversed the protective effects of gAd on the
viability reduced by PQ.
gAd reduced PQ-mediated apoptosis
The results indicated that there was a low level of
cell death in the control group (Fig.
2A). However, PQ was observed to induce apoptosis in ~37.5%
cells (Fig. 2B). However, gAd
significantly reversed the PQ-induced cytotoxicity, reducing the
percentage of apoptotic cells to 22.3% (P<0.01). However, the
pretreatment with compound C significantly reversed gAd's
protective effect (Fig. 2).
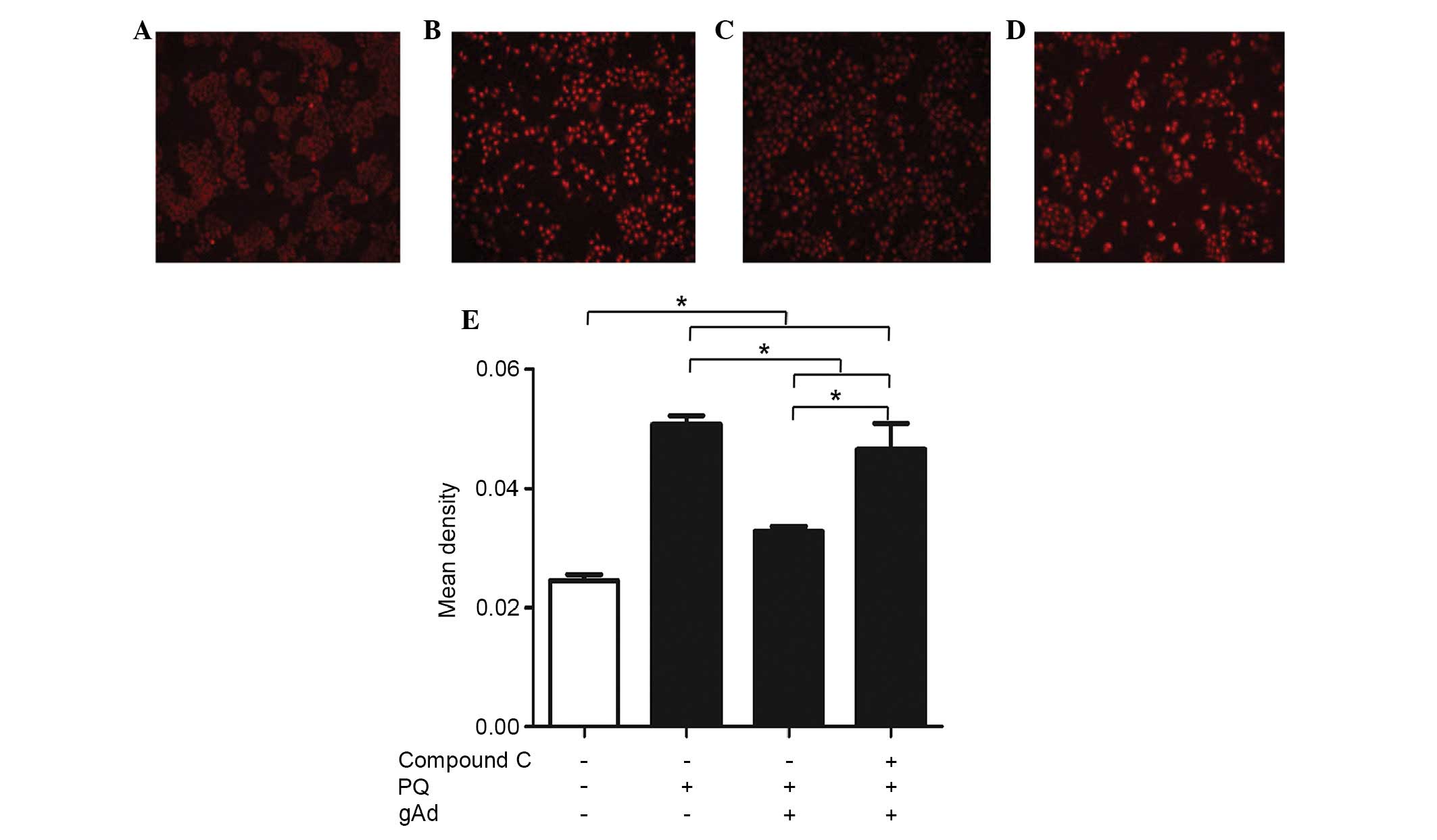
gAd reduced PQ-mediated superoxide
production
As presented in Fig.
3, control cultures generally exhibited low fluorescence
intensity while PQ-treated cultures exhibited bright red
fluorescence. This indicated that PQ significanlty increased
O2−. production in cells
compared with that of the controls (P<0.001). gAd inhibited the
O2−. production induced by PQ
significanlty. However, pre-treatment with compound C partly
reversed gAd's effects on O2−.
production.
gAd reduced PQ-mediated nitrite
production
Addition of PQ to the cultures significantly
increased nitrite production compared with the controls
(P<0.01). Administration of gAd or compound C significantly
reversed the increase, however pre-treatment with compound C did
not reverse the effects of gAd on nitrite production (Fig. 4).
gAd reduced PQ-mediated ΔΨ
depolarization
Incubation with PQ depolarized the ΔΨ significantly,
resulting in a low ratio of red/green. Treatment with gAd
repolarized the ΔΨ of the cells, resulting in a greater ratio of
red/green, compared with PQ group (P<0.01). However, compound C
reversed the repolarization of gAd, resulting in a lower ratio of
red/green as compared with the gAd group (P<0.01; Fig. 5).
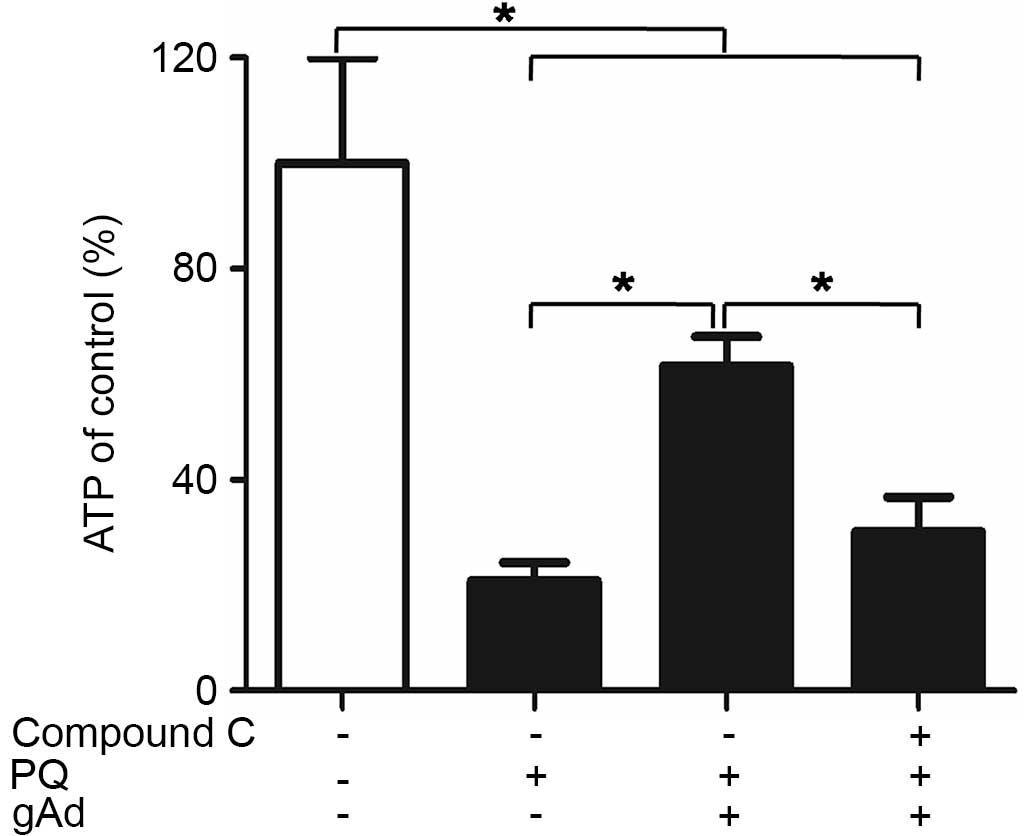
gAd partly reversed the PQ-mediated
reduction in ATP
Addition of PQ significantly reduced the
intracellular ATP concentration, as compared with the initial
control levels: 21% after 24 h, irrespective of the presence of
other medium supplements or stimulation (P<0.05). Conversely,
treatment with gAd increased the ATP level to ~50% of that of the
control group (P<0.05). However, compound C reversed the
gAd-mediated increase (P<0.05; Fig.
6).
Discussion
The most prevalent cause of morbidity and mortality
in patients with PQ poisoning is organ injury, in particular,
injury to the lungs (22). It has
been demonstrated that excessive ROS/RNS stress resulting from
impaired mitochondria, is an important feature of the injured lung
epithelium in PQ poisoning, though the underlying mechanism
responsible for the mitochondrial dysfunction remains unclear
(23). Therefore, studies have
focussed upon clarifying the underlying mechanisms for
PQ-associated mitochondria injury of alveolar type II cells
(24,25) and developing novel strategies for
the treatment of PQ-poisoned patients (26). In the present study, an established
PQ poisoning model with A549 cells cultured with PQ for 24 h was
used, and it was confirmed that apoptosis and cell death were
exacerbated in the presence of PQ. Furthermore, it was indicated
that PQ markedly disturbed mitochondrial function, depolarizing ΔΨ
and reducing ATP production, resulting in progressive increases of
O2−. and NO. This may be
explained by the fact that PQ2+ induced the release of
Ca2+ from cytoplasm (27), which may induce the depolarization
of mitochondria and reduce ΔΨ (28). This results in reductions of ATP
synthesis to less than 50% of the normal level, thus activating
pro-apoptotic and pro-necrotic proteins, including Bax and caspase
3/7/8/9 (29,30). As a result, the injured
mitochondria produce excessive O2−. and NO, which attached to the
mitochondrial membrane in turn, and resulted in increases in
ROS/RNS production and ultimately inducing apoptosis (31).
Injury resulting from PQ poisoning was however,
reversed by treatment with gAd in the current study. Although the
associated mechanisms remain to be fully elucidated, a previous
study demonstrated the potential cellular protective effects of gAd
against apoptosis via inhibiting mitochondrial dysfunction and
ROS/RNS attachment (32).
Therefore, the current study suggested that gAd may protect the
alveolar type II cells aginst PQ-induced cell injury via inhibiting
mitochondrial dysfunction and reducing oxidative/nitrative
injury.
In order to further elucidate the underlying
mechanisms for the cellular-protective effects of gAd in PQ
poisoning against mitochondrial dysfunction and oxidative/nitrative
injury, the signaling of AMPK, a protective kinase in
oxidative/nitrative injury and a downstream mediator of gAd's
mitochondrial protective actions, was investigated (33). It was identified that the
protective effects of gAd could be reversed by an AMPK inhibitor,
compound C, which has been previously identified to block the
cardiac protective benefits of gAd treatment against cardiac
myocardial hypoxia/reoxygenation injury (15). This result clarified the critical
role of AMPK in the pneumonocyte-protective effects of gAd,
indicating it may be used in the therapy for PQ poisoning. This
observation is consistent with a previous study by Yan et al
(15), which demonstrated that gAd
was able to activate AMPK-peroxisome proliferator-activated
receptor γ coactivator 1-α, resulting in increased ATP production,
improving ΔΨ, thus leading to return to normal function. These
results indicate that AMPK may be one of the key molecules in gAd's
inhibition of mitochondrial dysfunction induced by PQ. However,
interspecies differences (34,35),
animal model establishment (36,37)
and the injury procedure (38) may
affect whether AMPK signaling serves a beneficial or detrimental
role in the mitochondria. Experiments using animal models should be
conducted in order to further clarify the mechanisms.
There were limitations in the present study. The
inactivation of AMPK was not introduced when PQ was added to the
cells, due to the high rate of mortality, thus the ability to
directly demonstrate the role of AMPK in PQ-poisoned mitochondria
following oxidative/nitrative injury was compromised. In addition,
gAd treatment in PQ poisoning may modulate numerous other key
molecules in addition to AMPK-ATP (39–41).
Thus, investigation of these molecules in oxidative/nitrative
injury requires additional investigation.
In conclusion, it was demonstrated that the ΔΨ
exhibited progressive reductions with increased oxidative/nitrative
injury in PQ-induced apoptosis. In addition, it was identified that
in PQ poisoning, ΔΨ is increased in response to gAd treatment,
which significantly attenuated mitochondrial dysfunction and
oxidative/nitrative injury in PQ-poisoned alveolar type II cells.
The present study provided indirect evidence for the possible role
of the AMPK-ATP signaling pathway in gAd against
oxidative/nitrative injury originating from the mitochondria
following PQ poisoning. To the best of our knowledge, this is the
first study to demonstrate the ability of gAd in attenuating
oxidative/nitrative injury in PQ poisoning of lung cells beyond its
metabolic actions. These observations add to the current
understanding of the pathophysiology of PQ poisoning and the
mechanisms of the widely used gAd therapy. This may aid in the
development of future therapeutic strategies capable of assisting
gAd in protecting against PQ-induced lung injury.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant nos. 30900493 and
81471836) and the Science and Technology Department Foundation of
Sichuan Province (grant no. 2013JY0011). The authors would
additionally like to acknowledge Professor Xin-liang Ma of Thomas
Jefferson University (Philadelphia, PA, USA) for his help and
advice aiding in the completion of the present study.
Abbreviations:
|
ROS/RNS
|
reactive oxygen/nitrogen species
|
|
PQ
|
paraquat
|
|
gAd
|
globular adiponectin
|
|
ATP
|
adenosine 5′-triphosphate
|
|
AMPK
|
adenosine 5′-monophosphate-activated
protein kinase
|
References
|
1
|
Vieira DN and de Azevedo-Bernarda R:
Paraquat poisoning. Pulmonary lesions. J Toxicol Clin Exp.
9:177–186. 1989.In French. PubMed/NCBI
|
|
2
|
Cai Q, Lu Z, Hong G, Jiang X, Wu Z, Zheng
J, Song Q and Chang Z: Recombinant adenovirus Ad-RUNrf2 reduces
paraquat-induced A549 injury. Hum Exp Toxicol. 31:1102–1112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Golbidi S, Botta A, Gottfred S, Nusrat A,
Laher I and Ghosh S: Glutathione administration reduces
mitochondrial damage and shifts cell death from necrosis to
apoptosis in ageing diabetic mice hearts during exercise. Br J
Pharmacol. 171:5345–5360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Haagsman HP, Schuurmans EA, Batenburg JJ
and Van Golde LM: Phospholipid synthesis in isolated alveolar type
II cells exposed in vitro to paraquat and hyperoxia. Biochem J.
245:119–126. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kurokawa H, Sugiyama S, Nozaki T, Sugamura
K, Toyama K, Matsubara J, Fujisue K, Ohba K, Maeda H, Konishi M, et
al: Telmisartan enhances mitochondrial activity and alters cellular
functions in human coronary artery endothelial cells via
AMP-activated protein kinase pathway. Atherosclerosis. 239:375–385.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Barreto-Torres G, Hernandez JS, Jang S,
Rodríguez-Muñoz AR, Torres-Ramos CA, Basnakian AG and Javadov S:
The beneficial effects of AMP kinase activation against oxidative
stress are associated with prevention of PPARα-cyclophilin D
interaction in cardiomyocytes. Am J Physiol Heart Circ Physiol.
308:H749–H758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berg AH, Combs TP and Scherer PE:
ACRP30/adiponectin: An adipokine regulating glucose and lipid
metabolism. Trends Endocrinol Metab. 13:84–89. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okamoto Y, Kihara S, Funahashi T,
Matsuzawa Y and Libby P: Adiponectin: A key adipocytokine in
metabolic syndrome. Clin Sci (Lond). 110:267–278. 2006. View Article : Google Scholar
|
|
9
|
Robinson K, Prins J and Venkatesh B:
Clinical review: Adiponectin biology and its role in inflammation
and critical illness. Crit Care. 15:2212011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kadowaki T, Yamauchi T and Kubota N: The
physiological and pathophysiological role of adiponectin and
adiponectin receptors in the peripheral tissues and CNS. FEBS Lett.
582:74–80. 2008. View Article : Google Scholar
|
|
11
|
Fang X and Sweeney G: Mechanisms
regulating energy metabolism by adiponectin in obesity and
diabetes. Biochem Soc Trans. 34:798–801. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tao L, Gao E, Jiao X, Yuan Y, Li S,
Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ and Ma XL:
Adiponectin cardioprotection after myocardial ischemia/reperfusion
involves the reduction of oxidative/nitrative stress. Circulation.
115:1408–1416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim JE, Song SE, Kim YW, Kim JY, Park SC,
Park YK, Baek SH, Lee IK and Park SY: Adiponectin inhibits
palmitate-induced apoptosis through suppression of reactive oxygen
species in endothelial cells: Involvement of cAMP/protein kinase A
and AMP-activated protein kinase. J Endocrinol. 207:35–44. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei CD, Li Y, Zheng HY, Sun KS, Tong YQ,
Dai W, et al: Globular adiponectin protects H9c2 cells from
palmitate-induced apoptosis via Akt and ERK1/2 signaling pathways.
Lipids Health Dis. 11:1352012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yan W, Zhang H, Liu P, Wang H, Liu J, Gao
C, Liu Y, Lian K, Yang L, Sun L, et al: Impaired mitochondrial
biogenesis due to dysfunctional adiponectin-AMPK-PGC-1α signaling
contributing to increased vulnerability in diabetic heart. Basic
Res Cardiol. 108:3292013. View Article : Google Scholar
|
|
16
|
Kim JE, Ahn MW, Baek SH, Lee IK, Ki YW,
Kim JY, Dan JM and Park SY: AMPK activator, AICAR, inhibits
palmitate-induced apoptosis in osteoblast. Bone. 43:394–404. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Krysko DV, Vanden Berghe T, D'Herde K and
Vandenabeele P: Apoptosis and necrosis: Detection, discrimination
and phagocytosis. Methods. 44:205–221. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Radad K, Rausch WD and Gille G: Rotenone
induces cell death in primary dopaminergic culture by increasing
ROS production and inhibiting mitochondrial respiration. Neurochem
Int. 49:379–386. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Green LC, Wagner DA, Glogowski J, Skipper
PL, Wishnok JS and Tannenbaum SR: Analysis of nitrate, nitrite and
15 N.nitrate in biological fluids. Anal Biochem. 126:131–138. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Akao M, Ohler A, O'Rourke B and Marbán E:
Mitochondrial ATP-sensitive potassium channels inhibit apoptosis
induced by oxidative stress in cardiac cells. Circ Res.
88:1267–1275. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yeh ST, Guo HR, Su YS, Lin HJ, Hou CC,
Chen HM, Chang MC and Wang YJ: Protective effects of
N-acetylcysteine treatment post acute paraquat intoxication in rats
and in human lung epithelial cells. Toxicology. 223:181–190. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Onyon LJ and Volans GN: The epidemiology
and prevention of paraquat poisoning. Hum Toxicol. 6:19–29. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gaudreault P, Karl PI and Friedman PA:
Paraquat and putrescine uptake by lung slices of fetal and newborn
rats. Drug Metab Dispos. 12:550–552. 1984.PubMed/NCBI
|
|
24
|
Wang GY, Hirai K and Shimada H:
Mitochondrial breakage induced by the herbicide paraquat in
cultured human lung cell. J Electron Microsc (Tokyo). 3:181–184.
1992.
|
|
25
|
Chen YW, Yang YT, Hung DZ, Su CC and Chen
KL: Paraquat induces lung alveolar epithelial cell apoptosis via
Nrf-2-regulated mitochondrialdysfunction and ER stress. Arch
Toxicol. 86:1547–58. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shokrzadeh M, Shaki F, Mohammadi E,
Rezagholizadeh N and Ebrahimi F: Edaravone decreases paraquat
toxicity in a549 cells and lung isolated mitochondria. Iran J Pharm
Res. 13:675–681. 2014.PubMed/NCBI
|
|
27
|
Chang X, Lu W, Dou T, Wang X, Lou D, Sun
X, et al: Paraquat inhibits cell viability via enhanced oxidative
stress and apoptosis in human neural progenitor cells. Chem Biol
Interact. 206:248–255. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang CL, Lee YC, Yang YC, Kuo TY and
Huang NK: Minocycline prevents paraquat-induced cell death through
attenuating endoplasmic reticulum stress and mitochondrial
dysfunction. Toxicol Lett. 209:203–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Helewski KJ, Kowalczyk-Ziomek GI and
Konecki J: Apoptosis and necrosis-two different ways leading to the
same target. Wiad Lek. 59:679–684. 2006.In Polish.
|
|
30
|
Kasahara A and Scorrano L: Mitochondria:
From cell death executioners to regulators of cell differentiation.
Trends Cell Biol. 24:761–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang Z, Wang J, Xie R, Liu R and Lu Y:
Mitochondria-derived reactive oxygen species play an important role
in Doxorubicin-induced platelet apoptosis. Int J Mol Sci.
16:11087–11100. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Park M, Youn B, Zheng XL, Wu D, Xu A and
Sweeney G: Globular adiponectin, acting via AdipoR1/APPL1, protects
H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One.
6:e191432011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin Z, Wu F, Lin S, Pan X, Jin L, Lu T,
Shi L, Wang Y, Xu A and Li X: Adiponectin protects against
acetaminophen-induced mitochondrial dysfunction and acute liver
injury by promoting autophagy in mice. J Hepatol. 61:825–831. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zheng A, Li H, Xu J, Cao K, Li H, Pu W,
Yang Z, Peng Y, Long J, Liu J and Feng Z: Hydroxytyrosol improves
mitochondrial function and reduces oxidative stress in the brain of
db/db mice: Role of AMP-activated protein kinase activation. Br J
Nutr. 113:1667–1676. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chen H, Wang JP, Santen RJ and Yue W:
Adenosine monophosphate activated protein kinase (AMPK), a mediator
of estradiol-induced apoptosis in long-term estrogen deprived
breast cancer cells. Apoptosis. 20:821–830. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen LJ, Na R, Gu M and Ran QT: Reduction
in mitochondria H2O2 protects against insulin resistance through a
mechanism of Akt and AMPK activation. Free Radic Biol Med.
47:S852009.
|
|
37
|
Pan JS, Huang L, Belousova T, Lu L, Yang
Y, Reddel R, Chang A, Ju H, DiMattia G, Tong Q, et al:
Stanniocalcin-1 inhibits renal ischemia/reperfusion injury via an
AMP-activated protein kinase-dependent pathway. J Am Soc Nephrol.
26:364–378. 2015. View Article : Google Scholar :
|
|
38
|
Wu SB, Wu YT, Wu TP and Wei YH: Role of
AMPK-mediated adaptive responses in human cells with mitochondrial
dysfunction to oxidative stress. Biochim Biophys Acta.
1840.1331–1344. 2014.
|
|
39
|
Qiao LP, Kinney B, Yoo HS, Lee B, Schaack
J and Shao JH: Adiponectin increases skeletal muscle mitochondrial
biogenesis by suppressing mitogen-activated protein kinase
phosphatase-1. Diabetes. 61:1463–1470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou M, Xu A, Tam PK, Lam KS, Chan L, Hoo
RL, Liu J, Chow KH and Wang Y: Mitochondrial dysfunction
contributes to the increased vulnerabilities of adiponectin
knockout mice to liver injury. Hepatology. 48:1087–1096. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yamauchi T, Iwabu M, Okada-Iwabu M and
Kadowaki T: Adiponectin receptors: A review of their structure,
function and how they work. Best Pract Res Clin Endocrinol Metab.
28:15–23. 2014. View Article : Google Scholar : PubMed/NCBI
|