Introduction
Subarachnoid hemorrhage (SAH) is a severe
neurological emergency, which accounts for 5% of all strokes
(1). Although the diagnosis and
treatment of SAH have improved in the past 20 years, SAH remains
one of the most life-threatening acute neurological diseases, and
is associated with a high rate of mortality and a poor prognosis.
The primary cause of mortality in patients with SAH is brain injury
that occurs during the early phase of SAH (within 48 h of SAH)
(2), which is characterized by an
initial sudden increase in intracranial pressure (ICP) and
reduction in cerebral blood flow (CBF) (3). These complications trigger focal or
global cerebral ischemia with various deleterious effects,
including inflammation and neuronal cell death.
Due to the difficulties in predicting and preventing
the occurrence of SAH, the management of SAH primarily focuses on
protecting the brain from secondary damages during the acute and
chronic phases that follow SAH. Mild hypothermia (MH) is well known
for its powerful neuroprotective effects against neuronal injury
following ischemia and traumatic brain injury, including stroke
(4). Several mechanisms have been
proposed to underlie the protective effects of MH. For example, MH
has been reported to reduce mitochondrial dysfunction and
per-ischemic production of reactive oxygen species (ROS) following
ischemic disorders, which is believed to be one of the major causes
of cell death and inflammation post-ischemia (5,6).
Furthermore, MH treatment has been demonstrated to reduce global
brain glucose and oxygen metabolic rates (7,8),
which may help the brain to deal with energy failure, and prevent
mitochondrial dysfunction and neuronal apoptosis. Other studies
have suggested that MH treatment stabilizes the blood-brain barrier
(9), reduces brain edema (10) and the release of excitatory amino
acids (11,12), and attenuates inflammatory
reactions (13) and lipid
peroxidation (14).
Due to the well-documented protective effects of MH
against ischemia and traumatic brain injury, MH has also been
investigated as a potential therapeutic strategy for the treatment
of SAH in humans and in animal models of SAH. Experimental studies
in patients with SAH and animal models of SAH have suggested that
MH treatment may improve ICP control (15), facilitate the resolution of
cerebral vasospasm through modulation of blood flow velocity
(16,17), and prevent neuronal damage and
apoptosis caused by cerebral ischemia. However, little is currently
known regarding the molecular mechanisms and signaling pathways
associated with the effects of MH on protection against early brain
injury following SAH.
Neurotrophic factors are required for the growth and
survival of developing neurons and the maintenance of mature
neurons. It has previously been reported that the tropomyosin
receptor kinase B (TrkB)-mediated neurotrophic pathway has an
important role in neuronal survival following ischemic stroke
(18,19). Pharmacological activation of
TrkB-cAMP response element binding protein (CREB) may ameliorate
ischemic neuronal injury via the prevention of neuronal apoptosis,
and therefore may improve functional recovery following stroke
(18,19).
The present study aimed to determine the effects and
mechanisms of MH on SAH development. The present study demonstrated
that treatment with MH induced strong protective effects against
neuronal injury in a rat model of SAH. Rats treated with MH
exhibited a marked reduction in ROS production and caspase-3
activation following SAH. Furthermore, the TrkB/extracellular
signal-regulated kinases (ERK)/CREB pathway mediated the protective
effects of MH. Suppression of the TrkB/ERK/CREB pathway using an
ERK inhibitor markedly abrogated the protective effects of MH in
SAH rats. These findings indicated that activation of the
TrkB/ERK/CREB pathway may be an essential mechanism underlying the
protective effects of MH against early brain injury following SAH
in vivo.
Materials and methods
Animal study
All experiments were approved by the Animal Care
Committee at Harbin Medical University (Harbin, China). Male Wistar
rats (weight, 200–250 g; n=45) were housed in a temperature and
humidity-controlled environment under a 12:12-h light/dark cycle
(light phase, 7:00 a.m.–7:00 p.m.), and were given ad
libitum access to food and water. The rat brain tissues were
surgically removed following overdose with 5% isoflurane.
Rat model of SAH
The SAH model was established in male rats as
described previously (20–22). Briefly, the rats were anesthetized.
A sharpened 3–0 monofilament was introduced into the right internal
carotid artery through the external carotid artery until resistance
was felt (10–12 mm from the common carotid bifurcation). The
monofilament was subsequently pushed further to perforate the
bifurcation of the internal carotid artery, and was then withdrawn
immediately. In the sham group, the monofilament was inserted into
the carotid artery; however, no perforation was performed.
Following removal of the monofilament, the incision was closed.
Endovascular occlusion by perforation lasted <5 min in each
animal.
MH treatment
SAH was induced in 25 rats (except for 2 rats that
had succumbed and the 3 which were excluded due to low weight, 30
rats purchased in total). Then, 5 rats were randomly selected and
sacrificed at five different time points (0, 0.5, 4, 24 and 72 h)
after induction.
A total of 20 rats (except for rats that had
succumbed or did not qualify) were randomly divided into four
groups: Sham, SAH, SAH + MH and SAH + MH + PD98059 groups. MH was
conducted for 120 min as previously described (16), commencing 60 min after SAH. PD98059
(5 mg/kg/day dissolved in 0.2 ml dimethyl sulfoxide; New England
Biolabs, Ipswich, MA, USA) was administered intravenously 0.5 h
prior to SAH, in order to inhibit TrkB/ERK signaling as described
previously (23). Mice were
sacrificed 3 days after SAH for further evaluation.
Measurement of body weight and brain
water content
The body weight and brain water content were
measured on the third day following establishment of the SAH model.
For brain water content, the brain tissues were removed, and the
hemispheres were separated and weighed to assess their wet weight.
After the wet weight of the brain tissues was quantified, the
hemispheres were desiccated for 36 h at 110°C, until the weight was
constant. Hemispheric water content (%) was calculated as follows:
(Wet weight − dried weight) / wet weight × 100%.
ROS detection
Intracellular ROS levels were determined in brain
tissue homogenates using 2′,7′-dichlorodihydrofluorescein diacetate
(DCFH-DA; Beyotime Institute of Biotechnology, Beijing, China)
according to manufacturer's protocol. Briefly, the rat brain
extracts were incubated with DCFH-DA at 37°C in the dark for 30
min. The fluorescence intensity was then quantified using a
multi-detection microplate reader at 485 nm excitation and 530 nm
emission wavelengths.
Caspase-3 activity assay
Caspase-3 activity was determined using a CaspACE
assay system (Promega Corporation, Madison, WI, USA) was conducted
according to the manufacturer's protocol. Rat brain tissues were
lysed on ice for 30 min and were centrifuged at 12,000 × g
for 15 min at 4°C. The levels of caspase-3 were expressed relative
to the amounts in the control group. The caspase-3 activities in
the supernatant were measured at 405 nm.
Western blot analysis
The rat brains were surgically collected and cut
into pieces. Subsequently, the brain tissues were directly
homogenized in ice-cold lysis buffer [62.5 mM Tris-HCl, 2% (w/v)
sodium dodecyl sulfate (SDS), 5% (w/v) β-mercaptoethanol, 10% (v/v)
glycerol, 0.002% (w/v) bromophenol blue] for 30 min. The lysates
were then centrifuged for 15 min at 12,000 × g at 4°C, and
the resulting supernatants were collected and boiled. Protein
concentrations were measured in the extracts using a bichinchoninic
acid assay. Protein samples (20–40 µg/lane) were separated
by 10% SDS-polyacrylamide gel electrophoresis and were transferred
to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). The membranes were then blocked overnight with 5% bovine
serum albumin (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany)
in Tris-buffered saline containing 0.1% Tween-20 (TBST), and were
incubated with antibodies against cleaved caspase-3 (cat. no.
ab2302; 1:1,000; Abcam, Cambridge, UK), phosphorylated (p)-TrkB
(sc-7987, 1: 500; Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
TrkB (cat. no. sc-377218; 1:1,000; Santa Cruz Biotechnology, Inc.),
p-ERK1/2 (cat. no. sc-136521; 1:1,000; Santa Cruz Biotechnology,
Inc.), ERK1/2 (cat. no. sc-292838; 1:1,000; Santa Cruz
Biotechnology, Inc.) p-CREB (cat. no. sc-7978, 1:1,000; Santa Cruz
Biotechnology, Inc.), CREB (cat. no. sc-377154; 1:1,000; Santa Cruz
Biotechnology, Inc.) and glyceraldehyde 3-phosphate dehydrogenase
(GAPDH; cat. no. sc-47724; 1:1,000; Santa Cruz Biotechnology, Inc.)
at 4°C overnight, followed by washing with TBST. The membranes were
then incubated for 2 h at room temperature with horseradish
peroxidase-conjugated secondary antibody (Wuhan Boster Biological
Technology Co., Ltd., Wuhan, China). The blots were detected using
an Enhanced Chemiluminescence Plus reagent kit (Wuhan Boster
Biological Technology Co., Ltd.). GAPDH was used as a loading
control. The bands were quantified using Image J version 1.37
software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence staining
The brain tissues were fixed in 4% paraformaldehyde,
and were sliced using a cryostat. The sections (20 µm) were
stored in anti-freeze solution (15% glucose and 30% ethylene glycol
in 50 mM phosphate buffer, pH 7.4) at −20°C and were used for
immunofluorescence staining. Frozen sections were incubated
overnight at 4°C with primary antibodies against cleaved caspase-3
(cat. no. ab2302; 1:100; Abcam) and p-CREB (cat. no. sc-7987;
1:100; Santa Cruz Biotechnology, Inc.). The slices were then washed
three times with phosphate-buffered saline and were incubated with
the corresponding fluorescence dye-conjugated bovine anti-rabbit
IgG-FITC (cat. no. sc-2365; 1:100; Santa Cruz Biotechnology, Inc.)
and donkey anti-goat IgG-TR (cat. no. sc-2783; 1:100; Santa Cruz
Biotechnology, Inc.) secondary antibodies for 2 h. After being
washed and counterstained with 4′,6-diamidino-2-phenylindole,
immunofluorescence was observed under a fluorescence microscope
(Olympus Corporation, Tokyo, Japan).
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Group differences were analyzed using one-way analysis
of variance followed by Tukey's honest significant difference test.
Statistical analyses were conducted using GraphPad Prism version
5.0 statistical software (GraphPad Software, Inc., La Jolla, CA,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
MH protects against early brain injury in
rats following SAH
To investigate the protective function of MH in SAH,
a rat model of SAH was generated, and the rats were treated with MH
1 h after the model was established. Consistent with previous
observations regarding this model (20–22),
the induction of SAH reduced body weight and increased brain water
content in the rats. These detrimental effects caused by SAH were
significantly attenuated by MH, as indicated by the reduction in
body weight loss (Fig. 1A) and
brain water accumulation (Fig.
1B). Given that mitochondrial dysfunction and the activation of
apoptotic cascades are key pathological events in early brain
injury following SAH (24,25), the present study investigated
whether MH was able to protect neurons from mitochondrial
dysfunction and apoptosis in early brain injury. The results
clearly demonstrated that MH improved mitochondrial function
following SAH, as evidenced by the reduction in ROS production
(Fig. 1C). Furthermore, the
activation of caspase-3 was markedly reduced by MH (Fig. 1D and E). These data indicate that
MH treatment in the early phase of SAH may reduce ROS release and
neuronal apoptosis, thus improving the outcome of SAH in
vivo.
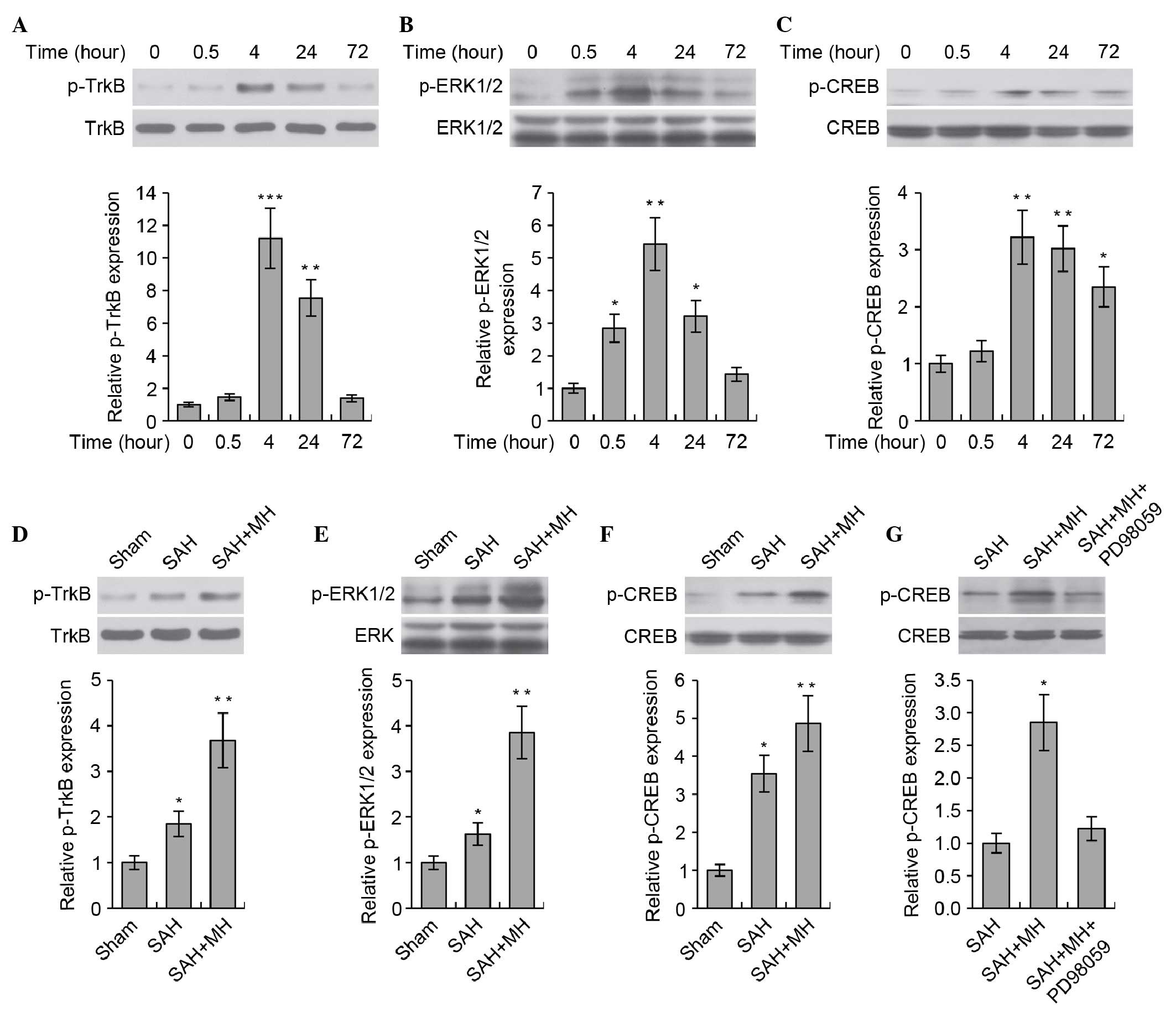
MH promotes TrkB/ERK/CREB signaling
The present study aimed to determine the molecular
mechanism underlying the protective effects of MH on SAH The
TrkB-mediated neurotrophic pathway has critical roles in neuronal
survival and growth. Previous studies have demonstrated that MH was
able to induce the expression of brain-derived neurotrophic factor
(BDNF) in rat brains following cerebral ischemia; BDNF functions as
a ligand of the TrkB receptor (26,27).
Therefore, the present study hypothesized that MH would prevent
neuronal injury via activation of the TrkB-mediated neurotrophic
pathway. The activity of TrkB, and the downstream ERK1/2/CREB
pathway, was analyzed at various time points (0, 0.5, 4, 24 and 72
h) following SAH. SAH markedly stimulated TrkB/ERK/CREB signaling
(Fig. 2A–C). The levels of p-TrkB,
p-ERK1/2 and p-CREB in the rat brain peaked at 4 h, and then
decreased gradually. Treatment with MH markedly enhanced the
phosphorylation of TrkB (Fig. 2D),
ERK1/2 (Fig. 2E) and CREB
(Fig. 2F) following SAH.
Furthermore, MH-induced phosphorylation of CREB was shown to be
dependent on the activation of TrkB and ERK, since inhibition of
ERK using the small molecule inhibitor PD98059 markedly abrogated
CREB phosphorylation (Fig. 2G).
These data indicate that the TrkB/ERK/CREB signaling pathway may be
involved in the progress of SAH, and that MH promotes activation of
the TrkB/ERK/CREB pathway following SAH.
Inhibition of the TrkB/ERK/CREB signaling
pathway reduces the protective effects of MH in SAH-induced early
brain injury
To examine whether the TrkB/ERK/CREB signaling
pathway is required for the protective effects of MH, the effects
of MH on early brain injury were detected following inhibition of
the TrkB/ERK/CREB pathway using PD98059. Notably, inhibition of the
TrkB/ERK/CREB pathway almost completely abrogated the beneficial
effects of MH on SAH. The reductions in body weight loss and brain
water content were reversed by PD98059. There was no significant
difference in body weight (Fig.
3A) and brain water content (Fig.
3B) between the SAH and SAH + MH + PD98059 groups. Treatment
with PD98059 also abrogated the effects of MH on the prevention of
ROS production (Fig. 3C) and
caspase-3 activation (Fig. 3D)
following SAH.
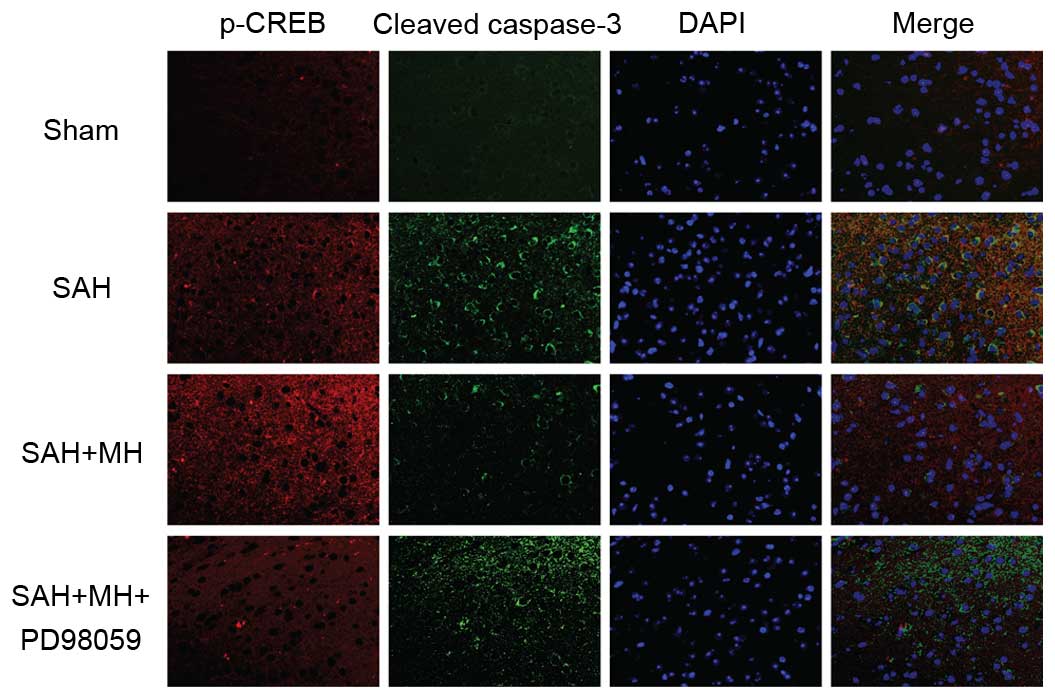
To determine the role of CREB in SAH and MH-treated
SAH, fluorescence staining of p-CREB and cleaved caspase-3 was
performed on brain sections. SAH induced extensive cell apoptosis
and promoted CREB phosphorylation (Fig. 4). In addition, MH increased the
levels of p-CREB, and decreased the levels of cleaved caspase-3.
These effects were abrogated by inhibition of TrkB/ERK signaling.
These results indicate that MH may protect the brain from
SAH-induced neuronal injury by activating the TrkB/ERK/CREB
signaling pathway.
Discussion
MH has been revealed to be effective in minimizing
neuronal damage and improving the functional outcome of SAH;
however, the molecular mechanisms underlying the beneficial effects
of MG remain unclear. Using a rat SAH perforation model, the
present study demonstrated that MH was able to attenuate
mitochondrial dysfunction and activation of apoptosis in the brain
following the induction of SAH. These protective effects were
mediated by enhanced activity of the TrkB/ERK/CREB pathway. These
observations identify a potential mechanism by which MH protects
rats from early brain damage following SAH.
Chronic post-SAH pathological consequences,
characterized by delayed cerebral ischemia and vasospasm of the
major cerebral arteries (3–7 days after SAH), have been extensively
studied and treated; however, these efforts have not resulted in an
effective treatment to prevent or ameliorate brain injury following
SAH (28,29). The early brain injury that occurs
within 48 h of SAH has gained more attention as a novel target for
improving SAH patient outcome, since >60% of patients with SAH
succumb due to early brain injury during the first 48 h after SAH
(30,31). In addition, the majority of chronic
secondary injuries are initiated by early brain injury. It has
previously been indicated that mitochondrial dysfunction and
extensive neuronal apoptosis are the key events associated with
early brain injury following SAH (32). Following SAH-induced global
ischemia, apoptosis has been observed in several regions of the
brain, including the hippocampus, blood-brain barrier (BBB) and
vasculature (24). Activation of
apoptotic cascades may lead to severe pathological complications,
including BBB disruption (33) and
vasospasm (34). Therefore,
anti-apoptosis may be considered a potential therapeutic
intervention for the treatment of SAH.
MH is a neuroprotective approach that has been
employed in various clinical scenarios, particularly in the
treatment of ischemic stroke and traumatic brain injury. Based on
the pathophysiological similarity between stroke and SAH, MH has
been tested as a potential therapy for SAH in humans and animal
models of SAH. Several studies have indicated that MH may reduce
ICP and improve CBF in the early phase of SAH, and minimize the
detrimental effects of delayed cerebral ischemia and vasospasm in
the chronic secondary injuries following SAH, thus improving the
functional outcome in patients with SAH (35,36).
Although the exact molecular mechanisms underlying the protective
effects of MH on SAH are largely unknown, it is generally believed
that MH protects against neuronal damage via several mechanisms.
The present study demonstrated that MH reduced ROS generation and
activation of apoptotic cascades during the early phase following
SAH. These results suggested that MH may reduce neuronal damage, at
least partially, through improving mitochondrial function and
promoting neuronal survival.
BDNF/TrkB signaling is critical for neuronal
survival, morphogenesis and plasticity. It is well-known that
activation of the TrkB receptor elicits various intracellular
signaling pathways, including the mitogen-activated protein
kinase/ERK pathway, the phosphoinositide 3-kinase pathway, and CREB
transcription (37). All of these
pathways have been reported to participate in the regulation of
neuronal growth and survival (37). Beyond its physiological function,
pharmaceutical activation of the TrkB pathway promotes neuronal
survival following ischemic brain injury (19,38).
The present study established a previously unappreciated link
between MH and the neurotrophic pathway. MH enhanced activity of
the TrkB/ERK/CREB pathway in vivo in a rat model of SAH.
Notably, the TrkB/ERK/CREB pathway is essential for the
neuroprotective effects of MH on SAH, since inhibition of this
pathway using a small molecule inhibitor almost fully abolished the
beneficial effects of MH. These results indicated a novel
protective mechanism for MH in the context of early brain injury
following SAH. Based on these findings, MH may reduce neuronal loss
not only through inhibiting cell death activators (such as c-Jun
N-terminal kinase) (39) but also
through enhancing pro-survival signaling pathways. Notably, studies
in other animal models of ischemic brain injury have suggested that
treatment with MH could induce BDNF expression in the hippocampus
(26,27), which may be a potential mechanism
that explains how MH treatment activates the TrkB/ERK/CREB pathway.
The phosphorylated TrkB, ERK and CREB were stumilated by SAH and
reached peak at 4 h after hemorrhage. The levels of phosphorylated
protein then decreased due to short half-lives. Downstream target
genes were then elevated (40–42).
In conclusion, using a rat model of SAH, the present
study demonstrated that MH ameliorates early brain injury through
the prevention of mitochondrial dysfunction and inhibition of
apoptotic cascades following SAH. The beneficial effects of MH are
largely dependent on activation of the TrkB/ERK/CREB pathway. In
the past few decades, early brain injury has evolved to be a
promising therapeutic target for SAH. The present study indicated
that MH is an effective strategy that may be used to reduce
neuronal damage in the early phase of SAH. This mechanistic study
of MH action in SAH revealed that the TrkB/ERK/CREB pathway may
represent a novel therapeutic target for the intervention of early
brain injury following SAH.
Acknowledgments
The present study was supported by the Postdoctoral
Grant of Harbin Medical University (grant no. 2014M561373).
References
|
1
|
van Gijn J, Kerr RS and Rinkel GJ:
Subarachnoid haemorrhage. Lancet. 369:306–318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ostrowski RP, Colohan AR and Zhang JH:
Molecular mechanisms of early brain injury after subarachnoid
hemorrhage. Neurol Res. 28:399–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bederson JB, Levy AL, Ding WH, Kahn R,
DiPerna CA, Jenkins AL III and Vallabhajosyula P: Acute
vasoconstriction after subarachnoid hemorrhage. Neurosurgery.
42:352–362. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thome C, Schubert GA and Schilling L:
Hypothermia as a neuroprotective strategy in subarachnoid
hemorrhage: A pathophysiological review focusing on the acute
phase. Neurol Res. 27:229–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tissier R, Chenoune M, Pons S, Zini R,
Darbera L, Lidouren F, Ghaleh B, Berdeaux A and Morin D: Mild
hypothermia reduces per-ischemic reactive oxygen species production
and preserves mitochondrial respiratory complexes. Resuscitation.
84:249–255. 2013. View Article : Google Scholar
|
|
6
|
Lee SM, Zhao H, Maier CM and Steinberg GK:
The protective effect of early hypothermia on PTEN phosphorylation
correlates with free radical inhibition in rat stroke. J Cereb
Blood Flow Metab. 29:1589–1600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Milde LN: Clinical use of mild hypothermia
for brain protection: A dream revisited. J Neurosurg Anesthesiol.
4:211–215. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Erecinska M, Thoresen M and Silver IA:
Effects of hypothermia on energy metabolism in Mammalian central
nervous system. J Cereb Blood Flow Metab. 23:513–530. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith SL and Hall ED: Mild pre- and
posttraumatic hypothermia attenuates blood-brain barrier damage
following controlled cortical impact injury in the rat. J
Neurotrauma. 13:1–9. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Karibe H, Zarow GJ, Graham SH and
Weinstein PR: Mild intraischemic hypothermia reduces postischemic
hyperperfusion, delayed postischemic hypoperfusion, blood-brain
barrier disruption, brain edema, and neuronal damage volume after
temporary focal cerebral ischemia in rats. J Cereb Blood Flow
Metab. 14:620–627. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Maeda T, Katayama Y, Kawamata T and
Yamamoto T: Mechanisms of excitatory amino acid release in contused
brain tissue: Effects of hypothermia and in situ administration of
Co2+ on extracellular levels of glutamate. J Neurotrauma.
15:655–664. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mori K, Maeda M, Miyazaki M and Iwase H:
Effects of mild and moderate hypothermia on cerebral metabolism and
glutamate in an experimental head injury. Acta Neurochir Suppl.
71:222–224. 1998.PubMed/NCBI
|
|
13
|
Chatzipanteli K, Yanagawa Y, Marcillo AE,
Kraydieh S, Yezierski RP and Dietrich WD: Posttraumatic hypothermia
reduces polymorphonuclear leukocyte accumulation following spinal
cord injury in rats. J Neurotrauma. 17:321–332. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Busto R, Globus MY, Dietrich WD, Martinez
E, Valdes I and Ginsberg MD: Effect of mild hypothermia on
ischemia-induced release of neurotransmitters and free fatty acids
in rat brain. Stroke. 20:904–910. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gasser S, Khan N, Yonekawa Y, Imhof HG and
Keller E: Long-term hypothermia in patients with severe brain edema
after poor-grade subarachnoid hemorrhage: Feasibility and intensive
care complications. J Neurosurg Anesthesiol. 15:240–248. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thome C, Schubert G, Piepgras A, Elste V,
Schilling L and Schmiedek P: Hypothermia reduces acute vasospasm
following SAH in rats. Acta Neurochir Suppl. 77:255–258.
2001.PubMed/NCBI
|
|
17
|
Bishop CC, Powell S, Rutt D and Browse NL:
Transcranial Doppler measurement of middle cerebral artery blood
flow velocity: A validation study. Stroke. 17:913–915. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hasegawa Y, Suzuki H, Altay O and Zhang
JH: Preservation of tropomyosin-related kinase B (TrkB) signaling
by sodium orthovanadate attenuates early brain injury after
subarachnoid hemorrhage in rats. Stroke. 42:477–483. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He Q, Wang S, Liu X, Guo H, Yang H, Zhang
L, Zhuang P, Zhang Y, Ye Z and Hu L: Salvianolate lyophilized
injection promotes post-stroke functional recovery via the
activation of VEGF and BDNF-TrkB-CREB signaling pathway. Int J Clin
Exp Med. 8:108–122. 2015.PubMed/NCBI
|
|
20
|
Doczi T, Laszlo FA, Szerdahelyi P and Joo
F: Involvement of vasopressin in brain edema formation: Further
evidence obtained from the brattleboro diabetes insipidus rat with
experimental subarachnoid hemorrhage. Neurosurgery. 14:436–441.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Piepgras A, Elste V, Frietsch T, Schmiedek
P, Reith W and Schilling L: Effect of moderate hypothermia on
experimental severe subarachnoid hemorrhage, as evaluated by
apparent diffusion coefficient changes. Neurosurgery. 48:1128–1135.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun BL, Zhang SM, Xia ZL, Yang MF, Yuan H,
Zhang J and Xiu RJ: The effects of nimodipine on regional cerebral
blood flow, brain water and electrolyte contents in rats with
subarachnoid hemorrhage. Clin Hemorheol Microcirc. 29:337–344.
2003.
|
|
23
|
Wojcicka G, Jamroz-Wisniewska A, Widomska
S, Ksiazek M and Bełtowski J: Role of extracellular
signal-regulated kinases (ERK) in leptin-induced hypertension. Life
Sci. 82:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cahill J, Calvert JW and Zhang JH:
Mechanisms of early brain injury after subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 26:1341–1353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Niizuma K, Endo H and Chan PH: Oxidative
stress and mitochondrial dysfunction as determinants of ischemic
neuronal death and survival. J Neurochem. 109(Suppl 1): S133–S138.
2009. View Article : Google Scholar
|
|
26
|
D'Cruz BJ, Fertig KC, Filiano AJ, Hicks
SD, DeFranco DB and Callaway CW: Hypothermic reperfusion after
cardiac arrest augments brain-derived neurotrophic factor
activation. J Cereb Blood Flow Metab. 22:843–851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boris-Moller F, Kamme F and Wieloch T: The
effect of hypothermia on the expression of neurotrophin mRNA in the
hippocampus following transient cerebral ischemia in the rat. Brain
Res Mol Brain Res. 63:163–173. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGirt MJ, Garces Ambrossi GL, Huang J and
Tamargo RJ: Simvastatin for the prevention of symptomatic cerebral
vasospasm following aneurysmal subarachnoid hemorrhage: A
single-institution prospective cohort study. J Neurosurg.
110:968–974. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vajkoczy P, Meyer B, Weidauer S, Raabe A,
Thome C, Ringel F, Breu V and Schmiedek P: Clazosentan
(AXV-034343), a selective endothelin A receptor antagonist, in the
prevention of cerebral vasospasm following severe aneurysmal
subarachnoid hemorrhage: Results of a randomized, double-blind,
placebo-controlled, multicenter phase IIa study. J Neurosurg.
103:9–17. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Broderick JP, Brott TG, Duldner JE,
Tomsick T and Leach A: Initial and recurrent bleeding are the major
causes of death following subarachnoid hemorrhage. Stroke.
25:1342–1347. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Le Roux PD and Winn HR: Management of the
ruptured aneurysm. Neurosurg Clin N Am. 9:525–540. 1998.PubMed/NCBI
|
|
32
|
Abe Y, Sakairi T, Kajiyama H, Shrivastav
S, Beeson C and Kopp JB: Bioenergetic characterization of mouse
podocytes. Am J Physiol. 299:464–476. 2010. View Article : Google Scholar
|
|
33
|
Germanò A, d'Avella D, Imperatore C,
Caruso G and Tomasello F: Time-course of blood-brain barrier
permeability changes after experimental subarachnoid haemorrhage.
Acta Neurochir (Wien). 142:575–581. 2000. View Article : Google Scholar
|
|
34
|
Zhou C, Yamaguchi M, Colohan AR and Zhang
JH: Role of p53 and apoptosis in cerebral vasospasm after
experimental subarachnoid hemorrhage. J Cereb Blood Flow Metab.
25:572–582. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kirkman MA and Smith M: Intracranial
pressure monitoring, cerebral perfusion pressure estimation, and
ICP/CPP-guided therapy: A standard of care or optional extra after
brain injury? Br J Anaesth. 112:35–46. 2014. View Article : Google Scholar
|
|
36
|
Haddad SH and Arabi YM: Critical care
management of severe traumatic brain injury in adults. Scand J
Trauma Resusc Emerg Med. 20:122012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Numakawa T, Suzuki S, Kumamaru E, Adachi
N, Richards M and Kunugi H: BDNF function and intracellular
signaling in neurons. Histol Histopathol. 25:237–258. 2010.
|
|
38
|
Tian X, Guo J, Zhu M, Li M, Wu G and Xia
Y: δ-Opioid receptor activation rescues the functional TrkB
receptor and protects the brain from ischemia-reperfusion injury in
the rat. PLoS One. 8:e692522013. View Article : Google Scholar
|
|
39
|
Xu L, Yenari MA, Steinberg GK and Giffard
RG: Mild hypothermia reduces apoptosis of mouse neurons in vitro
early in the cascade. J Cereb Blood Flow Metab. 22:21–28. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fujioka A, Terai K, Itoh RE, Aoki K,
Nakamura T, Kuroda S, Nishida E and Matsuda M: Dynamics of the
Ras/ERK MAPK cascade as monitored by fluorescent probes. J Biol
Chem. 281:8917–8926. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang T, Massa SM and Longo FM: LAR protein
tyrosine phosphatase receptor associates with TrkB and modulates
neurotrophic signaling pathways. J Neurobiol. 66:1420–1436. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sánchez-Huertas C and Rico B:
CREB-dependent regulation of GAD65 transcription by BDNF/TrkB in
cortical interneurons. Cereb Cortex. 21:777–788. 2011. View Article : Google Scholar
|