Introduction
There has been an increase in the number of studies
examining the possible detrimental effects of anesthesia in the
developing brain. Propofol (2,6-diisopropylphenol) is a
sedative-hypnotic agent widely used for the induction and
maintenance of anesthesia in surgery, and sedation in intensive
care units. It has been reported that propofol exposure can induce
cell death in neural precursor or stem cells (1), immature hippocampal neurons (2) and cortical cells (3) in vitro. Several in vivo
studies have also demonstrated that propofol can cause neuronal
cell apoptosis in the developing brain of rodents (4,5) and
non-human primates (NHPs) (6).
Therefore, it is important to develop promising strategies for
protection of the developing brain from the potentially deleterious
effects of propofol.
Dexmedetomidine is an α-2 adrenoceptor agonist with
high selectivity, and with sympatholytic, sedative and analgesic
properties. It is considered to offer potential benefits towards
neuroprotection (7). In a murine
model of perinatal excitotoxic brain damage, dexmedetomidine has
been found to provide potent neuroprotection (8). Previous in vitro and in
vivo observations have demonstrated that dexmedetomidine
protects against neuroapoptosis induced by isoflurane in the
hippocampus of neonatal rats (9,10).
Isoflurane is a type of volatile anesthetic and it is reported to
cause a similar pattern of neuronal apoptosis as propofol in the
neonatal brain (6).
Anesthesia-induced apoptotic damage in the developing brain is
regulated, at least in part, by the brain-derived neurotrophic
factor (BDNF)-modulated apoptotic cascade (11). In addition, it has been reported
that the neuroprotective effects of dexmedetomidine are mediated by
upregulating the levels of BDNF, phosphorylated-cyclic-AMP response
element binding protein (p-CREB) (12,13)
and the antiapoptotic factor, B-cell lymphoma 2 (Bcl-2) (14).
The present study used neuronal cultures from the
rat hippocampus to investigate whether dexmedetomidine pretreatment
is able to effectively attenuate propofol-induced neurotoxicity
in vitro. The study also aimed to examine alterations in the
expression levels of p-CREB, Bcl-2 and BDNF following exposure to
propofol and dexmedetomidine.
Materials and methods
Hippocampal neuronal culture and drug
treatment
The experimental procedure was approved by the
Animal Use and Care Committee of Guangxi Medical University
(Guangxi, China) and performed in strict accordance with the
guidelines of the National Institutes of Health Guide for the Use
of Laboratory Animals. Primary hippocampal cultures were prepared,
as described previously (15). In
brief, 40 female Sprague-Dawley rats (age, 4 months; weight,
400–450 g) in advanced pregnancy (Guangxi Medical University
Laboratory Animal Co.; permission no. SCXK 2009–0002) were housed
under standard conditions with a 12-h light/dark cycle at 23±2°C,
50±5% relative humidity and free access to food and water. The rats
were then anesthetized using 10% (w/v) chloral hydrate (3.5 ml/kg;
Sigma-Aldrich, Merck Millipore, Darmstadt, Germany) and embryonic
day 16–18 fetuses were removed. Next, the pregnant rats were
sacrificed by cervical dislocation. The fetuses were sacrificed by
rapid decapitation, followed by immediate removal of the brain and
its surrounding membranes. The hippocampus was then rapidly
dissected from the cortex. The meningeal tissues were removed and
the hippocampus was dissociated mechanically into 1 mm3
pieces. Equal volume of 0.25% trypsin solution (Sigma-Aldrich,
Merck Millipore) was added to the dissected tissue and incubated at
37°C for 15 min, mixing every 5 min. Following the removal of the
trypsin solution, 1 ml precooled fetal calf serum (FCS; Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) was added into
the tissue, mixed gently, and incubated in a 37°C water bath for 5
min to stop the digestion. The tissues were centrifuged for 5 min
at 106 × g and the supernatant was discarded. Cells were washed
three times and resuspended in plating medium [Neurobasal medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% FCS,
0.2 mM glutamine, 100 U/ml penicillin and 100 U/ml streptomycin]
and were transferred as a 0.5 ml aliquot to a tube that contained
0.5 ml of 4% Typan blue. The number of Trypan blue-excluding cells
were counted by inverted microscope (Olympus Corporation, Tokyo,
Japan). The number of Trypan blue-excluding cells were counted, and
the cells were plated onto six-well culture plates (Corning, Inc.,
Acton, MA, USA) previously coated with poly-L-lysine (0.1 mg/ml,
Gibco; Thermo Fisher Scientific, Inc.) at a density of
5×104 cells/ml. The cultures were maintained at 37°C
with 5% CO2, supplemented with Neurobal medium with 2%
B-27 (Gibco; Thermo Fisher Scientific, Inc.), 2 mmol/ml glutamine,
penicillin (100 U/ml) and streptomycin (100 µg/ml). Half the
cell culture media was replaced every 3 days. Immunocytochemical
staining was performed on neurons maintained for 8 days in
vitro (DIV 8) using mouse monoclonal antibody against NeuN
(cat. no. ABN78; Chemicon, Temecula, CA, USA). The cells adhered to
and grew on the coverslips. Following fixing with 4%
paraformaldehyde, coverslips bearing the neuronal cultures were
pre-incubated in 5% goat serum in phosphate-buffered saline (PBS)
supplemented with 0.2% Triton-X 100 for 1 h at room temperature,
followed by incubation with the primary antibody (1:500 dilution)
overnight at 4°C. Binding of the NeuN antibody was detected with a
goat anti-mouse IgG biotinylated secondary antibody (1:5,000; cat.
no. ab32096; Abcam, Cambridge, MA, USA). 3,3′-Diaminobenzidine
tetrahydrochloride was used as the substrate. Staining for NeuN was
then visualized using an AxioM1 light microscope (BX53; Olympus
Corporation).
The DIV 8 primary hippocampal cultures were used for
the drug exposure experiments in the present study. The cells were
seeded at a density of 5.0×104 cells/well and were
assigned to a control group, propofol group and dexmedetomidine +
propofol groups. The cells in the control group were incubated
without drugs in an intralipid vehicle (Baxter, Guangzhou, China)
at 37°C for 3 h, whereas the cells in the propofol group were
incubated with 100 µM propofol for 3 h at 37°C in the
absence of dexmedetomidine pretreatment. The cells in the
dexmedetomidine + propofol groups were incubated with 0.001, 0.01,
0.1, 1, 10 or 100 µM dexmedetomidine, respectively, at 37°C
for 30 min, following which 100 µM propofol was added to the
culture medium at 37°C for 3 h. The viability and apoptotic rate of
the neurons were then detected using a Cell Counting Kit-8 (CCK-8)
assay and flow cytometry. The expression levels of BDNF, Bcl-2 and
p-CREB were detected using semiquantitative reverse
transcription-polymerase chain reaction (RT-PCR) and western blot
analyses.
Cell viability assay
Cell viability was assessed using a CCK-8 assay
(Dojindo Molecular Technologies, Inc., Kumamoto, Japan). The cells
were incubated with 100 µM propofol for 3 h in the absence
or presence of dexmedetomidine pretreatment, following which CCK-8
solution was added and the culture was incubated for 2 h under 5%
CO2 at 37°C. The absorbance was read at 450 nm on a
microplate reader (Thermo Fisher Scientific, Inc.), with the value
directly proportional to the number of viable cells in the culture
medium.
Flow cytometric analysis
Flow cytometric analysis was performed using an
Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit
(KeyGen, Nanjing, China) according to the manufacturer's protocol.
Primary hippocampal neurons were briefly trypsinized and then
washed twice with cold 1X PBS. The cells were pelleted by
centrifugation at 425 × g for 5 min at 4°C and resuspended in 1X
binding buffer followed by incubation with staining solution of
annexin V-FITC and propidium iodide (PI) for 10 min in the dark at
4°C. The samples were maintained on ice during the entire procedure
and analyzed immediately using flow cytometry. The cells from each
sample (10,000 cells) were scanned and analyzed using a FACS
Calibur flow cytometer (Becton Dickinson; BD Biosciences, San
Diego, CA, USA). Necrosis and apoptosis were determined by PI (FL2)
and annexin V-FITC (FL1) fluorescence, respectively. The
percentages of apoptotic cells in each sample were estimated.
Semiquantitative RT-PCR
Total RNA from primary hippocampal neurons in each
of the different groups was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The reagents for semiquantitative RT-PCR
were those supplied with the RevertAid™ First Strand cDNA Synthesis
kit (Fermentas; Thermo Fisher Scientific, Inc.). First-strand cDNA
was generated from the total RNA (2 µg) by reverse
transcription. PCR was performed in a total volume of 25 µl
(2 µl cDNA, 0.5 µl of 10 µM each primer, 10
µl PCR master mix and 12 µl sterilized, deionized
water). The PCR reaction conditions were as follows: Denaturation
at 94°C for 45 sec, annealing at 59.1°C (BDNF), 55.6°C (Bcl-2) or
57.5°C (GAPDH) for 30 sec, and extension at 72°C for 30 sec. Primer
sequences were as follows: BDNF, forward
5′-AGCCTCCTCTGCTCTTTCTGCTGGA-3′ and reverse
5′-CTTTTGTCTATGCCCCTGCAGCCTT-3′; Bcl-2, forward
5′-GGTGGTGGAGGAACTCTTCA-3′ and reverse 5′-CTCACTTGTGGCCCAGGTAT-3′;
GAPDH, forward 5′-ACAGCAACAGGGTGGTGGAC-3′ and reverse
5′-TTTGAGGGTGCAGCGAACTT-3′. Following PCR amplification, the
products were electrophoresed and separated on a 1.5% agarose gel
stained with ethidium bromide. Densitometric analyses of bands for
specific genes were performed and normalized to the level of the
endogenous control mRNA (GAPDH) using Quantity One v5.0 software
(Bio-Rad, Berkeley, CA, USA).
Western blot analysis
Following incubation, the cell proteins were
extracted on ice for 60 min in Western and IP lysis buffer
(Beyotime Institute of Biotechnology, Shanghai, China), which was
added to PMSF (1 mM) prior to use and supplemented with protease
inhibitor cocktail. Following centrifugation at 4°C at 17,254 × g
for 10 min, the total lysates were separated on 10% SDS-PAGE gels
(30 mg/lane; Solarbio, Beijing, China), and electrophoretically
transferred onto polyvinylidene fluoride membranes (0.22 µm;
EMD Millipore, Billerica, MA, USA). The membranes were blocked in
Tris-buffered saline/0.1% Tween-20/5% non-fat milk for 1 h at room
temperature, and were then incubated with primary antibodies
against Bcl-2 (1:1,000; cat. no. ab32096; Abcam), BDNF (1:1,000;
cat. no. ab108319; Abcam), pCREB (1:1,000; cat. no. ab32096; Abcam)
and GAPDH (1:5,000; cat. no. sc-25778; Santa Cruz Biotechnology,
Inc.) overnight at 4°C. The membranes were then rinsed in PBS with
0.1% Tween 20 and incubated with horseradish peroxidase-conjugated
secondary antibody (1:5,000; cat. no. sc-25778; Santa Cruz
Biotechnology, Inc. Dallas, TX, USA) for 30 min at room
temperature. Images of the immunoblots were analyzed using Quantity
One V5.0 software (Bio-Rad). The level of each protein was
normalized with respect to GAPDH, the domestic loading control.
Statistical analysis
SPSS 19 (IBM SPSS, Armonk, NY, USA) and Origin 7.5
(OriginLab, Northampton, MA, USA) were used for statistical
analysis. The data are expressed as the mean ± standard deviation,
and one-way analysis of variance was performed to estimate
significant differences among groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Hippocampal neuron culture
In the present study, immunocytochemical staining
with NeuN was used to identify putative neurons, which revealed a
population of neuronal cells with >90% purity. These results
showed that the hippocampal neurons had been cultured
successfully.
Propofol reduces neuronal viability and
induces apoptosis in neuronal cultures from the rat
hippocampus
The DIV 8 primary hippocampal neurons in the
propofol group were exposed to propofol (100 µM for 3 h) in
the absence of dexmedetomidine pretreatment. Neuronal injury was
then determined using a CCK-8 assay and flow cytometric analysis.
The results of the CCK-8 assay revealed that the neuronal viability
in the propofol-treated group was reduced by 57.4%, compared with
that in the control group (Fig.
1). An increase in the percentage of apoptotic cells was also
observed in the propofol-treated cells (31.05±4.33%), compared with
the control cells (12.26±2.48%), as shown in Fig. 2A and B.
 | Figure 1Dex attenuates Pro-induced reductions
in neuronal viability in neuronal cultures from the rat
hippocampus. Cells were exposed to Pro (100 µM; 3 h) in the
absence or presence of Dex pretreatment (0.001, 0.01, 0.1, 1, 10
and 100 µM). The viability of the cells was detected using a
Cell Counting Kit-8 assay. Data are presented as the mean ±
standard deviation of values obtained from four culture wells per
experiment, determined in three independent experiments.
**P<0.01, compared with group C,
#P<0.05 and ##P<0.01, compared with
group Pro. Dex, dexmedetomidine; C, control; Pro, propofol. |
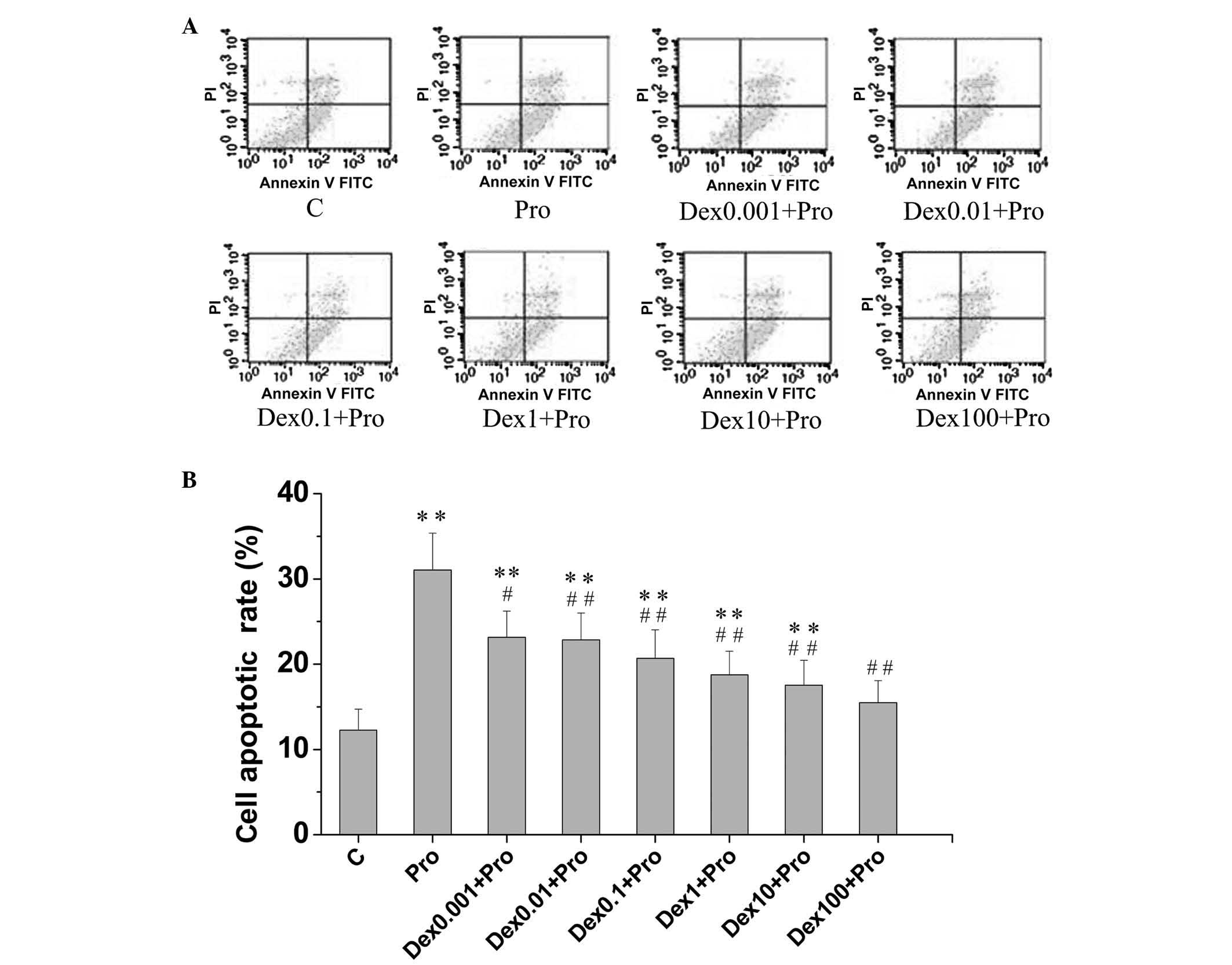 | Figure 2Dex attenuates Pro-induced apoptosis
in immature hippocampal neurons. The cells (8 days in vitro)
were exposed to Pro (100 µM, 3 h) in the absence or presence
of Dex pretreatment (0.001, 0.01, 0.1, 1, 10 and 100 µM).
Apoptotic cells were analyzed using flow cytometry. (A) Hippocampal
neurons were stained with annexin V-FITC and PI. (B) Percentages of
apoptotic neurons in the control cultures and each separate
experimental culture were determined. Data are presented as the
means ± standard deviation of values from triplicate independent
experiments.**P<0.01, compared with group C;
#P<0.05 and ##P<0.01, compared with
group Pro. Dex, dexmedetomidine; C, control; Pro, propofol; FITC,
fluorescein isothiocyanate; PI. propidium iodide. |
Dexmedetomidine attenuates
propofol-induced apoptosis and increases neuronal viability in
neuronal cultures from the rat hippocampus
The DIV 8 primary hippocampal neurons in the
propofol + dexmedetomidine groups were pretreated with different
concentrations (0.001, 0.01, 0.1, 1, 10 and 100 µM) of
dexmedetomidine, prior to propofol exposure (100 µM for 3
h), and subjected to a CCK-8 assay and flow cytometric analysis.
Dexmedetomidine significantly increased neuronal viability, by up
110%, compared with propofol exposure without dexmedetomidine
pretreatment (P<0.05; Fig. 1).
No significant differences in cell viability were found among the
10–100 µM dexmedetomidine-pretreated cells and the control
cells (P>0.05; Fig. 1). In the
cells pretreated with 0.001, 0.01, 0.1, 1, 10 and 100 µM
dexmedetomidine, the apoptotic rates of the cells were 23.16±3.06,
22.83±3.17, 20.67±3.35, 18.75±2.76, 17.53±2.92 and 15.47±2.59%,
respectively. Pretreatment with dexmedetomidine at all
concentration levels assessed in the present study resulted in
fewer apoptotic cells, compared with propofol exposure in the
absence of dexmedetomidine pretreatment (31.05±4.33%; P<0.05).
No significant difference was observed in apoptotic rates between
the 100 µM dexmedetomidine-pretreated cells and the control
cells (P>0.05; Fig. 2B).
Propofol decreases the levels of BDNF,
Bcl-2 and p-CREB in hippocampal neurons
The present study evaluated the effects of propofol
exposure on the mRNA expression levels of BDNF and Bcl-2, known to
be important in cell survival, using a semiquantitative RT-PCR
method. Compared with the control cells, the mRNA levels of Bcl-2
(Fig. 3A and B) and BDNF (Fig. 3C and D) in the cells treated with
propfol were reduced by 72.4 and 60.3%, respectively (P<0.05).
Consistent with the results of the semiquantitative RT-PCR
analysis, western blot analysis (Fig.
4A) revealed that the protein levels of BDNF (Fig. 4B) and Bcl-2 (Fig. 4C) in the cells treated with
propofol were reduced by 63.5 and 64.5%, respectively, compared
with control cells (P<0.05). Propofol also significantly
decreased the protein level of p-CREB, by 59.0%, compared with the
control (P<0.05; Fig. 4D).
 | Figure 3Dex attenuates Pro-induced reductions
in the mRNA expression levels of BDNF and Bcl-2 in immature
hippocampal neurons. The cells (8 days in vitro) were
exposed to Pro (100 µM, 3 h) in the absence or presence of
Dex pretreatment (0.001, 0.01, 0.1, 1, 10 and 100 µM). The
mRNA levels of (A and B) BDNF and (C and D) Bcl-2, were determined
using semiquantitative reverse transcription-quantitative
polymerase chain reaction analysis, in which the densities of bands
for BDNF and Bcl-2 were determined and standardized to the mRNA
levels of GAPDH. Data are presented as the mean ± standard
deviation of at least three independent experiments.
*P<0.05 and **P<0.01, compared with
group C; #P<0.05 and ##P<0.01, compared
with group Pro. Dex, dexmedetomidine; C, control; Pro, propofol;
BDNF; brain-derived neurotrophic factor; Bcl-2, B-cell lymphoma
2. |
 | Figure 4Dex attenuates Pro-induced reductions
in the protein expression levels of p-CREB, BDNF and Bcl-2 in
immature hippocampal neurons. The cell (8 days in vitro)
were exposed to Pro (100 µM, 3 h) in the absence or presence
of Dex pretreatment (0.001, 0.01, 0.1, 1, 10 and 100 µM).
Protein levels of Bcl-2, BDNF and p-CREB were examined using
western blot analysis. (A) Western blotting bands of all groups of
hippocampal neurons. Protein levels of (B) p-CREB, (C) BDNF and (D)
Bcl-2 were standardized by the expression of GAPDH. Data are
presented as the mean ± standard deviation and compared using
one-way analysis of variance. *P<0.05 and
**P<0.01, compared with group C;
#P<0.05 and ##P<0.01, compared with
group Pro. Dex, dexmedetomidine; C, control; Pro, propofol; BDNF;
brain-derived neurotrophic factor; Bcl-2, B-cell lymphoma 2;
phosphorylated-cyclic-AMP response element binding protein. |
Dexmedetomidine attenuates the
propofol-induced reduction in the expression levels of BDNF, Bcl-2
and p-CREB in hippocampal neurons
The semiquantitative RT-PCR analysis revealed that
the treatment of DIV 8 primary hippocampal neurons with
dexmedetomidine at concentrations of 0.001,0.01,0.1,1,10 and 100
µM, prior to propofol exposure (100 µM for 3 h),
significantly increased the mRNA expression of Bcl-2 by 87.2,
132.0, 139.6, 143.4, 164.2 and 165.3%, respectively, compared with
the cells exposed to propofol without dexmedetomidine pretreatment
(P<0.05; Fig. 3A and B).
Similarly, increases of 35.1, 38.8, 58.0, 90.4, 108.1 and 135.1%,
respectively, were detected in the mRNA levels of BDNF in the
dexmedetomidine-pretreated cells, compared to the propofol-treated
cells (P<0.05; Fig. 3C and D).
Consistent with the results of the semiquantitative RT-PCR
analysis, dexmedetomidine significantly increased the protein
levels of BDNF and Bcl-2, compared with the cells exposed to
propofol without dexmedetomidine pretreatment (P<0.05; Fig. 4C and D). The protein levels of
p-CREB in the dexmedetomidine-pretreated cells were also
significantly upregulated by 18.4, 54.7, 60.4, 80.7, 116.3 and
121.2%, respectively, compared with those in the propofol-treated
cells in the absence of dexmedetomidine pretreatment (P<0.05;
Fig. 4B).
Discussion
The present study used neuronal cultures from the
rat hippocampus to investigate the neuroprotective effect of
dexmedetomindine against propofol-induced neurotoxicity in
vitro. The results showed that 100 µM propofol triggered
immature hippocampal neuron (DIV 8) apoptosis, reduced neuronal
viability and caused a reduction in the levels of p-CREB, BDNF and
Bcl-2. Pretreatment of the DIV 8 neurons with 0.001–100 µM
dexmedetomindine prior to propofol exposure attenuated the
propofol-induced neuronal apoptosis and reduction in neuronal
viability, and was accompanied by increases in the levels of
p-CREB, Bcl-2 and BDNF. These in vitro data indicated that
dexmedetomidine prevented propofol-induced neurotoxicity in
neuronal cultures from the rat hippocampus, which was associated
with upregulation in the expression levels of p-CREB, BDNF and
Bcl-2.
Several studies have demonstrated that the
administration of anesthetics, including isoflurane, ketamine and
propofol, trigger neuronal apoptosis in developing rodent and NHP
brains (6,16–18).
In the present study DIV 8 hippocampal neurons were used to
represent a model of immature and developing neurons. Primary
neurons at DIV7–8 are sensitive to anesthetics, and it has been
shown that high doses of propofol induce marked neuroapoptosis in
the neonatal brain (4,19). In aggregated cell cultures,
propofol at a high concentration of 10 µg/ml has been
reported to produce neurotoxic effects on neurons (20). In the present study, the
concentration of propofol selected (100 µM) was sufficiently
high enough to induce neurotoxicity, which was confirmed by
preliminary experiments. Therefore, the hippocampal neurons at DIV
8 in the present study were treated with 100 µM propofol in
the follow-up experiment. The present study found that exposure of
immature neurons to 100 µM propofol for 3 h resulted in a
significant reduction in neuronal viability and increase in the
percentage of apoptotic cells, as indicated using the CCK-8 assay
and flow cytometric analysis. This indicated that propofol exposure
induced neuronal injury and apoptosis. These results are in
accordance with previous in vitro reports on the
neurotoxicity of propofol (3,21).
In the present study, propofol exposure provoked neuronal damage
in vitro and demonstrated that this damage was attenuated by
dexmedetomidine.
Dexmedetomidine has been shown to exert
neuroprotective effects against neurotoxicity induced by
anesthetics, including isoflurane and ketamine, in the developing
brain (9,10,22,23).
However, there is no data available on whether dexmedetomidine
provides neuroprotective properties against propofol-induced
neurotoxicity in the developing brain. The present study assessed
the neuroprotective effect of dexmedetomidine on neuronal cultures
from the rat hippocampus. Based on the previous studies of Sanders
et al (22) and Dahmani
et al (24), the
concentration range of dexmedetomidine was extended (0.001–100
µM) in the present study. A marked increase in neuronal
viability and reduction in the rate of neuronal apoptosis were
observed in the cells pretreated with dexmedetomidine at
concentrations between 0.001 and 100 µM. This suggested that
dexmedetomidine attenuated the negative effects of propofol on
neuronal viability and survival in the rat hippocampal neuronal
cultures. Unlike a previous study by Laudenbach et al
(25), which found that
dexmedetomidine concentrations of 10 and 100 µM provided
less neuroprotection, compared with lower concentrations in
vitro, maximal neuroprotection was observed at the highest
concentration of dexmedetomidine (100 µM) in the present
study, as revealed using an CCK-8 assay and flow cytometric
analysis. At this concentration, no significant differences in
neuronal viability or apoptosis were found between the
dexmedetomidine + propofol group and the control group, which
indicated that 100 µM dexmedetomidine caused complete
reversal of the neuronal injury induced by propofol in
vitro. Other in vivo studies have reported that
dexmedetomidine at a high concentration attenuates the
neurotoxicity caused by ketamine completely (23), but it is unable to completely
attenuate the cortical injury caused by isoflurane (22). This discrepancy may be due to
differences in the experimental approach and anesthetics used in
these studies. The present study also investigated whether
alterations in the levels of p-CREB, Bcl-2 and BDNF are involved in
the neuroprotective effects of dexmedetomidine.
The mechanisms underlying the neuroprotective
effects of dexmedetomidine remain to be fully elucidated. Previous
studies have indicated that dexmedetomidine-induced neuroprotection
is mediated partially by the activation of α2-adrenergic receptors
(9), the c-Jun N-terminal kinase
and p38 mitogen-activated protein kinase pathways (26), the phosphoinositide 3-kinase/Akt
pathway (10), and the
extracellular signal-regulated kinase (ERK)1/2 pathways (27). CREB, a transcription factor family
member, can be activated by activation of ERK1/2 and Akt pathways
(28). The activation of CREB was
has been suggested to regulate the expression of the genes
associated with neuronal survival, synaptic plasticity and memory
maintenance (29,30). Whether CREB pathways are involved
in the neuroprotective effects of dexmedetomidine remain to be
fully elucidated. CREB is located upstream of neurotrophins. The
phosphorylation of CREB at Ser-133 (p-CREB) regulates the
transcription of pro-survival factors, including BDNF and Bcl-2.
Increases in the levels of BDNF and p-CREB are observed in the
process of neuroprotection following neuronal injury (31,32).
Bcl-2, as a key apoptosis inhibitory protein, determines the
mitochondrial response to apoptotic stimuli, and protects cells
from apoptosis (33). In the
present study, semiquantitative RT-PCR and western blot analyses
revealed decreases in the expression levels of p-CREB, BDNF and
Bcl-2 following propofol administration. Dexmedetomidine treatment
prior to propofol exposure significantly attenuated the
propofol-induced neuroapoptosis and increased the expression levels
of p-CREB, BDNF and Bcl-2 compared with the propofol only group.
These findings suggested that propofol-induced neurotoxicity was
associated with lower levels of p-CREB, BDNF and Bcl-2, and
revealed a link between the upregulation of p-CREB, BDNF and Bcl-2
and the protective effect of dexmedetomindine against
propofol-induced neuronal damage caused in developing neurons.
The present study had a number of limitations.
Firstly, whether dexmedetomidine itself is directly toxic to
immature neurons and whether it inhibits developmental
neuroapoptosis, a normal physiologic process in the developing
brain, were not investigated. However, several studies have shown
that dexmedetomidine itself does not induce neuronal apoptosis in
the developing brain of rodents (9,22) or
NHPs (34). Secondly, due to the
size of the subjects, in vivo experiments were not performed
immediately following the in vitro experiments. Therefore,
careful in vivo experiments are required to confirm the
in vitro findings obtained in the present study to determine
the correlation between the concentrations and protective efficacy
of dexmedetomidine, and to investigate the underlying molecular
mechanisms in the developing brain.
In conclusion, the present study demonstrated that
dexmedetomidine attenuated propofol-induced neurotoxicity in
neuronal cultures from the rat hippocampus, and this was associated
with an increase in the levels of p-CREB, BDNF and Bcl-2. The
results of the present study contribute to the increasing body of
evidence that dexmedetomidine exerts neuroprotective
properties.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 81373498 and 81060277), the
Science Study and Technology Development Program of Guangxi (grant
no. 1355005-4-2) and the Science and Technology Research Project of
Guangxi University (grant no. 2013ZD014).
References
|
1
|
Sall JW, Stratmann G, Leong J, Woodward E
and Bickler PE: Propofol at clinically relevant concentrations
increases neuronal differentiation but is not toxic to hippocampal
neural precursor cells in vitro. Anesthesiology. 117:1080–1090.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kahraman S, Zup SL, McCarthy MM and Fiskum
G: GABAergic mechanism of propofol toxicity in immature neurons. J
Neurosurg Anesthesiol. 20:233–240. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spahr-Schopfer I, Vutskits L, Toni N,
Buchs PA, Parisi L and Muller D: Differential neurotoxic effects of
propofol on dissociated cortical cells and organotypic hippocampal
cultures. Anesthesiology. 92:1408–1417. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cattano D, Young C, Straiko MM and Olney
JW: Subanesthetic doses of propofol induce neuroapoptosis in the
infant mouse brain. Anesth Analg. 106:1712–1714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pesić V, Milanović D, Tanić N, Popić J,
Kanazir S, Jevtović-Todorović V and Ruzdijić S: Potential mechanism
of cell death in the developing rat brain induced by propofol
anesthesia. Int J Dev Neurosci. 27:279–287. 2009. View Article : Google Scholar
|
|
6
|
Creeley C, Dikranian K, Dissen G, Martin
L, Olney J and Brambrink A: Propofol-induced apoptosis of neurones
and oligodendrocytes in fetal and neonatal rhesus macaque brain. Br
J Anaesth. 110(Suppl 1): i29–i38. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Afonso J and Reis F: Dexmedetomidine:
Current role in anesthesia and intensive care. Rev Bras Anestesiol.
62:118–133. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paris A, Mantz J, Tonner PH, Hein L, Brede
M and Gressens P: The effects of dexmedetomidine on perinatal
excitotoxic brain injury are mediated by the alpha2A-adrenoceptor
subtype. Anesth Analg. 102:456–461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sanders RD, Xu J, Shu Y, Januszewski A,
Halder S, Fidalgo A, Sun P, Hossain M, Ma D and Maze M:
Dexmedetomidine attenuates isoflurane-induced neurocognitive
impairment in neonatal rats. Anesthesiology. 110:1077–1085. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Zeng M, Chen W, Liu C, Wang F, Han
X, Zuo Z and Peng S: Dexmedetomidine reduces isoflurane-induced
neuroapoptosis partly by preserving PI3K/Akt pathway in the
hippocampus of neonatal rats. PloS One. 9:e936392014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu LX, Yon JH, Carter LB and
Jevtovic-Todorovic V: General anesthesia activates BDNF-dependent
neuroapoptosis in the developing rat brain. Apoptosis.
11:1603–1615. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hwang L, Choi IY, Kim SE, Ko IG, Shin MS,
Kim CJ, Kim SH, Jin JJ, Chung JY and Yi JW: Dexmedetomidine
ameliorates intracerebral hemorrhage-induced memory impairment by
inhibiting apoptosis and enhancing brain-derived neurotrophic
factor expression in the rat hippocampus. Int J Mol Med.
31:1047–1056. 2013.PubMed/NCBI
|
|
13
|
Yan M, Dai H, Ding T, Dai A, Zhang F, Yu
L, Chen G and Chen Z: Effects of dexmedetomidine on the release of
glial cell line-derived neurotrophic factor from rat astrocyte
cells. Neurochem Int. 58:549–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bell MT, Puskas F, Bennett DT, Herson PS,
Quillinan N, Fullerton DA and Reece TB: Dexmedetomidine, an α-2a
adrenergic agonist, promotes ischemic tolerance in a murine model
of spinal cord ischemia-reperfusion. J Thorac Cardiovasc Surg.
147:500–506. 2014. View Article : Google Scholar
|
|
15
|
Lesuisse C, Qiu D, Böse CM, Nakaso K and
Rupp F: Regulation of agrin expression in hippocampal neurons by
cell contact and electrical activity. Brain Res Mol Brain Res.
81:92–100. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bercker S, Bert B, Bittigau P,
Felderhoff-Müser U, Bührer C, Ikonomidou C, Weise M, Kaisers UX and
Kerner T: Neurodegeneration in newborn rats following propofol and
sevoflurane anesthesia. Neurotox Res. 16:140–147. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brambrink AM, Evers AS, Avidan MS, Farber
NB, Smith DJ, Zhang X, Dissen GA, Creeley CE and Olney JW:
Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque
brain. Anesthesiology. 112:834–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Young C, Jevtovic-Todorovic V, Qin YQ,
Tenkova T, Wang H, Labruyere J and Olney JW: Potential of ketamine
and midazolam, individually or in combination, to induce apoptotic
neurodegeneration in the infant mouse brain. Br J Pharmacol.
146:189–197. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fredriksson A, Pontén E, Gordh T and
Eriksson P: Neonatal exposure to a combination of
N-methyl-D-aspartate and gamma-aminobutyric acid type A receptor
anesthetic agents potentiates apoptotic neurodegeneration and
persistent behavioral deficits. Anesthesiology. 107:427–436. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Honegger P and Matthieu JM: Selective
toxicity of the general anesthetic propofol for GABAergic neurons
in rat brain cell cultures. J Neurosci Res. 45:631–636. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vutskits L, Gascon E, Tassonyi E and Kiss
JZ: Clinically relevant concentrations of propofol but not
midazolam alter in vitro dendritic development of isolated
gamma-aminobutyric acid-positive interneurons. Anesthesiology.
102:970–976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sanders RD, Sun P, Patel S, Li M, Maze M
and Ma D: Dexmedetomidine provides cortical neuroprotection: Impact
on anaesthetic-induced neuroapoptosis in the rat developing brain.
Acta Anaesthesiol Scand. 54:710–716. 2010. View Article : Google Scholar
|
|
23
|
Duan X, Li Y, Zhou C, Huang L and Dong Z:
Dexmedetomidine provides neuroprotection: Impact on
ketamine-induced neuroapoptosis in the developing rat brain. Acta
Anaesthesiol Scand. 58:1121–1126. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dahmani S, Rouelle D, Gressens P and Mantz
J: Characterization of the postconditioning effect of
dexmedetomidine in mouse organotypic hippocampal slice cultures
exposed to oxygen and glucose deprivation. Anesthesiology.
112:373–383. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Laudenbach V, Mantz J, Lagercrantz H,
Desmonts JM, Evrard P and Gressens P: Effects of
alpha(2)-adrenoceptor agonists on perinatal excitotoxic brain
injury: Comparison of clonidine and dexmedetomidine.
Anesthesiology. 96:134–141. 2002. View Article : Google Scholar
|
|
26
|
Liao Z, Cao D, Han X, Liu C, Peng J, Zuo
Z, Wang F and Li Y: Both JNK and P38 MAPK pathways participate in
the protection by dexmedetomidine against isoflurane-induced
neuroapoptosis in the hippocampus of neonatal rats. Brain Res Bull.
107:69–78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu YM, Wang CC, Chen L, Qian LB, Ma LL,
Yu J, Zhu MH, Wen CY, Yu LN and Yan M: Both PI3K/Akt and ERK1/2
pathways participate in the protection by dexmedetomidine against
transient focal cerebral ischemia/reperfusion injury in rats. Brain
Res. 1494:1–8. 2013. View Article : Google Scholar
|
|
28
|
Lonze BE and Ginty DD: Function and
regulation of CREB family transcription factors in the nervous
system. Neuron. 35:605–623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Impey S, McCorkle SR, Cha-Molstad H, Dwyer
JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G and Goodman
RH: Defining the CREB regulon: A genome-wide analysis of
transcription factor regulatory regions. Cell. 119:1041–1054.
2004.PubMed/NCBI
|
|
30
|
Kandel ER: The molecular biology of memory
storage: A dialogue between genes and synapses. Science.
294:1030–1038. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang L, Zhao H, Zhang X, Chen L, Zhao X,
Bai X and Zhang J: Nobiletin protects against cerebral ischemia via
activating the p-Akt, p-CREB, BDNF and Bcl-2 pathway and
ameliorating BBB permeability in rat. Brain Res Bull. 96:45–53.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang W, Cao J, Liu X, Meng F, Li M, Chen
B and Zhang J: AMPK plays a dual role in regulation of CREB/BDNF
pathway in mouse primary hippocampal cells. J Mol Neurosci.
56:782–788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cory S, Huang DC and Adams JM: The Bcl-2
family: Roles in cell survival and oncogenesis. Oncogene.
22:8590–8607. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koo E, Oshodi T, Meschter C, Ebrahimnejad
A and Dong G: Neurotoxic effects of dexmedetomidine in fetal
cynomolgus monkey brains. J Toxicol Sci. 39:251–262. 2014.
View Article : Google Scholar : PubMed/NCBI
|














