Introduction
Natural herbal medicines have long been used to
treat cancer. Biomedical research and cancer treatment clinical
trials have provided evidence regarding the use of herbal
medicines; therefore, they are increasingly being accepted as a
complementary and alternative treatment (1). In addition, natural medicines have
been reported to have an important role in human health,
particularly certain well-studied plants, including Taxus
chinensis (Taxus madia) (2), Radix Sophorae flavescentis
(Sophora flavescens) (3),
Alkanna tinctoria, Lithospermum erythrorhizon and licorice
(Glycyrrhiza L) (4).
Licorice is one of the most commonly prescribed herbs in Chinese
Traditional Medicine, and has been used for >2,000 years. The
effects of licorice have been reported on various diseases, ranging
from microbial infection to cancer (5–7).
However, since herbs usually contain numerous chemical
compositions, the mechanism of action of these herbs is currently
unclear. Recently, several chemical ingredients have been isolated
and proved to contribute to the activities of licorice. The main
bioactive constituents of licorice include triterpene saponins and
various types of flavonoids (8).
Licochalcone A (LCA) is a chalcone compound isolated
from licorice root (Radix Glycyrrhizae), which is known for
its numerous biological activities, including anti-inflammatory
(9) antimicrobial (10), antioxidant (11) and anticancer effects (12).
Apoptosis, or 'programmed cell death' (PCD), is a
normal event that occurs in multicellular organisms (13). Previous studies have suggested that
reactive oxygen species (ROS) have an important role in the
mitochondrial apoptotic pathway (14), and have been shown to be involved
in endoplasmic reticulum (ER) stress-induced apoptosis (15,16).
In addition, increases in intracellular Ca2+ levels may
contribute to ROS accumulation and ER stress (17).
Ca2+, which is one of the most versatile
second messengers, regulates various cellular functions, including
contraction, secretion, metabolism, gene expression, cell survival
and PCD (18). Calpains are
Ca2+-activated non-lysosomal cysteine proteases, which
have been reported to promote cysteinyl aspartate specific
proteinase (caspase)-4 activation during ER stress-induced
apoptosis (19–21). Furthermore, it has been reported
that LCA induces apoptosis in HepG2 hepatocellular carcinoma cells
through induction of ER stress via a phospholipase C gamma 1-,
Ca2+- and ROS-dependent pathway (22).
Our previous studies have demonstrated that LCA may
significantly increase ROS levels and induce apoptosis of T24 human
bladder cancer cells (23).
However, the underlying molecular mechanism remains unclear. The
present study aimed to further elucidate the mechanism by which LCA
induces apoptosis of T24 cells.
Materials and methods
Reagents
LCA (purity ≥98%) was purchased from Zhongxin
Pharmaceutical Group Corp., Ltd. (Tianjin, China). Roswell Park
Memorial Institute (RPMI) 1640 medium was obtained from Gibco
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Super neonatal
bovine serum (NBS) was purchased from Hangzhou Sijiqing Biological
Engineering Materials Co., Ltd. (Hangzhou, China). Annexin
V/propidium iodide (PI) apoptosis kit was purchased from Nanjing
KeyGen Biotech. Co. Ltd. (Nanjing, China). Molecular Probes
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Thermo Fisher Scientific, Inc.,), ethylene glycol tetraacetic acid
(EGTA), Fluo-3 AM ester, and Hoechst 33258 were purchased from
Sigma-Aldrich (St. Louis, MO, USA). BAPTA-AM was purchased from AAT
Bioquest, Inc. (Sunnyvale, CA, USA).
Cell culture and treatment
The T24 human bladder cancer cells were purchased
from the Cell Bank of the Committee on Type Culture Collection of
the Chinese Academy of Sciences (Shanghai, China). The cells were
maintained in RPMI 1640 medium supplemented with 10% NBS (v/v), 100
U/ml penicillin and 100 μg/ml streptomycin at 37°C in a
humidified atmosphere containing 5% CO2. Cells were
allowed to attach for 24 h prior to treatment. LCA was dissolved in
dimethyl sulfoxide (DMSO) and diluted with fresh medium to achieve
the desired concentration. The final concentration of DMSO did not
exceed 0.2% in the fresh medium, and DMSO at this concentration is
known to have no significant effect on cell viability.
Cell viability assay
Cell viability was measured using the sulforhodamine
B (SRB) assay (24). Briefly, T24
cells were trypsinized and seeded into 96-well plates at
1.0×105 cells/ml. Subsequently, the cells were exposed
to LCA (0, 20, 40 and 80 μM) for 24 h, followed by a further
24 h incubation in fresh medium. The T24 cells were then fixed with
trichloroacetic acid and stained for 30 min with 0.4% (wt/vol) SRB
(Sigma-Aldrich) dissolved in 1% acetic acid. Unbound dye was
removed by four washes with 1% acetic acid, and protein-bound dye
was extracted with 150 ml DMSO for determination of optical density
was detected at a wavelength of 490 nm using a Varioskan Flash 3001
plate reader (Thermo Fisher Scientific, Inc.).
Quantification of apoptosis by flow
cytometry
Apoptosis was assessed using Annexin V-fluorescein
isothiocyanate (FITC) and PI labeling, as previously described
(25). Cells were treated with 0,
20, 40 and 80 μM LCA for 24 h. Subsequently, the cells were
washed twice with phosphate-buffered saline (PBS), and were
resuspended in staining buffer containing 5 μl (1 mg/ml) PI
and 5 μl Annexin V-FITC. Double-labeling was performed at
room temperature for 10 min in the dark prior to flow cytometric
analysis. Cell staining was detected using a FACStar flow cytometer
(BD Biosciences, Franklin Lakes, NJ, USA).
Determination of morphological
alterations
Alterations in the nuclear morphology of apoptotic
cells were observed by labeling the cells with the nuclear stain
Hoechst 33258, and examining them under a fluorescent microscope.
After treatment with 0, 20, 40 and 80 μM LCA for 24 h, the
cells were fixed in formaldehyde (40 g/l) in PBS for 20 min,
followed by Hoechst 33258 (10 mg/l) staining for 30 min in the dark
at 37°C. Nuclear morphology was subsequently observed under a
fluorescence microscope (MIC00266; Zeiss, Oberkochen, Germany)
(26).
Detection of Ca2+
concentration
Ca2+ concentration was measured by Fluo-3
AM staining and microscopy. Briefly, T24 cells were incubated with
or without Ca2+ chelators (200 μM EGTA and 10
μM BAPTA-AM) for 1 h prior to 5 μM LCA treatment for
24 h. Subsequently, the cells were harvested and washed twice, and
were resuspended in Fluo 3 AM (5 μM) at 37°C for 30 min.
After washing three times, the stained cells were observed under a
computer-assisted microscope (MIC00266; Carl Zeiss AG, Oberkochen,
Germany) at an excitation wavelength of 488 nm and an emission
wavelength of 525 nm.
Detection of intracellular ROS
levels
The intracellular levels of ROS in T24 cells were
assessed using H2DCFDA (27). Briefly, cells were incubated with
55 μM LCA or with 10 μM BAPTA-AM (a chelator of
cytosolic Ca2+) for 1 h prior to LCA treatment. The
treated cells were then washed in PBS and incubated with 30
μM H2DCFDA for 30 min at 37°C. Fluorescence was
detected using a fluorescent plate reader at 485/525 nm
excitation/emission wavelengths (Varioskan Flash 3001; Thermo
Fisher Scientific, Inc.) and data were expressed as median
fluorescence intensity. The stained cells were then observed under
a computer-assisted microscope (MIC00266; Carl Zeiss AG).
Measurement of mitochondrial membrane
potential (MMP)
The MMP was assessed using a dual-emission
potential-sensitive probe, 5,5′,6, 6′-tetra-chloro-1,
1′,3,3′-tetraethyl-imidacarbocyanine iodide (JC-1; KeyGen Biotech
Co., Ltd., Nanjing, China). The ratio of red to green fluorescence
of JC-1 depends solely on membrane potential, with a decrease being
indicative of membrane depolarization (28). Briefly, the cells were exposed to
LCA (0, 20, 40 or 80 μM) for 4, 8, 16 or 24 h. Subsequently,
the cells were loaded with 2 mg/l JC-1 at 37°C for 20 min, and were
analyzed using a plate reader (Varioskan Flash 3001; Thermo Fisher
Scientific, Inc.).
Reverse transcription semiquantitative
and quantitative polymerase chain reaction (PCR)
Total RNA was extracted from T24 cells using TRIzol
(Sangon Biotech Co., Ltd., Shanghai, China). RNA quality was
determined using the A260/A280 ratio and 1.5% agarose gel
electrophoresis. cDNA synthesis was performed using Moloney murine
leukemia virus reverse transcriptase with a First Strand cDNA
Synthesis kit (Fermentas; Thermo Fisher Scientific, Inc.). The
synthesized cDNA was amplified by Riboloek Nase-free
ddH2O (8 μl), template RNA (3 μl),
Oligo(dT)18 (1 μl) to a final volume of 12 μl. Tubes
were placed into the C1000 Thermal Cycler (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) at 70°C for 5 min, following which 5X
reaction buffer (4 μl), Riboloek Nase Indihitor (1
μl), dNTP M-M RT RTase (1 μl) and RNase DEPC-treated
water to a final volume of 20 μl were added and incubated at
42°C 1 h for denaturation and 70°C 5 min for annealing. The
synthesized cDNA was amplified by Oligo(dT)18, according
to the instructions of a PCR Amplification kit (Fermentas; Thermo
Fisher Scientific, Inc.). The PCR primers (synthesized by Sangon
Biotech Co., Ltd.; presented in Table
I) and their cycling conditions were set as indicated. The PCR
reaction volume consisted of 12.5 μl 2X PCR Master (Sangon
Biotech Co., Ltd.), 3 μl cDNA template and 0.5 μl of
each primer. The cycling conditions were as follows:
Pre-denaturation at 94°C for 3 min, with 30–35 cycles of
denaturation at 94°C for 30 sec, annealing for 30 sec, extension at
72°C and a final extension at 72°C for 10 min, using a C1000
Thermal Cycler (Bio-Rad Laboratories, Inc.). The gene products were
quantified using agarose gel electrophoresis (Biodee Biotechnology
Co., Ltd., Beijing, China) and a Bio-Rad gel imaging system
(Bio-Rad Laboratories, Inc.).
 | Table IPolymerase chain reaction primer
sequences. |
Table I
Polymerase chain reaction primer
sequences.
| Primer | Forward | Reverse |
|---|
| GAPDH |
CAAGGTCATCCATGACAACTTTG |
GTCCACCACCCTGTTGCTGTAG |
| Bcl-xL |
GCATATCAGAGCTTTGAACAGGT |
TAGGTGGTCATTCAGGTAAGTGG |
| Bax |
ACGAACTGGACAGTAACATGGAG |
CAGTTTGCTGGCAAAGTAGAAAAG |
| Bim |
CACATGAGCACATTTCCCTCT |
AAGGCACAAAACCTGCAGTAA |
| Caspase-3 |
CTGGACTGTGGCATTGAGAC |
ACAAAGCGACTGGATGAACC |
| Caspase-4 |
TGAACTGGAAGGAAGAGGAA |
GCGGTTGTTGAATATCTGGA |
| Caspase-9 |
CAGTGGGCTCACTCTGAAGACC |
ACGCGTTACTGGCATTGAGG |
| Apaf-1 |
TGGAATGGCAGGCTGTGGGA |
TGCACTCCCCCTGGGAAACA |
| Calpain 2 |
GCAGCCATTGCCTCCCTCAC |
ACCTCCACCCACTCGCCGTA |
The quantitative PCR analysis was carried out
according to the manufacturer's protocol of the Taqman One-Step PCR
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.).
Total cDNA (2 μl) was added per 25 μl reaction,
alongside 0.5 μl sequence-specific primers and 12.5
μl SYBR Premix Ex Taq. All target gene primers and probes
were purchased commercially (Sangon Biotech Co., Ltd.).
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an
internal control. The cycling conditions were as follows:
Pre-denaturation at 95°C for 4 min, denaturation at 95°C for 5 sec,
annealing for 30 sec, extension at 72°C and a final extension at
72°C for 10 min, using a Rotor-Gene Q Real time PCR machine (Qiagen
China Co., Ltd, Shanghai, China). Relative expression levels of the
target genes were calculated based on the 2−ΔΔCq method
of relative quantification (29),
according to the following equation: Relative expression level =
2(Cq value of GAPDH - Cq value gene of interest). The
primer sequences are presented in Table I.
Statistical analysis
Data are presented as the mean ± standard from at
least three independent experiments. Data were evaluated by one-way
analysis of variance followed by Student-Newman-Keuls test, uding
Origin software, version 8.0 (OriginLab, Northampton, MA, USA). In
all cases, P<0.05 was considered to indicate a statistically
significant difference.
Results
LCA induces cell apoptosis in T24 human
bladder cancer cells
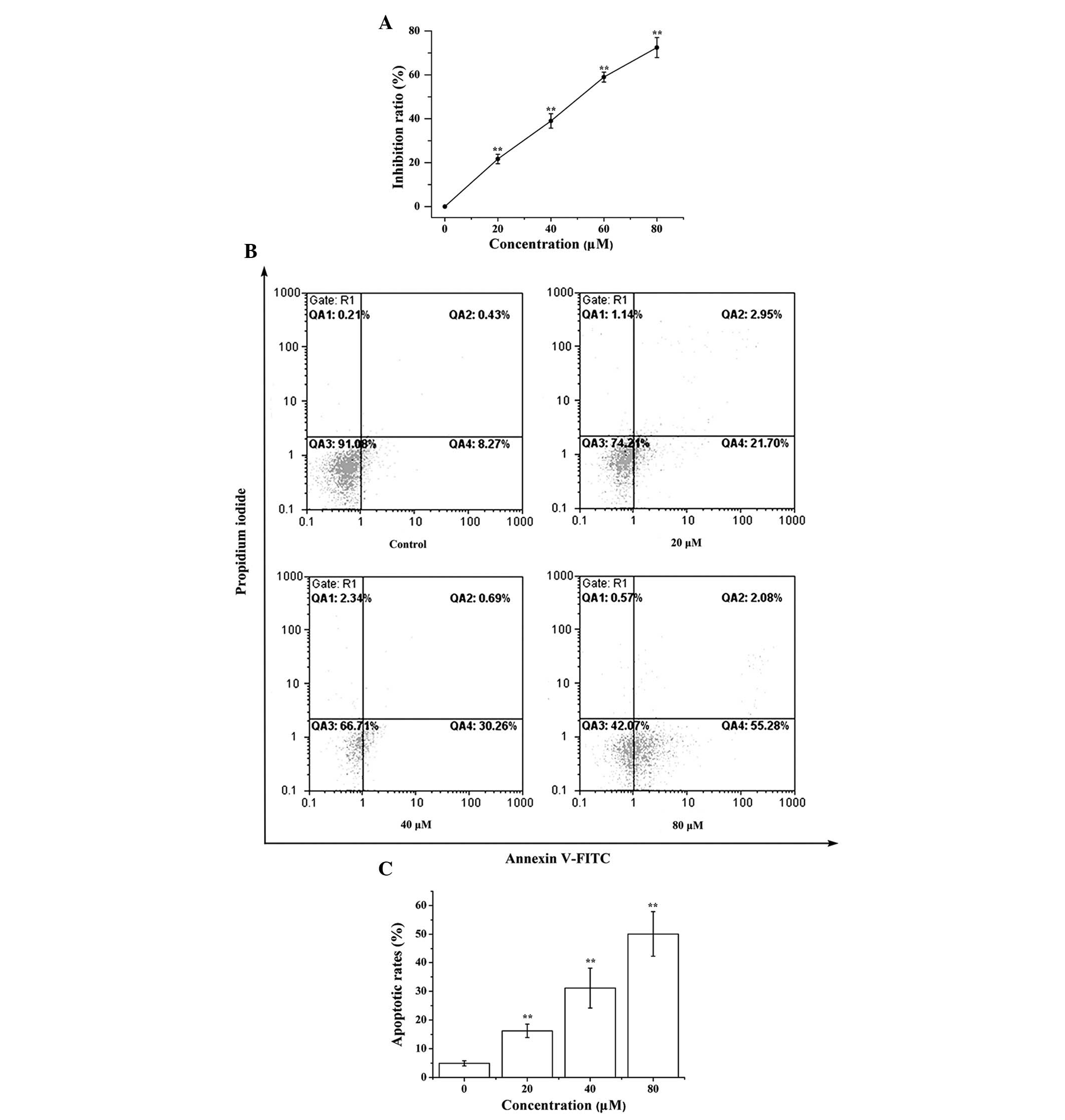
To examine cell viability in vitro, the SRB
assay was used to determine the inhibitory effects of LCA on
proliferation. A total of 24 h post-LCA treatment, LCA reduced the
proliferation of T24 cells in a dose-dependent manner; the half
maximal inhibitory concentration was ~55 μM
(P=4.3×10−13; Fig. 1A).
Subsequently, it was investigated whether LCA was able to induce
cell death through an apoptotic mechanism. Annexin V-FITC and PI
double-labeling was used for the detection of phosphatidylserine
externalization, a hallmark of early phase apoptosis. Compared with
the control group, a high proportion of Annexin V+
labeling was detected in cells treated with LCA, thus indicating
that they were in the early phase of apoptosis
(P=1.3×10−5; Fig. 1B and
C). These results indicate that LCA significantly induced
apoptosis in T24 cells.
LCA induces alterations in nuclear
morphology
Typical apoptotic morphological alterations, as
indicated by condensed nuclei and nuclear fragmentation, were
apparent after exposure to 40 μM LCA. Apoptotic nuclear
alterations were markedly increased in the cells pretreated with 80
μM LCA (Fig. 2).
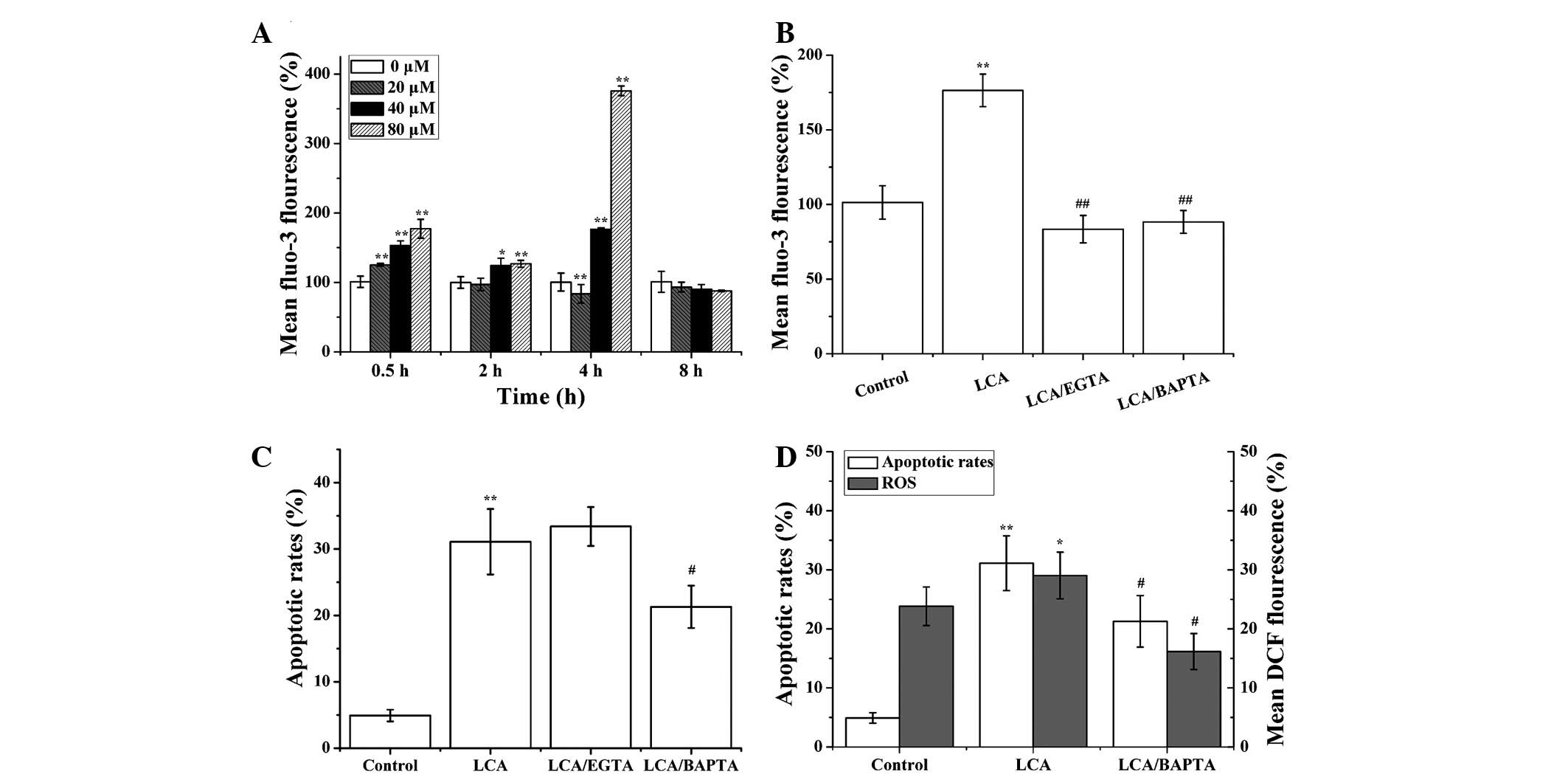
LCA induces Ca2+ release
Following treatment of T24 cells with LCA,
Ca2+ levels were significantly increased compared with
the control group. These results indicate that LCA promoted
Ca2+ release in a time-dependent manner (30 min, P=
4.3×10−13; 2 h, P=1.2×10−5; 4 h, P=0.0084;
Fig. 3A). In order to investigate
whether LCA could induce an increase in cytosolic Ca2+
through extracellular or intracellular Ca2+ pools, the
T24 cells were pretreated with EGTA (an extracellular
Ca2+ chelator) and BAPTA-AM (an intracellular
Ca2+ chelator). EGTA and BAPTA-AM significantly
suppressed LCA-induced Ca2+ release
(P=1.9×10−6 and P=2.2×10−6, respectively;
Fig. 3B). Furthermore, BAPTA-AM
could attenuate LCA-induced apoptosis, whereas EGTA had no
inhibitory effects on apoptosis (Fig.
3C). Collectively, these results indicate that LCA mainly
induced release of intracellular Ca2+ to promote
apoptosis.
To determine whether cytosolic Ca2+
release is a signal leading to ROS accumulation in LCA-induced
apoptosis, T24 cells were pretreated with BAPTA-AM, then treated
with LCA for 1 h, or the cells were treated with LCA alone. The
levels of ROS generation were lower in the LCA and BAPTA-AM treated
group, as compared with in the LCA-treated group (apoptotic rates,
P= 0.0241; ROS, P= 0.0128; Fig.
3D). These results suggest that BAPTA-AM may decrease
LCA-induced apoptosis and ROS generation, thus indicating that
cytosolic Ca2+ release may act upstream of ROS
generation in LCA-treated T24 cells.
Intracellular Ca2+ has a
critical role in LCA-induced apoptosis of T24 cells
As shown in Fig.
4A, treatment of T24 cells with LCA for 4, 8, 16 and 24 h
induced a time-dependent reduction in MMP (P=6.0×10−8,
P=9.5×10−7, P=0.0793, P=1.1×10−5,
respectively), thus suggesting that LCA-induced cell apoptosis may
be associated with mitochondrial dysfunction.
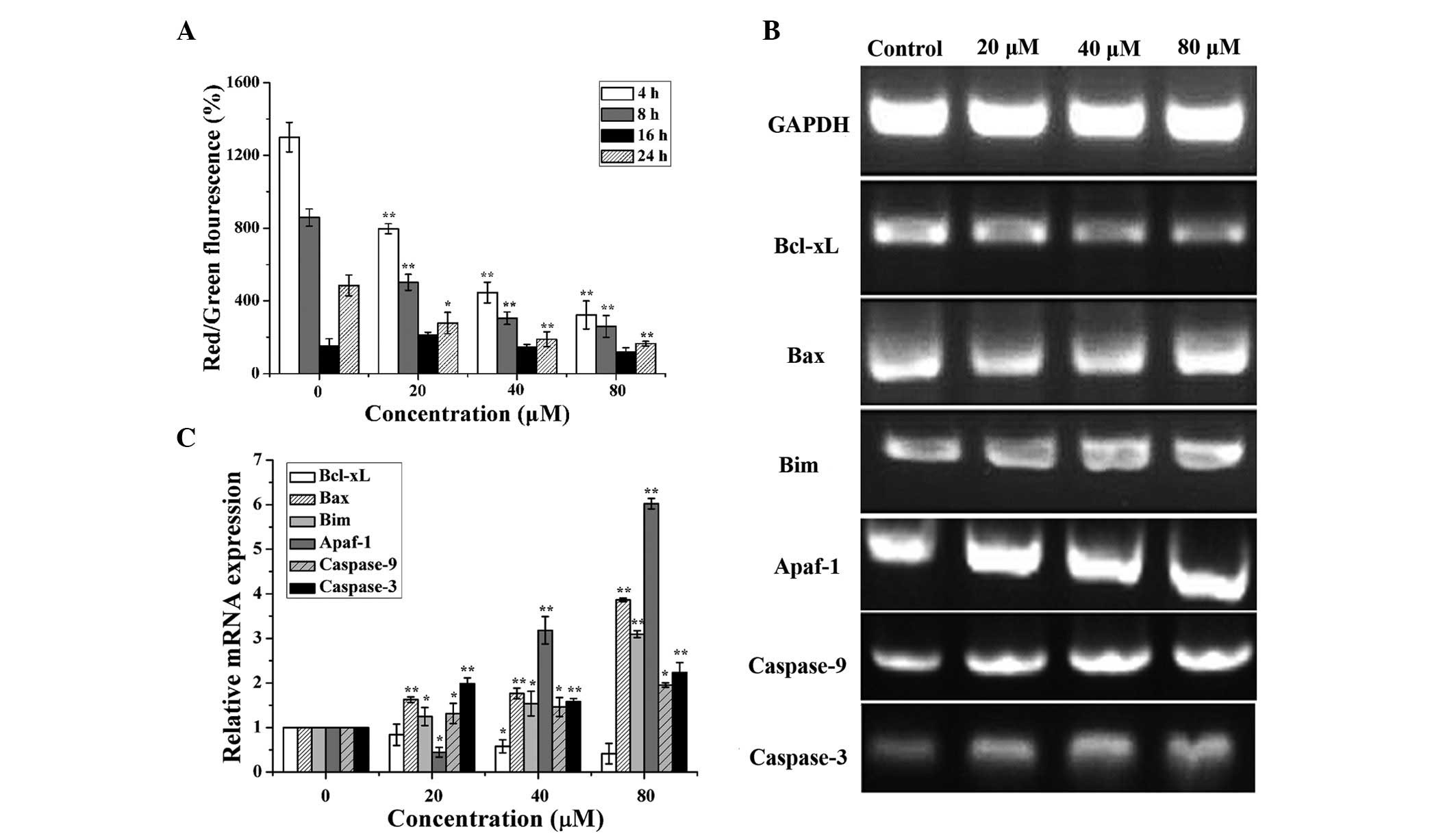 | Figure 4Licochalcone A (LCA) induced
mitochondrial dysfunction in T24 cells. (A) T24 cells were
incubated with 0, 20, 40 and 80 μM LCA for 4, 8, 16 and 24
h, and the mitochondrial membrane potential was determined. The
number of cells with normal polarized mitochondrial membranes (red)
compared with the number of cells with depolarized mitochondrial
membranes (green) is expressed as a percentage of the total cell
number. (B) B-cell lymphoma (Bcl)-extra large (Bcl-xL),
Bcl-2-associated X protein (Bax), Bcl-2-interacting mediator of
cell death (Bim), apoptotic protease activating factor-1 (Apaf-1),
caspase-9 and caspase-3 expression levels were detected by
semiquantitative polymerase chain reaction (PCR) following
treatment with 0, 20, 40 and 80 μM LCA for 24 h. (C) Changes
in the mRNA expression levels of Bcl-xL, Bax, Bim, Apaf-1,
caspase-9 and caspase-3 were examined by quantitative PCR analysis.
T24 cells were incubated with 0, 20, 40 and 80 μM LCA for 24
h. Data are presented as the mean ± standard deviation of three
separate experiments. *P<0.05, **P<0.01
compared with the control group. GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
To further explore whether LCA induces apoptosis via
the regulation of mitochondrial apoptosis-associated genes, B-cell
lymphoma (Bcl)-2-associated X protein (Bax), Bcl-2-interacting
mediator of cell death (Bim), Bcl-extra large (xL), apoptotic
protease activating factor-1 (Apaf-1), caspase-9 and caspase-3 were
detected in T24 cells. The cells were treated with various
concentrations of LCA for 24 h. As shown in Fig. 4B and C, the mRNA expression levels
of Bcl-xL were downregulated (P=0.052), whereas Bax (P=
4.1×10−12), Bim (P=2.4×10−8), Apaf-1
(P=1.8×10−14), caspase-9 (P= 0.0107) and caspase-3 (P=
0.0001) expression levels were upregulated in a
concentration-dependent manner.
To determine whether intracellular Ca2+
levels exert a critical role on LCA-induced mitochondrial
apoptosis, BAPTA-AM, an intracellular Ca2+ chelator, was
used. T24 cells were treated with or without BAPTA-AM for 1 h prior
to LCA treatment for 24 h. As shown in Fig. 5, compared with the LCA-treated
group, Bax (P=0.0374), Apaf-1 (P=5.7×10−5), caspase-9
(P=0.0211) and caspase-3 (P=0.0095) expression were markedly
downregulated. However, there was no significant difference in
Bcl-xL mRNA expression between the LCA-treated and LCA +
BAPTA-AM-treated groups. These data suggest that intracellular
Ca2+ has a critical role in mitochondrial apoptosis.
 | Figure 5BAPTA-AM mediated the expression of
mitochondrial apoptosis-associated genes. (A) B-cell lymphoma
(Bcl)-extra large (Bcl-xL), Bcl-2-associated X protein (Bax),
Bcl-2-interacting mediator of cell death (Bim), apoptotic protease
activating factor-1 (Apaf-1), caspase-9 and caspase-3 expression
levels were detected by semiquantitative polymerase chain reaction
(PCR). (B) Changes in the mRNA expression levels of Bcl-xL, Bax,
Bim, Apaf-1, caspase-9 and caspase-3 were examined by quantitative
PCR analysis. T24 cells were pretreated for 24 h with licochalone A
(LCA; 55 μM) followed by stimulation with BAPTA-AM (10
μM). Data are presented as the mean ± standard deviation of
three separate experiments. *P<0.05,
**P<0.01 compared with the control group;
#P<0.05, ##P<0.01 compared with the
LCA-treated group. GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
LCA induces apoptosis via the ER stress
pathway in T24 cells
The involvement of ER stress signaling in the
responses triggered by LCA-induced apoptosis was evaluated based on
the expression of calpain 2 and caspase-4. As shown in Fig. 6, cells were treated with LCA for 24
h, and calpain 2 and caspase-4 expression levels were increased in
a concentration-dependent manner. Subsequently, the T24 cells were
treated with or without BAPTA-AM for 1 h prior to LCA treatment for
24 h,. As shown in Fig. 7,
compared with the LCA-treated group, calpain 2 (P=0.0418) and
caspase-4 (P=3.8×10−5) expression levels were
downregulated in response to BAPTA-AM treatment. These results
suggest that Ca2+ is involved in the ER stress-related
apoptotic pathway.
Discussion
LCA has been reported to inhibit proliferation and
induce apoptosis in various cancer cells, including MCF-7 human
breast cancer cells (30) and
colon cancer cells (22). We
previously reported that LCA induced an increase in cytoplasmic ROS
levels, by sensing inner mitochondrial ROS production, and
activated caspase-3/caspase-9-mediated mitochondrial apoptotic
signaling pathways (31). The
present study demonstrated that: i) LCA induced Ca2+
release in T24 human bladder cancer cells; ii) LCA predominantly
induced an increase in intracellular Ca2+ release to
promote apoptosis; iii) intracellular Ca2+ may cause
upstream ROS accumulation in LCA-treated T24 cells; iv) increased
intracellular Ca2+ levels are involved in LCA-induced
T24 cell apoptosis via mitochondrial dysfunction and the ER
stress-related pathway. The mechanism by which LCA induces
apoptosis may be mediated through increased levels of intracellular
Ca2+. Notably, LCA enhanced intracellular
Ca2+, induced mitochondrial dysfunction, and activated
the apoptotic cascade and ER stress in T24 cells. These findings
indicated that intracellular Ca2+ may have a prominent
role in LCA-induced T24 cell apoptosis via he
mitochondria-dependent and ER stress-activated apoptotic
signals.
It has been indicated that increased Ca2+
levels may be associated with the apoptotic process (32). Apoptosis is often accompanied by
increased Ca2+ levels, and the addition of calcium
regulators or calmodulin inhibitors can directly induce apoptosis.
These results suggested that apoptosis is closely associated with
intracellular Ca2+ (33). In the present study, LCA induced
apoptosis by the release of intracellular Ca2+, but not
extracellular Ca2+, thus suggesting that intracellular
Ca2+ is closely associated with apoptosis in T24 cells.
Our previous study demonstrated that LCA inhibited proliferation by
inducing ROS production in T24 cells (31). The present study demonstrated that
when intracellular Ca2+ was inhibited, LCA-induced
apoptosis and ROS generation were suppressed. These results
indicated that Ca2+ may act upstream of ROS generation
in T24 cells (Fig. 3).
Mitochondria have been demonstrated to have a
crucial role in cell apoptosis, and the mitochondria-dependent
apoptotic pathway is involved in LCA-induced apoptosis (7). The present study examined whether
apoptosis is mediated through mitochondrial dysfunction, and the
MMP was analyzed using the mitochondrion-sensitive dye JC-1. The
results indicated that LCA was capable of inducing T24 cell
apoptosis by decreasing MMP (Fig.
4A). In addition, mitochondrial apoptosis-related genes Bax,
Bim, Apaf-1, caspase-9 and caspase-3 were activated by LCA, whereas
Bcl-xL was inhibited. These data suggested that LCA induced
apoptosis through mitochondrial dysfunction (Fig. 4B and C). Furthermore, when
intracellular Ca2+ levels were decreased, Bax, Bim,
Apaf-1 caspase-9 and caspase-3 expression levels were inhibited,
whereas Bcl-xL expression was not altered. These results indicated
that it was the increased levels of Ca2+ that regulated
the LCA-induced mitochondrial apoptotic pathway (Fig. 5).
Calpain is necessary for reconstruction of the
cytoskeleton, intracellular signal transduction, regulation of the
cell cycle, and apoptosis. Previous studies have reported that high
cellular concentrations of Ca2+ can activate m-calpain,
which can subsequently activate caspase-4, thus inducing the
caspase cascade reaction and leading to ER stress-associated
apoptosis (34–36). The present study, as expected,
demonstrated that treatment of T24 cells with LCA induced an
upregulation in the expression levels of calpain 2 and caspase-4.
Conversely, calpain 2 and caspase-4 expression levels were reduced
alongside a reduction in the concentration of Ca2+.
These findings indicated that intracellular Ca2+ is
associated with LCA-induced ER stress-associated apoptosis in T24
cells (Figs. 6 and 7).
In conclusion, the present study demonstrated that
LCA induces apoptosis in T24 cells, and its mechanism may be
associated with an intracellular imbalance in calcium homeostasis.
LCA was able to induce intracellular Ca2+ release,
promote ROS accumulation, and regulate the expression of
mitochondrial apoptosis-related genes (Bax, Bim, Apaf-1, caspase-9
and caspase-3 expression) and ER stress-induced apoptosis-related
genes (calpain2 and caspase-4), thus resulting in apoptosis. These
data provide further support for the notion that LCA should be
further explored as a possible chemopreventive modality, as well as
in terms of its possible effectiveness in the treatment of bladder
cancer.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31471338 and
81260338), the Science and Technology project of Shihezi City, and
the Xinjiang Production and Construction Corps Funds for Innovation
Team in Key Areas (to Q.S. Zheng).
References
|
1
|
Wang X, Feng Y, Wang N, Cheung F, Tan HY,
Zhong S, Li C and Kobayashi S: Chinese medicines induce cell death:
The molecular and cellular mechanisms for cancer therapy. Biomed
Res Int. 2014:5303422014.PubMed/NCBI
|
|
2
|
Lu C and Mei X: Study on anti-tumor
activity of extracts from cultured cells of Taxus chinensis. Zhong
Yao Cai. 26:335–337. 2003.In Chinese. PubMed/NCBI
|
|
3
|
Wang Q, Du H, Geng G, Zhou H, Xu M, Cao H,
Zhang B, Song G and Hu T: Matrine inhibits proliferation and
induces apoptosis via BID-mediated mitochondrial pathway in
esophageal cancer cells. Mol Biol Rep. 41:3009–3020. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao H, Jiang JT and Zheng QS: Advance in
studies on pharmacological effects of licochalcone A. Zhongguo
Zhong Yao Za Zhi. 38:3814–3818. 2013.In Chinese.
|
|
5
|
Fukai T, Marumo A, Kaitou K, Kanda T,
Terada S and Nomura T: Anti-Helicobacter pylori flavonoids from
licorice extract. Life Sci. 71:1449–1463. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Funakoshi-Tago M, Nakamura K, Tsuruya K,
Hatanaka M, Mashino T, Sonoda Y and Kasahara T: The fixed structure
of Licochalcone A by alpha, beta-unsaturated ketone is necessary
for anti-inflammatory activity through the inhibition of NF-kappaB
activation. Int Immunopharmacol. 10:562–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xiao XY, Hao M, Yang XY, Ba Q, Li M, Ni
SJ, Wang LS and Du X: Licochalcone A inhibits growth of gastric
cancer cells by arresting cell cycle progression and inducing
apoptosis. Cancer Lett. 302:69–75. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng Q and Ye M: Chemical analysis of
Chinese herbal medicine Gan-Cao (licorice). J Chromatogr A.
1216:1954–1969. 2009. View Article : Google Scholar
|
|
9
|
Feldman M and Grenier D: Cranberry
proanthocyanidins act in synergy with licochalcone A to reduce
Porphyromonas gingivalis growth and virulence properties, and to
suppress cytokine secretion by macrophages. J Appl Microbiol.
113:438–447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Messier C and Grenier D: Effect of
licorice compounds licochalcone A, glabridin and glycyrrhizic acid
on growth and virulence properties of Candida albicans. Mycoses.
54:e801–e806. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Furusawa J, Funakoshi-Tago M, Mashino T,
Tago K, Inoue H, Sonoda Y and Kasahara T: Glycyrrhiza
inflata-derived chalcones, Licochalcone A, Licochalcone B and
Licochalcone D, inhibit phosphorylation of NF-kappaB p65 in LPS
signaling pathway. Int Immunopharmacol. 9:499–507. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee CS, Kwak SW, Kim YJ, Lee SA, Park ES,
Myung SC, Kim W, Lee MS and Lee JJ: Guanylate cyclase activator
YC-1 potentiates apoptotic effect of licochalcone A on human
epithelial ovarian carcinoma cells via activation of death receptor
and mitochondrial pathways. Eur J Pharmacol. 683:54–62. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kerr JF, Wyllie AH and Currie AR:
Apoptosis: A basic biological phenomenon with wide-ranging
implications in tissue kinetics. Br J Cancer. 26:239–257. 1972.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim J, Choi TG, Ding Y, Kim Y, Ha KS, Lee
KH, Kang I, Ha J, Kaufman RJ, Lee J, et al: Overexpressed
cyclophilin B suppresses apoptosis associated with ROS and
Ca2+ homeostasis after ER stress. J Cell Sci.
121:3636–3648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Santos CX, Tanaka LY, Wosniak J and
Laurindo FR: Mechanisms and implications of reactive oxygen species
generation during the unfolded protein response: Roles of
endoplasmic reticulum oxidoreductases, mitochondrial electron
transport, and NADPH oxidase. Antioxid Redox Signal. 11:2409–2427.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yan Y, Wei CL, Zhang WR, Cheng HP and Liu
J: Cross-talk between calcium and reactive oxygen species
signaling. Acta Pharmacol Sin. 27:821–826. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Berridge MJ, Lipp P and Bootman MD: The
versatility and universality of calcium signalling. Nat Rev Mol
Cell Biol. 1:11–21. 2000. View
Article : Google Scholar
|
|
19
|
Nakagawa T and Yuan J: Cross-talk between
two cysteine protease families. Activation of caspase-12 by calpain
in apoptosis. J Cell Biol. 150:887–894. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Martinez JA, Zhang Z, Svetlov SI, Hayes
RL, Wang KK and Larner SF: Calpain and caspase processing of
caspase-12 contribute to the ER stress-induced cell death pathway
in differentiated PC12 cells. Apoptosis. 15:1480–1493. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tan Y, Dourdin N, Wu C, De Veyra T, Elce
JS and Greer PA: Ubiquitous calpains promote caspase-12 and JNK
activation during endoplasmic reticulum stress-induced apoptosis. J
Biol Chem. 281:16016–16024. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Choi AY, Choi JH, Hwang KY, Jeong YJ, Choe
W, Yoon KS, Ha J, Kim SS, Youn JH, Yeo EJ and Kang I: Licochalcone
A induces apoptosis through endoplasmic reticulum stress via a
phospholipase Cγ1-, Ca(2+)-, and reactive oxygen species-dependent
pathway in HepG2 human hepatocellular carcinoma cells. Apoptosis.
19:682–697. 2014. View Article : Google Scholar
|
|
23
|
Yuan X, Li D, Zhao H, Jiang J, Wang P, Ma
X, Sun X and Zheng Q: Licochalcone A-induced human bladder cancer
T24 cells apoptosis triggered by mitochondria dysfunction and
endoplasmic reticulum stress. Biomed Res Int. 2013:4742722013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Skehan P, Storeng R, Scudiero D, Monks A,
McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S and Boyd MR:
New colorimetric cytotoxicity assay for anticancer-drug screening.
J Natl Cancer Inst. 82:1107–1112. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hockenbery D, Nuñez G, Milliman C,
Schreiber RD and Korsmeyer SJ: Bcl-2 is an inner mitochondrial
membrane protein that blocks programmed cell death. Nature.
348:334–346. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jung JI, Lim SS, Choi HJ, Cho HJ, Shin HK,
Kim EJ, Chung WY, Park KK and Park JH: Isoliquiritigenin induces
apoptosis by depolarizing mitochondrial membranes in prostate
cancer cells. J Nutr Biochem. 17:689–696. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vanden Hoek TL, Li C, Shao Z, Schumacker
PT and Becker LB: Significant levels of oxidants are generated by
isolated cardiomyocytes during ischemia prior to reperfusion. J Mol
Cell Cardiol. 29:2571–2583. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reers M, Smith TW and Chen LB: J-aggregate
formation of a carbocyanine as a quantitative fluorescent indicator
of membrane potential. Biochemistry. 30:4480–4486. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carthagena L, Bergamaschi A, Luna JM,
David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C,
Pancino G and Nisole S: Human TRIM gene expression in response to
interferons. PLoS One. 4:e48942009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rafi MM, Vastano BC, Zhu N, Ho CT, Ghai G,
Rosen RT, Gallo MA and DiPaola RS: Novel polyphenol molecule
isolated from licorice root (Glycyrrhiza glabra) induces apoptosis,
G2/M cell cycle arrest, and Bcl-2 phosphorylation in tumor cell
lines. J Agric Food Chem. 50:677–684. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang J, Yuan X, Zhao H, Yan X, Sun X and
Zheng Q: Licochalcone A inhibiting proliferation of bladder cancer
T24 cells by inducing reactive oxygen species production. Biomed
Mater Eng. 24:1019–1025. 2014.
|
|
32
|
Kaiser N and Edelman IS: Calcium
dependence of glucocorticoid-induced lymphocytolysis. Proc Natl
Acad Sci USA. 74:638–642. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McConkey DJ and Orrenius S: The role of
calcium in the regulation of apoptosis. Biochem Biophys Res Commun.
239:357–366. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sergeev IN: Calcium as a mediator of
1,25-dihydroxyvitamin D3-induced apoptosis. J Steroid Biochem Mol
Biol. 89–90:419–425. 2004. View Article : Google Scholar
|
|
35
|
Mekahli D, Bultynck G, Parys JB, De Smedt
H and Missiaen L: Endoplasmic-reticulum calcium depletion and
disease. Cold Spring Harb Perspect Biol. 3:a0043172011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Matsuzaki S, Hiratsuka T, Kuwahara R,
Katayama T and Tohyama M: Caspase-4 is partially cleaved by calpain
via the impairment of Ca2+ homeostasis under the ER stress.
Neurochem Int. 56:352–356. 2010. View Article : Google Scholar
|