Introduction
Panax ginseng C.A
Meyer is a perennial herb from the Araliaceae
family, and its roots have been widely used in China for thousands
of years as a tonic or adaptogen as it stated to promote vitality,
enhance physical performance, and increase resistance to stress and
aging (1–3). The annual growth cycle of ginseng is
a process that begins with bud breakage and leaf expansion in the
spring, followed by flowering, fruit set, ripening, leaf loss and
root growth following fruiting in the autumn (4). From a root growing perspective, each
step in the process is vital in the development of ginseng,
particularly in the leaf-expansion period, as growth during this
period determines future quality and yield (5,6).
Ginseng grows well in semi-shade conditions due to
its sensitivity to high temperatures. Usually, it takes 5–6 years
for ginseng to grow a mature root under optimal conditions.
However, ginseng cultivation is difficult due to its vulnerability
to environmental stresses (7,8).
During long-term cultivation, plants may be attacked by several
abiotic and fungal pathogens, which can cause severe damage to the
plants (9,10). To survive under different stresses,
plants have developed mechanisms to perceive external signals and
manifest adaptive responses, which are accompanied by appropriate
physiological changes (11). These
clues can assist in identifying specific unknown genes in
ginseng.
Transcriptome analysis is useful and convenient for
the identification of novel genes, and provides information on gene
expression, gene regulation and the amino acid content of proteins
(12–14). Although RNA sequencing-based
transcriptome analysis of ginseng has been performed, its
transcriptome has not been comprehensively described, and the
stress-associated genes active during growth remain to be fully
elucidated (15,16).
In the present study, using Illumina technology,
>3,000,000,000 bases of high-quality cDNA sequence were
generated from ginseng in the leaf-expansion period without any
prior genome information. Environmental stress-associated proteins,
including pathogenesis-related proteins, antioxidants, metal stress
proteins and other stress-induced proteins, were also found for the
first time. The assembled, annotated transcriptome sequences and
gene expression profiles not only provide valuable information
closely associated with its medicinal effects, but are also
important for further investigations on gene identification,
variety selection, quality control, genetic resource development
and secondary metabolites.
Materials and methods
Plant materials
Panax ginseng roots in the leaf-expansion
period were used as experimental materials in the present study.
The samples were collected from Fusong County (Jilin, China). The
fresh tissues were cut into small sections of >1 cm following
cleaning, and these were immediately frozen in liquid nitrogen and
stored in −80°C freezers until use.
RNA and library preparation for
transcriptome analysis
The samples were homogenized to a fine powder in
liquid nitrogen and total RNA was isolated using the improved
method with TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA), according to the manufacturer's protocol.
The RNA quality was analyzed using an ethidium bromide-stained 1%
agarose gel, and RNA integrity was confirmed using an Agilent 2100
Bioanalyzer (Agilent Technologies, Inc., Santa Clara, CA, USA)
(17,18). The total RNA for transcriptome
analysis was purified using an Illumina kit (Takara Biotechnology,
Co., Ltd., Dalian, China), according to the manufacturer's
protocol. The mRNA was purified using oligo (dT) magnetic beads,
and then randomly segmented into small fragments using divalent
cations in a fragmentation buffer (Takara Biotechnology, Co., Ltd.)
(19). The cleaved RNA fragments
were used as templates to synthesize first-strand cDNA using random
hexamer primers. Second-strand cDNA was synthesized using
ribonuclease (RNase)H (Takara Biotechnology, Co., Ltd.) and DNA
polymerase I (Takara Biotechnology, Co., Ltd.) (20). The short fragments were purified
using a polymerase chain reaction (PCR) extraction kit (Takara
Biotechnology, Co., Ltd.) and connected with sequencing adapters
(Illumina, Inc., San Diego, CA, USA). Following agarose gel
electrophoresis, suitable fragments (250±25 bp) were selected for
PCR amplification as templates.
Sequencing, de novo assembly and
functional annotation
Library sequencing was performed on the Illumina
sequencing platform (HiSeq 2000) (21). The average fragment size of the
library was ~200 bp, and both ends of the fragments were sequenced
(22). The raw reads were cleaned
by removing the adaptor sequences, empty reads and low-quality
sequences (reads with unknown sequences; 'N'). These raw reads were
randomly clipped into 29-mers for sequence assembly using Trinity
software version 2.0 (github. com/trinityrnaseq/trinityrnaseq/wiki)
(23). Small K-mers resulted in
graphic outputs, which were too complex to be meaningful, whereas
large K-mers resulted in poor overlap in regions with low
sequencing depth (24). The
contigs were subjected to further processing by unigene clustering
to form longer sequences without N. To assemble all the unigenes
from the two samples and form a single set of non-redundant
unigenes, the unigenes were clustered using TIGR Gene Indices
Clustering (TGICL) tools (25).
Quantification of the transcript levels were in reads per kilobase
of exon mode per million mapped reads (RPKM) (26).
Unigenes are firstly aligned by blastx to protein
databases of Nr (ftp://ftp.ncbi.nih.gov/blast/db/FASTA) and Swiss-Prot
database (www.uniprot.org/). Blast2Go software
(www.blast2go.com) was used to obtain functional
annotations using Gene Ontology terms (GO; geneontology.org) (27,28).
The Clusters of Orthologous Groups (COG) database (www.ncbi.nlm.nih.gov/COG) was also used to
predict and classify the functions of the unigenes, and the Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway database
(www.genome.jp/kegg/kegg1.html) was
used to perform GO functional classification and a survey of the
biological pathways of the unigenes (29,30).
Reverse transcription-quantitative
(RT-q)PCR validation
The selected genes, which were identified in the
transcriptome sequencing analysis, were validated and quantified
using RT-qPCR. Primers were designed according to the Illumina
sequencing data with PrimerPremier 5.0 (www.premierbio-soft.com/primerdesign/index.html). The
quantitative reaction was performed using an Mx3000p Real-Time PCR
detection system with the One Step SYBR PrimeScript PLUS RT-PCR kit
(Takara Biotechnology Co., Ltd.). The PCR amplification was
performed in a 25 µl mixture containing 2 µl cDNA,
0.5 µl each primer, 12.5 µl SYBR Premix Ex Taq, 0.5
µl ROX reference dye II and 9 µl distilled water. The
reaction was performed under the following thermocycling
conditions: Initial denaturation, 95°C for 30 sec; 40 cycles of
denaturation at 95°C for 5 sec, annealing at 54°C for 15 sec and
extension at 72°C for 30 sec. The relative expression levels were
calculated by comparing the quantification cycle (Cq) value of the
target gene with that of the housekeeping gene, TH-t (data not
shown). Relative gene expression levels were calculated using the
2−ΔΔCq method (31).
All the primer sequences used for RT-qPCR are listed in Table I.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis.
| No. | Gene | Sequence |
|---|
| 0 | Thioredoxin
H-type | F:
CCGAAGAAGGACAGGTGATTAG
R: GGGTAATGAAACGGCAAGG |
| 1 |
Ethylene-forming-enzyme-like
dioxygenase | F:
CAGGACAAACAAGTGGAAGG
R: AACCCTGTGTAACGGGCTCT |
| 2 | Nodulin MtN3 family
protein | F:
TTCTGCCTGGTATGGTTTGC
R: GTGAGAATGAAGGAGAGGAGTC |
| 3 | Actin 7 | F:
GTGAAGGCTGGCTTTGCTG
R: GGATGCTCTTCAGGGGCAACAC |
| 4 | Cytokinin-regulated
kinase 1 | F:
TTGGGAGTGGAAGTTTTGGT
R: AATGCGTTCTTGTTGTCGTT |
| 5 | Jasmonate
ZIM-domain protein 3 | F:
AGACATCAAGTTCAGGCACC
R: TGACGAAACAGATGTAGGGC |
| 6 | Phytochrome | F:
TTCTGCCTGGTATGGTTTGC
R: GTGAGAATGAAGGAGAGGAGTC |
Results
Illumina sequencing, assembly and
sequence analysis
To develop a comprehensive overview of the ginseng
transcriptome in the leaf-expansion period, total RNA was extracted
from ginseng roots and reverse transcribed into cDNA. Following
cleaning and quality checks, 40,385,232 high-quality reads were
obtained, with an average length of 90 bp. The data were deposited
in the National Center for Biotechnology Information (NCBI)
ArrayExpress repository under the accession number E-MTAB-974. The
raw reads, which were clipped into 29-mers, were assembled into
161,176 contigs with a mean length of 277 bp. Using paired-end
joining and gap-filling, the contigs were further assembled into
unique sequences using Trinity software. Following clustering using
TGICL software, 100,533 unigenes were produced, with a mean length
of 452 bp (Table II). The length
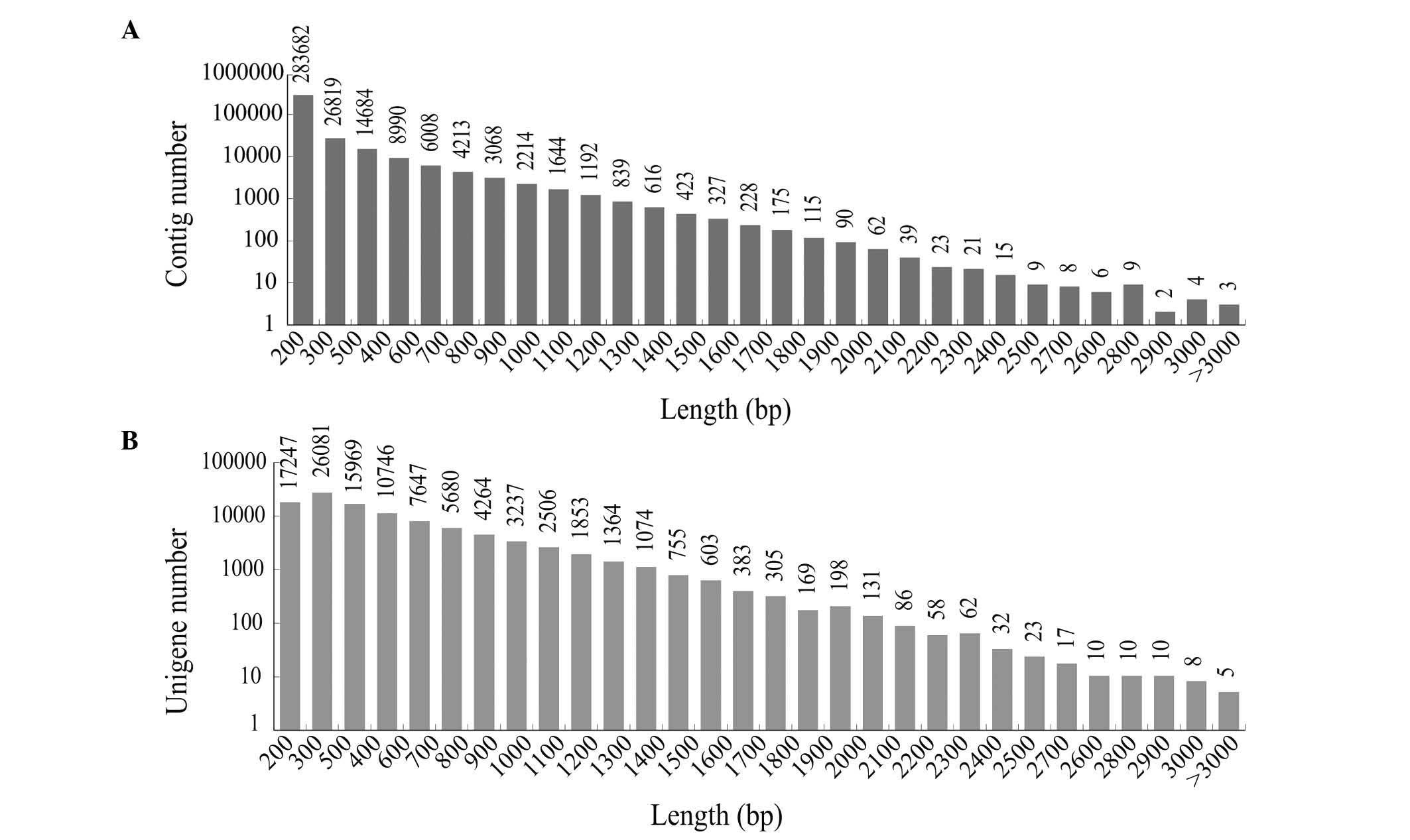
distribution of the contigs is shown in Fig. 1A, and of that of the unigenes is
shown in Fig. 1B. The results
showed that >41,236 unigenes (41.01%) were >500 bp in
length.
| Table IIOverview of the sequencing and
assembly. |
Table II
Overview of the sequencing and
assembly.
| Feature | Statistic |
|---|
| Total number of
reads (n) | 40,385,232 |
| Total base pairs
(bp) | 3,634,670,880 |
| Average read length
(bp) | 90 |
| Total number of
contigs (n) | 161,176 |
| Mean length of
contigs (bp) | 277 |
| Total number of
unigenes (n) | 100,533 |
| Mean length of
unigenes (bp) | 452 |
Functional annotation of the
transcriptome
A sequence similarity search was performed against
the NCBI non-redundant (nr) databases and Swiss-Prot protein
database. There were 61,599 gene matches (61.27% of all distinct
sequences) in the nr database, and 39,506 protein matches (39.30%
of all distinct sequences) in the Swiss-Prot database. GO
assignments based on sequence homology were used to classify the
functions of the distinct sequences; the 30,181 sequences in the
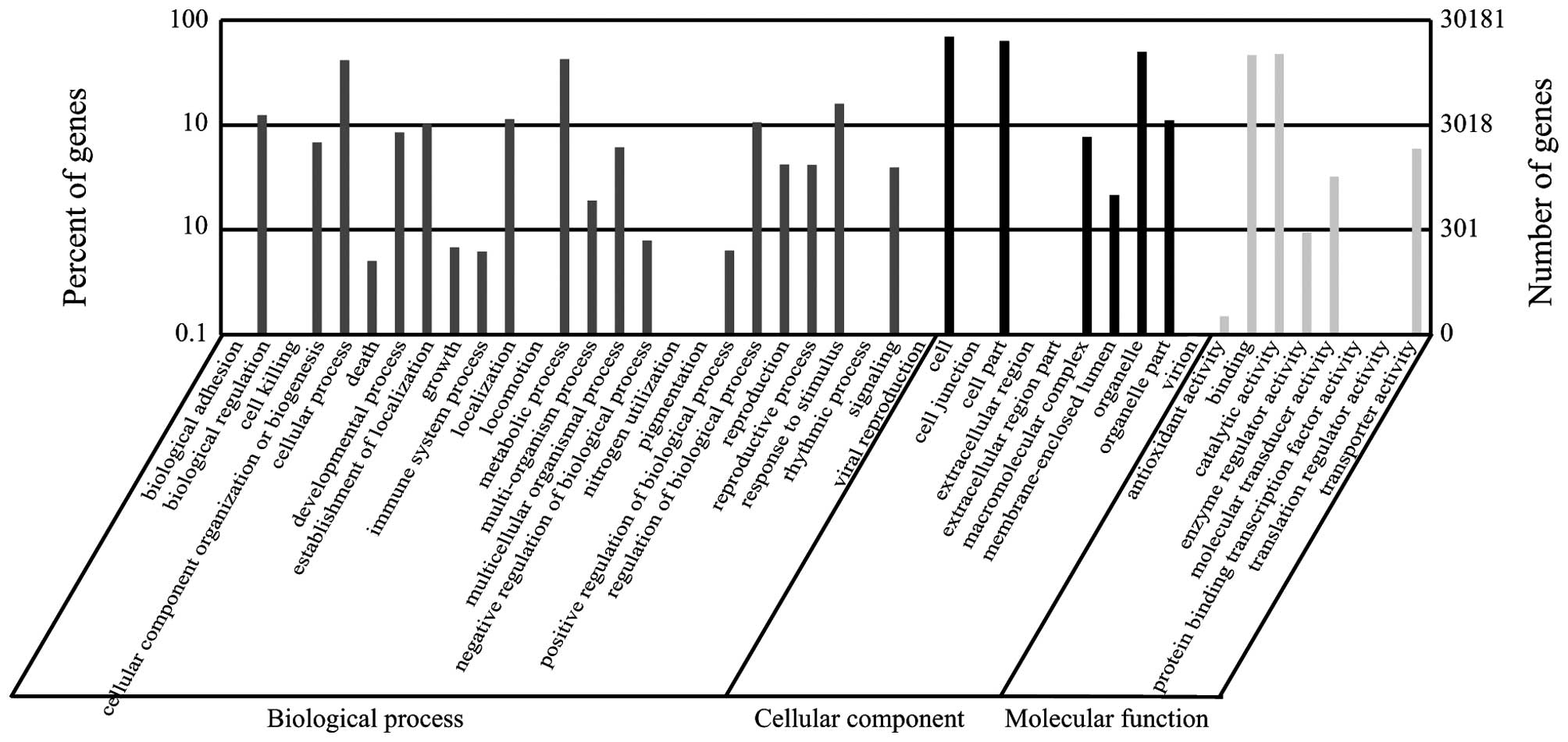
library were categorized into 44 functional groups (Fig. 2). In the three major categories
(biological process, cellular component and molecular function),
55,395, 61,279 and 31,495 GO terms were assigned, respectively. It
was noted the majority of the unigenes were annotated with
'metabolic process' (12,814 members; 23.13%), 'cellular process'
(12,520 members; 22.60%), and 'response to stimulus' (4,796
members; 8.66%) under the 'biological process' category. By
contrast, few genes were found in the clusters 'rhythmic process'
(seven members), 'cell killing' (four members), 'extracellular
region part' (four members), 'nitrogen utilization' (two members),
and 'translation regulator activity' (two members). In the
'cellular component' category, unigenes annotated with 'cell'
(20,899 members; 34.10%), 'cell part' (19,034 members; 31.06%), and
'organelle' (14,991 members; 24.46%) were predominant. The
'catalytic activity' (14,344 members; 45.54%) and 'binding' (14,014
members; 44.50%) classes were the most abundant in the 'molecular
function' category.
To evaluate the completeness of the transcriptome
library produced in the present study and the effectiveness of the
annotation process, all unigenes were subjected to a search against
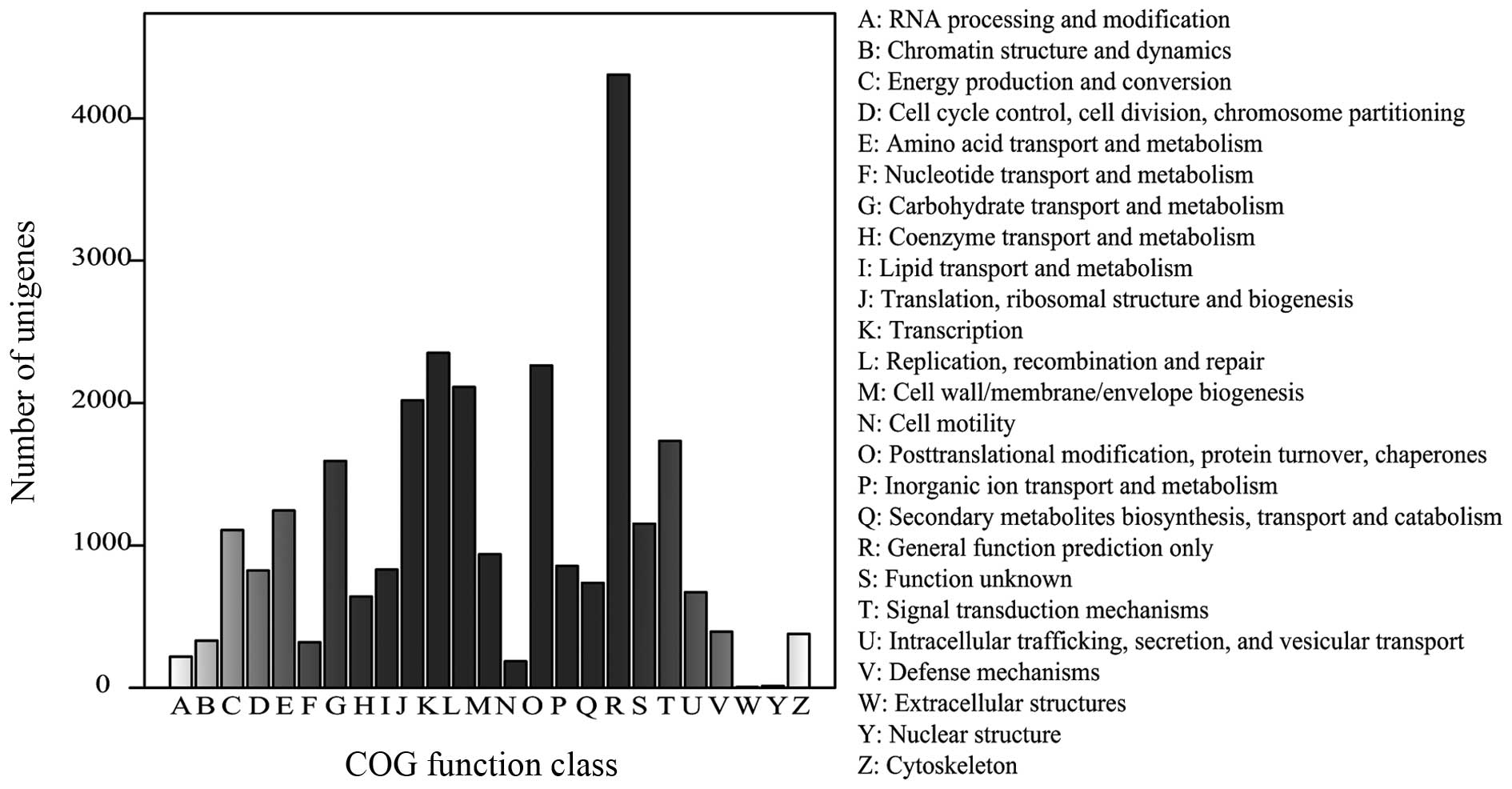
the COG database. From 61,599 nr hits, 27,232 genes were clustered
into 25 function categories (Fig.
3). The 'general function prediction only' cluster was found to
be the major COG category (4,306 members; 15.81%), followed by
'transcription' (2,352 members; 8.64%), 'posttranslational
modification, protein turnover, chaperones' (2,264 members; 8.31%),
and 'replication, recombination and repair' (2,113 members; 7.76%).
The 'nuclear structure' (13 members) and 'extracellular structures'
(five members) categories were the least-represented groups.
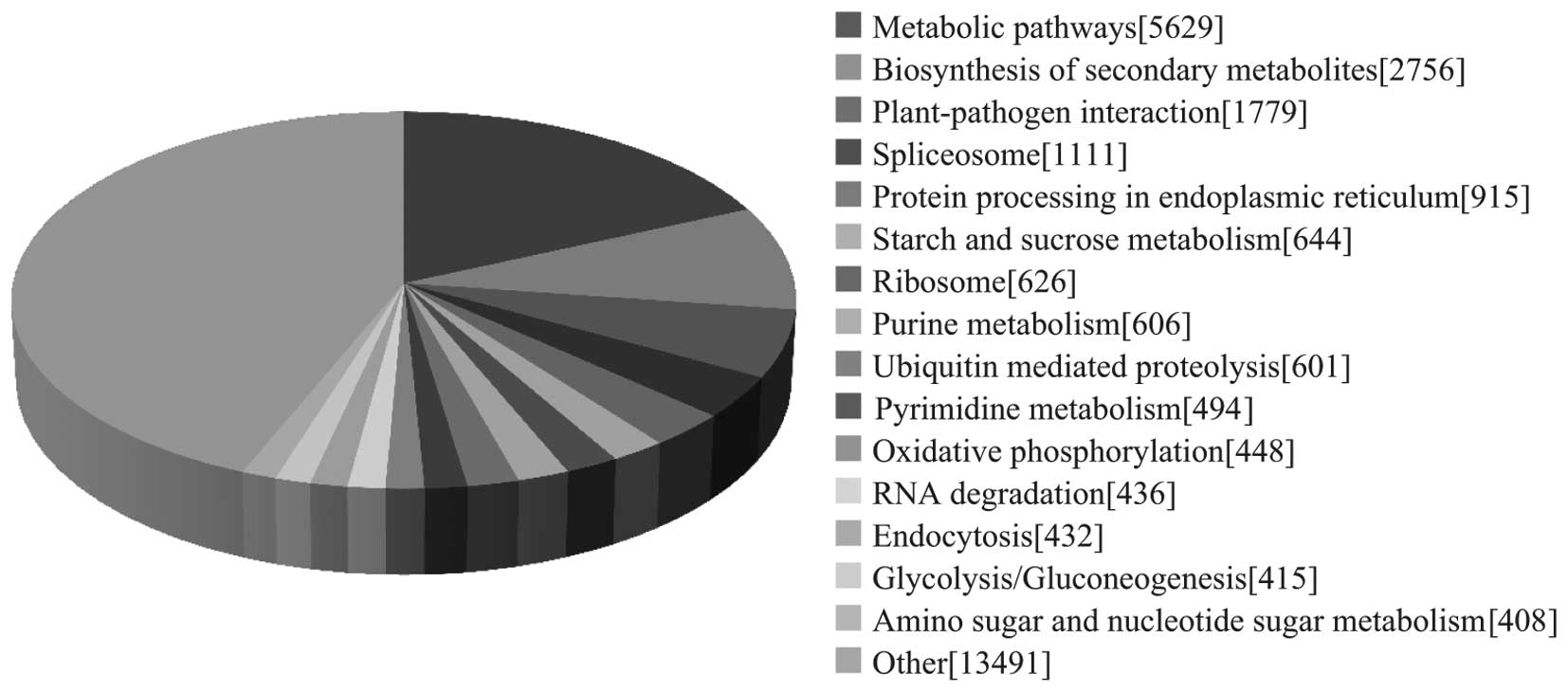
To identify the biological pathways active during
the ginseng growth stage investigated, the annotated sequences were
also mapped to the KEGG database. As a result, 24,486 sequences
were assigned to 121 KEGG pathways. The pathways with the most
representation by unique sequences were metabolic pathways
(18.28%), followed by those associated with biosynthesis of
secondary metabolites (8.95%) and plant-pathogen interaction
(5.78%; Fig. 4). These annotations
provide a valuable resource for investigating specific processes,
functions and pathways in ginseng.
Candidate genes involved in environmental
stress
The top 10 most abundant transcripts in the ginseng
transcriptome library are shown in Table III, with ribonuclease-like
storage protein being the most abundant. A number of environmental
stress genes were found among the transcripts in the library,
including pathogenesis-related, antioxidant-related and metal
stress proteins. There were several pathogenesis-related proteins
(PRs) in the data set, including PR1, PR2, PR4, PR5 and PR10, with
PR10 having the highest transcript level. A number of antioxidant
genes expressed at high levels, including catalase and superoxide
dismutase, were found in the present study (Tables IV and V). The present study also analyzed metal
stress proteins, heat shock proteins (HSPs), salt stress proteins
and water stress proteins, which may be involved in plant defense
responses (Table VI). The
accumulation of stress tolerance and pathogen response proteins in
ginseng roots showed that it was sensitive to external
environmental conditions in the leaf-expansion period.
| Table IIITranscripts expressed at high levels
in the ginseng library. |
Table III
Transcripts expressed at high levels
in the ginseng library.
| No. | ID | Non-redundant
annotation | RPKM |
|---|
| 1 |
Unigene48956_F1 | Ribonuclease-like
storage protein | 105,492.10 |
| 2 |
Unigene49972_F1 | Pathogensis-related
protein 10 (Panax ginseng) | 22,469.66 |
| 3 |
Unigene48226_F1 |
Metallothionein-like MT-3 (Jatropha
curcas) | 16,573.52 |
| 4 |
Unigene49842_F1 | Specific tissue
protein 2 (Cicer arietinum) | 10,068.34 |
| 5 |
Unigene49004_F1 | Basic blue copper
protein (Cicer arietinum) | 8,531.23 |
| 6 |
Unigene47883_F1 | Kirola
allergen | 8,292.52 |
| 7 |
Unigene16230_F1 | Hypothetical
protein (Vitis vinifera) | 6,682.50 |
| 8 |
Unigene48659_F1 | Catalase
(Solanum tuberosum) | 6,231.77 |
| 9 |
Unigene49760_F1 | Phloem protein 2-2
(Apium graveolens dulce group) | 4,874.98 |
| 10 |
Unigene48454_F1 | β-amylase
(Castanea crenata) | 4,849.63 |
| Table IVExpressed transcripts of
pathogenesis-related proteins in the ginseng library. |
Table IV
Expressed transcripts of
pathogenesis-related proteins in the ginseng library.
| No. | ID | Non-redundant
annotation | RPKM |
|---|
| 1 |
Unigene49972_F1 | Pathogensis-related
protein 10 (Panax ginseng) | 22,469.66 |
| 2 |
Unigene22713_F1 |
Pathogenesis-related protein 1
(Vitis hybrid cultivar) | 1,264.81 |
| 3 |
Unigene48216_F1 |
Pathogenesis-related protein (Lepidium
latifolium) | 654.54 |
| 4 |
Unigene49556_F1 |
Pathogenesis-related protein PR-4 type
(Sambucus nigra) | 481.79 |
| 5 |
Unigene47993_F1 |
Pathogenesis-related protein 2
(Petroselinum) | 317.11 |
| 6 |
Unigene41124_F1 |
Pathogenesis-related protein PR10a
(Nicotiana tabacum) | 15.70 |
| 7 | Unigene17748_F |
Pathogenesis-related protein 5 (Panax
ginseng) | 2.49 |
| Table VAntioxidant-associated transcripts
expressed in the ginseng library. |
Table V
Antioxidant-associated transcripts
expressed in the ginseng library.
| No. | ID | Non-redundant
annotation | RPKM |
|---|
| 1 |
Unigene49614_F1 | Ascorbate
peroxidase (Nicotiana tabacum) | 8,383.00 |
| 2 |
Unigene48959_F1 | Superoxide
dismutase (Cu-Zn) | 1,812.32 |
| 3 |
Unigene48521_F1 | Superoxide
dismutase (Mn) | 155.30 |
| 4 | Unigene8659_F1 | Chloroplast iron
superoxide dismutase (Dimocarpus longan) | 3.73 |
| 5 |
Unigene48659_F1 | Catalase
(Solanum tuberosum) | 6,231.77 |
| 6 |
Unigene49928_F1 | Glutathione
peroxidase 1 (Arachis hypogaea) | 1,049.05 |
| Table VIGenes involved in plant defense
responses in the ginseng library. |
Table VI
Genes involved in plant defense
responses in the ginseng library.
| Category | ID | Non-redundant
annotation | RPKM |
|---|
| Metal stress
protein |
Unigene48226_F1 |
Metallothionein-like MT-3 (Jatropha
curcas) | 16,573.52 |
|
Unigene48880_F1 | Metallothionein
type 2 (Sesbania drummondii) | 4,290.07 |
| Heat shock
protein |
Unigene49007_F1 | Heat shock protein
90 (Nicotiana tabacum) | 959.88 |
|
Unigene11322_F1 | Heat shock protein
70 (Arabidopsis thaliana) | 682.67 |
| Unigene492_F1 | Class II small heat
shock protein Le-HSP17.6 (Solanum lycopersicum) | 161.86 |
|
Unigene34233_F1 | Small molecular
heat shock protein 10 (Nelumbo nucifera) | 133.21 |
|
Unigene100483_F1 | Heat shock protein
60 (Ageratina adenophora) | 12.73 |
| Salt stress
protein |
Unigene21092_F1 | Salt tolerance
protein 1 (Beta vulgaris) | 48.08 |
| Unigene5170_F1 | Salt responsive
protein 2 (Solanum lycopersicum) | 141.48 |
| Unigene662_F1 | Salt tolerance
protein 3 (Beta vulgaris) | 14.96 |
| Unigene6719_F1 | Salt tolerance
protein 4 (Beta vulgaris) | 109.43 |
|
Unigene14324_F1 | Salt tolerance
protein 5-like protein (Solanum tuberosum) | 60.38 |
| Water stress
protein |
Unigene15597_F1 | Dehydrin 2
(Panax ginseng) | 82.85 |
|
Unigene21543_F1 | Dehydrin 3
(Panax ginseng) | 1,532.60 |
|
Unigene12318_F1 | Dehydrin 4
(Panax ginseng) | 13.96 |
|
Unigene36768_F1 | Dehydrin 5
(Panax ginseng) | 11.93 |
| Unigene4890_F1 | Dehydrin 6
(Panax ginseng) | 17.87 |
| Unigene1_F1 | Dehydrin 8
(Panax ginseng) | 70.96 |
|
Unigene54318_F1 | Dehydrin 9
(Panax ginseng) | 1.87 |
| Unigene6458_F1 | Lipid transfer
protein-like protein (Noccaea caerulescens) | 16.79 |
| Unigene5623_F1 | Late embryogenesis
abundant protein (LEA) family protein (Arabidopsis
thaliana) | 74.65 |
|
Unigene15942_F1 | 14 kDa proline-rich
protein DC2.15; Flags: Precursor | 360.01 |
RT-qPCR validation
The accuracy of the data obtained in the present
study for several genes was assessed by examining their expression
levels using RT-qPCR analysis. The RT-qPCR results were in
accordance with the RPKM values of the respective genes. The RPKM
values showed a progressive decrease, and the 2−ΔΔCq
values demonstrated a similar trend, as shown in Fig. 5.
Discussion
Illumina sequencing technology offers novel
potential in high-throughput sequencing for the majority of
species, as it provides an accurate, rapid and cost-effective
method for transcriptome analysis (32). In the present study, ginseng in the
leaf-expansion period was analyzed using this technology. A draft
sequence was successfully generated and assembled, and high
sensitivity in the coverage of weakly expressed genes was found.
Additionally, the present study provided a basis for the functional
analysis of genes involved in ginseng development without prior
genome annotation, and demonstrated the feasibility of using the
sequencing-based Illumina system for gene expression profiling.
Prior to the present study, ginseng was represented
by only 565 sequences in the NCBI protein database and 12,071
sequences in the NCBI Expressed Sequence Tags database. In the
present study, >40,000,000 reads with a mean length of 90 bp
were produced. The high-quality reads were assembled into 100,533
unique sequences with a mean length of 452 bp, and 61,599 (61.27%)
sequences showed a BLAST match above the cut-off level. These
findings represent a substantial contribution to the existing
sequence resources for ginseng and facilitate the acceleration of
investigations into pharmacologically active substances.
To evaluate the completeness of the transcriptome
library produced in the present study, and the effectiveness of the
annotation process, the sequences were annotated using GO
classifications, COG classifications and KEGG pathways. In total,
30,181 sequences were assigned to 44 GO classifications, 16,371
sequences to 25 COG classifications and 24,486 sequences to 121
KEGG pathways. These annotations provide a valuable resource for
investigating specific processes, functions and pathways in
ginseng, and facilitate comparisons of transcript levels within and
between samples. The expression levels of the genes in the data
were quantified by counting the number of RPKM. The RPKM measure of
read density reflects the molar concentration of a transcript in
the original sample by normalizing for RNA length and the total
number of reads in the measurement. This calculation normalizes the
read density measurement and, therefore, can be used for
comparisons within and between tissue samples (33).
Based on the de novo sequencing and analysis
of ginseng roots, the present study identified the top 10 most
frequent transcripts in the ginseng transcriptome library (Table III). The most abundant ginseng
transcript (RPKM=10,5492.1408) was annotated as a ribonuclease-like
storage protein. Of note, this type of protein has been identified
as the most abundant root protein of ginseng in comparative
proteome analysis (34). The
present study hypothesized that the ribonuclease-like storage
protein may accumulate in the leaf-expansion period to accompany
changes in the physiological characteristics of ginseng as the
aerial parts of ginseng grow rapidly during this stage, which
requires a high level of nutrients. Additionally, transcripts
encoding β-amylase were expressed at high levels as starch is the
most abundant component of ginseng roots.
Based on the frequencies of transcripts in the
ginseng transcriptome library, the present study found that several
of the candidate genes associated with environment stress were
expressed at high levels. To survive under different levels of
stress, plants have developed mechanisms to perceive external
signals and manifest adaptive responses, accompanied by appropriate
physiological changes (11). One
of the notable plant defense responses is the induction of PRs,
which have been classified into 17 families, based on their
structural differences, serological associations and biological
activities (35,36). In total, five groups of PRs were
found in the present study, including PR1, PR2, PR4, PR5 and PR10,
which have been characterized as β-1, 3-glucanases, chitinases, and
thaumatin-like and ribonuclease-like proteins (37,38).
The PR10 protein was first described in cultured parsley cells, and
this protein family consists of relatively diverse members, which
are sub-grouped into different functional classes (39). It has been suggested that PR10
proteins are involved in plant defense as the genes are usually
induced in environmental stress and attack by various pathogens. To
date, three previously isolated PR10 proteins from ginseng have
been characterized as ribonucleases (RNases), and several purified
PR10 proteins have exhibited in vitro RNase activity
(11,40). Other PRs were also expressed at
high levels in the present study, and high expression levels of PRs
during this growth period are associated with the growth
environment.
As shown in Table
V, several antioxidant enzymes expressed at high levels were
also identified. Catalase (CAT), the antioxidant enzyme with the
highest level of expression, was shown to be abundant in the
rhizome, and has also been identified in seedling shoots of
4-year-old ginseng and 11-year-old ginseng cultured in vitro
(41). Superoxide dismutase, found
in the mitochondrial intermembrane space, catalyzes dismutation of
the superoxide radical to H2O2 and oxygen
(42). H2O2
is converted to water and oxygen by catalase, ascorbate peroxidase
or glutathione peroxidase in peroxisomes; glutathione peroxidase
enzymes also have the ability to detoxify peroxides (43). Each of these enzymes has a
physiological function in the absence of stress, and they are all
increased under abiotic stress (42). CAT is significantly induced under
different stresses, including heavy metals, plant hormones, osmotic
agents, high light irradiance and abiotic stresses (44).
The present study found that certain metal stress
proteins were expressed at high levels, including
metallothionein-like MT-3 and metallothionein type 2, which have
metal-chaperoning and reactive oxygen species-scavenging functions
(45). Certain HSPs, including
HSP90, HSP70, HSP60 and small HSPs, were also abundant in the
library produced in the present study. In stressful conditions,
HSPs assist in the ability of individual cells to cope with stress
by maintaining important cellular processes (46). Although HSPs are active in cells
under normal circumstances, their functions are important in cells
under conditions of stress (47).
Five types of salt-responsive proteins were expressed at high
levels, including salt-responsive proteins 1–5, as well as water
stress-induced proteins, including dehydrin, lipid transfer, late
embryogenesis abundant and Dc2.15 proteins (48). Dehydrins, including Lea proteins,
typically accumulate in low temperatures or under conditions of
water-deficit, and are considered to be involved in freezing and
drought tolerance in plants (49,50).
Although ginseng is used worldwide, its sources are
limited. It grows in the shade, and has strict temperature and
water requirements; any changes in the environment affect its
growth. During extended cultivation, ginseng may be attacked by
several abiotic and biotic pathogens, resulting in severe damage
(51,52). In the present study, it was found
that genes associated with environmental stress are active during
ginseng growth. These genes respond to environmental changes,
affecting the growth of ginseng and the accumulation of products
during extended cultivation.
In conclusion, the present study performed de
novo transcriptome sequencing of ginseng in the leaf-expansion
period using the Illumina platform. In total, >40,000,000
sequencing reads were produced and assembled into 100,533 unique
sequences. By performing BLAST analysis of the unigenes against
public databases (Nr, Swiss-Prot, KEGG and COG), functional
annotations and classifications were obtained. The large number of
transcriptomic sequences produced and their functional annotations
provide valuable resources for molecular investigations of ginseng.
In addition, several candidate genes were identified, which are
potentially involved in growth and environmental stress responses;
identification of this set of candidate genes is valuable for
further investigations.
Acknowledgments
This study was supported by the grant from National
Natural Foundation of China (grant no. 81373937), the Scientific
and Technological Development Program of Jilin, China (grant no.
20140622003JC), and the Strategic Adjustment of the Economic
Structure of Jilin Province to Guide the Capital Projects (grant
no. 2014N155).
References
|
1
|
Briskin DP: Medicinal plants and
phytomedicines. Linking plant biochemistry and physiology to human
health. Plant Physiol. 124:507–514. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim YJ, Zhang D and Yang DC: Biosynthesis
and biotechnological production of ginsenosides. Biotechnol Adv.
33:717–735. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith I, Williamson EM, Putnam S,
Farrimond J and Whalley BJ: Effects and mechanisms of ginseng and
ginsenosides on cognition. Nutr Rev. 72:319–333. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu D, Austin RS, Zhou S and Brown D: The
root transcriptome for North American ginseng assembled and
profiled across seasonal development. BMC Genomics. 14:5642013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Munns R, Passioura JB, Guo J, Chazen O and
Cramer GR: Water relations and leaf expansion: Importance of time
scale. J Exp Bot. 51:1495–1504. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wei YC, Chen CB, Li YB, et al: Ginseng
development practical manual. Jilin Science and Technology Bureau;
Changchun: 2010
|
|
7
|
Peng Y, Lin W, Cai W and Arora R:
Overexpression of a Panax ginseng tonoplast aquaporin alters salt
tolerance, drought tolerance and cold acclimation ability in
transgenic Arabidopsis plants. Planta. 226:729–740. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pulla RK, Kim YJ, Parvin S, Shim JS, Lee
JH, Kim YJ, In JG, Senthil KS and Yang DC: Isolation of
S-adenosyl-L-methionine synthetase gene from Panax ginseng C.A.
meyer and analysis of its response to abiotic stresses. Physiol Mol
Biol Plants. 15:267–275. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee SK: Fusarium species associated with
ginseng (Panax ginseng) and their role in the root-rot of ginseng
plants. Res Plant Dis. 10:248–259. 2004. View Article : Google Scholar
|
|
10
|
Li Y, Liu SL, Huang XF and Ding WL:
Allelopathy of ginseng root exudates on pathogens of ginseng. Acta
Ecological Sinica. 29:161–167. 2009.
|
|
11
|
Lee OR, Pulla RK, Kim YJ, Balusamy SR and
Yang DC: Expression and stress tolerance of PR10 genes from Panax
ginseng C. A. Meyer. Mol Biol Rep. 39:2365–7234. 2012. View Article : Google Scholar
|
|
12
|
Gupta P, Goel R, Pathak S, Srivastava A,
Singh SP, Sangwan RS, Asif MH and Trivedi PK: De novo assembly,
functional annotation and comparative analysis of Withania
somnifera leaf and root transcriptomes to identify putative genes
involved in the withanolides biosynthesis. PLoS One. 8:e627142013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Margulies M, Egholm M, Altman WE, Attiya
S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, et
al: Genome sequencing in microfabricated high-density picolitre
reactors. Nature. 437:376–380. 2005.PubMed/NCBI
|
|
14
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View
Article : Google Scholar
|
|
15
|
Chen S, Luo H, Li Y, Sun Y, Wu Q, Niu Y,
Song J, Lv A, Zhu Y, Sun C, et al: 454 EST analysis detects genes
putatively involved in ginsenoside biosynthesis in Panax ginseng.
Plant Cell Rep. 30:1593–1601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Choi DW, Jung J, Ha YI, Park HW, In DS,
Chung HJ and Liu JR: Analysis of transcripts in methyl
jasmonate-treated ginseng hairy roots to identify genes involved in
the biosynthesis of ginsenosides and other secondary metabolites.
Plant Cell Rep. 23:557–566. 2005. View Article : Google Scholar
|
|
17
|
Fleige S and Pfaffl MW: RNA integrity and
the effect on the real-time qRT-PCR performance. Mol Aspects Med.
27:126–139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Metzker ML: Sequencing technologies-the
next generation. Nat Rev Genet. 11:31–46. 2010. View Article : Google Scholar
|
|
19
|
King M, Reeve W, Van der Hoek MB, Williams
N, McComb J, O'Brien PA and Hardy GE: Defining the
phosphite-regulated transcriptome of the plant pathogen
Phytophthora cinnamomi. Mol Genet Genomics. 284:425–435. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jiang Q, Wang F, Tan HW, Li MY, Xu ZS, Tan
GF and Xiong AS: De novo transcriptome assembly, gene annotation,
marker development and miRNA potential target genes validation
under abiotic stresses in Oenanthe javanica. Mol Genet Genomics.
290:671–683. 2015. View Article : Google Scholar
|
|
21
|
Yu M, Yu J, Gu C, Nie Y, Chen Z, Yin X and
Liu Y: De novo sequencing and transcriptome analysis of
Ustilaginoidea virens by using Illumina paired-end sequencing and
development of simple sequence repeat markers. Gene. 547:202–210.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tsanakas GF, Manioudaki ME, Economou AS
and Kalaitzis P: De novo transcriptome analysis of petal senescence
in Gardenia jasminoides Ellis. BMC Genomics. 15:5542014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grabherr MG, Haas BJ, Yassour M, Levin JZ,
Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et
al: Full-length transcriptome assembly from RNA-Seq data without a
reference genome. Nat Biotechnol. 15:644–652. 2011. View Article : Google Scholar
|
|
24
|
Wang Z, Zhang J, Jia C, Liu J, Li Y, Yin
X, Xu B and Jin Z: De novo characterization of the banana root
transcriptome and analysis of gene expression under Fusarium
oxysporum f. sp Cubense tropical race 4 infection. BMC Genomics.
13:6502012. View Article : Google Scholar
|
|
25
|
Lee Y, Tsai J, Sunkara S, Karamycheva S,
Pertea G, Sultana R, Antonescu V, Chan A, Cheung F and Quackenbush
J: The TIGR Gene Indices: Clustering and assembling EST and known
genes and integration with eukaryotic genomes. Nucleic Acids Res.
33(Database Issue): D71–D74. 2005. View Article : Google Scholar :
|
|
26
|
Mortazavi A, Williams BA, McCue K,
Schaeffer L and Wold B: Mapping and quantifying mammalian
transcriptomes by RNA-Seq. Nat Methods. 5:621–628. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Conesa A, Götz S, García-Gómez JM, Terol
J, Talón M and Robles M: Blast2GO: A universal tool for annotation,
visualization and analysis in functional genomics research.
Bioinformatics. 15:3674–3676. 2005. View Article : Google Scholar
|
|
28
|
Harris MA, Clark J, Ireland A, Lomax J,
Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C,
et al: The gene ontology (GO) database and informatics resource.
Nucleic Acids Res. 32(Database Issue): D258–D261. 2004. View Article : Google Scholar
|
|
29
|
Kanehisa M, Goto S, Kawashima S, Okuno Y
and Hattori M: The KEGG resource for deciphering the genome.
Nucleic Acids Res. 32(Database Issue): D277–D280. 2004. View Article : Google Scholar :
|
|
30
|
Tatusov RL, Natale DA, Garkavtsev IV,
Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY,
Fedorova ND and Koonin EV: The COG database: New developments in
phylogenetic classification of proteins from complete genomes.
Nucleic Acids Res. 29:22–28. 2001. View Article : Google Scholar :
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta DeltaC (T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
32
|
Hudson ME: Sequencing breakthroughs for
genomic ecology and evolutionary biology. Mol Ecol Resour. 8:3–17.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wagner GP, Kin K and Lynch VJ: Measurement
of mRNA abundance using RNA-seq data: RPKM measure is inconsistent
among samples. Theory Biosci. 131:281–285. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim SI, Kweon SM, Kim EA, Kim JY, Kim S,
Yoo JS and Park YM: Characterization of RNase-like major storage
protein from the ginseng root by proteomic approach. J Plant
Physiol. 161:837–845. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sels J, Mathys J, De Coninck BM, Cammue BP
and De Bolle MF: Plant pathogenesis-related (PR) proteins: A focus
on PR peptides. Plant Physiol Biochem. 46:941–950. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Van Loon LC, Rep M and Pieterse CM:
Significance of inducible defense-related proteins in infected
plants. Annu Rev Phytopathol. 44:135–162. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sticher L, Mauch-Mani B and Métraux JP:
Systemic acquired resistance. Annu Rev Phytopathol. 35:235–270.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
van Loon LC: Pathogenesis-related
proteins. Plant Mol Biol. 4:111–116. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Somssich IE, Schmelzer E, Kawalleck P and
Hahlbrock K: Gene structure and in situ transcript localization of
pathogenesis-related protein 1 in parsley. Mol Gen Genet.
213:93–98. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moiseyev GP, Fedoreyeva LI, Zhuravlev YN,
Yasnetskaya E, Jekel PA and Beintema JJ: Primary structures of two
ribo-nucleases from ginseng calluses. New members of the PR-10
family of intracellular pathogenesis-related plant proteins. FEBS
Lett. 28:207–210. 1997. View Article : Google Scholar
|
|
41
|
Purev M, Kim YJ, Kim MK, Pulla RK and Yang
DC: Isolation of a novel catalase (Cat1) gene from Panax ginseng
and analysis of the response of this gene to various stresses.
Plant Physiol Biochem. 48:451–460. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sathiyaraj G, Lee OR, Parvin S,
Khorolragchaa A, Kim YJ and Yang DC: Transcript profiling of
antioxidant genes during biotic and abiotic stresses in Panax
ginseng C. A. Meyer. Mol Biol Rep. 38:2761–2769. 2011. View Article : Google Scholar
|
|
43
|
Hernandez JA, Jimenez A, Mullineaux P and
Sevilia F: Tolerance of pea (Pisumsativum L) to long-term salt
stress is associated with induction of antioxidant defenses. Plant
Cell Environ. 23:853–862. 2000. View Article : Google Scholar
|
|
44
|
Apel K and Hirt H: Reactive oxygen
species: Metabolism, oxidative stress, and signal transduction.
Annu Rev Plant Biol. 55:373–399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Brkljacić JM, Samardzić JT, Timotijević GS
and Maksimović VR: Expression analysis of buckwheat (Fagopyrum
esculentum Moench) metallothionein-like gene (MT3) under different
stress and physiological conditions. J Plant Physiol. 161:741–746.
2004. View Article : Google Scholar
|
|
46
|
Mohamed H and Al-Whaibi: Plant heat-shock
proteins: A mini review. Journal of King Saud University Science.
23:139–150. 2011. View Article : Google Scholar
|
|
47
|
Jung JD, Park HW, Hahn Y, Hur CG, In DS,
Chung HJ, Liu JR and Choi DW: Discovery of genes for ginsenoside
biosynthesis by analysis of ginseng expressed sequence tags. Plant
Cell Rep. 22:224–230. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ingram J and Bartels D: The molecular
basis of dehydration tolerance in plants. Annu Rev Plant Physiol
Plant Mol Biol. 47:377–403. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pockley AG and Muthana M: Heat shock
proteins and allograft rejection. Contrib Nephrol. 148:122–134.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xu D, Duan X, Wang B, Hong B, Ho T and Wu
R: Expression of a late embryogenesis abundant protein gene, HVA1,
from barley confers tolerance to water deficit and salt stress in
transgenic rice. Plant physiol. 110:249–257. 1996.PubMed/NCBI
|
|
51
|
Shen L, Xu J, Dong LL, Li XW and Chen SL:
Cropping system and research strategies in Panax ginseng. Zhongguo
Zhong Yao Za Zhi. 40:3367–3373. 2015.In Chinese.
|
|
52
|
Liu Y, Zhao D, Liu M, Hu CY, Li Y and Ding
WL: Investigation of pests and diseases occurrence and pesticides
application in main producing areas of Panax ginseng. Chinese
Agricultural Science Bulletin. 30:294–298. 2014.
|