Introduction
Individuals with certain occupations, including
those in the textile industry, mining and clay manufacturing, are
at high risk for the occurrence of pulmonary silicosis (1). As one of the typical pathogens,
crystalline silica inhalation can lead to silicosis, which is one
of the most frequent forms of occupational lung disease (2). Silicosis is prevalent in developing
countries and areas where workers lack appropriate professional
protection (3). The pathological
characteristics of silicosis are considered to be pulmonary
inflammation and fibrosis (4,5). As
the fibrosis of silicosis develops, aggravation of the
ventilation-perfusion imbalance leads to hypoxemia and respiratory
failure (6). Thus, novel
therapeutic agents against fibrosis are of clinical
significance.
Pulmonary immune cells, particularly alveolar
macrophages, are considered to be important in the occurrence and
pathogenesis of silicosis (7).
When the silica particles are inhaled, macrophages target and
enclose the particles by endocytosis. The transcription of
inflammatory cytokines, including interleukin-1β (IL-1β), tumor
necrosis factor-α (TNF-α) and transforming growth factor-β1
(TGF-β1) are initiated (8).
Following the binding of TGF-β1 and its receptor in lung
fibroblasts, the downstream receptor-activated small mothers
against decapentaplegic (R-smads), including Smad3, are activated
and eventually translocated to the nucleus to initiate the
transcription of extracellular matrix (ECM) gene transcription
(9).
Emodin, also known as
1,3,8-trihydroxy-6-methylanthraquinone, is one of the agents
extracted from several herbaceous plants, including Reynoutria
japonica Houtt, and Rheum officinale Baill, which have
been used in traditional medicines in Eastern and Southern Asian
countries (10). Studies in
previous decades have indicated the anti-inflammatory, anticancer
and antifibrotic effects of emodin (11–13).
A previously published study suggested the therapeutic effect of
emodin against airway inflammation and fibrosis in an asthma animal
model (14).
As a member of a conserved family of
NAD+-dependent deacetylases, which are considered to
execute multiple fundamental biological functions by deacetylating
the lysine residues of several nuclear factors to affect their
transcriptional regulatory activities (15,16).
It has been suggested that the activation of Sirt1 exerts a
negative regulatory effect on TGF-β1/Smad3 signaling (15). Evidence has confirmed that Sirt1
can deacetylate Smad3, and subsequently promote the ubiquitination
and eventual degradation of R-Smad proteins (16). Therefore, the downstream
fibrosis-inducing effect of TGF-β1 is inhibited.
Accordingly, the present hypothesized the possible
involvement of activation of TGF-β1/Smad signaling in pulmonary
fibrosis in silicosis. Whether Sirt1 was involved in the
antifibrotic effects of emodin on silica-induced lung fibrosis was
investigated in the present study. It is anticipated that the
results from the present study can improve current knowledge of
pulmonary fibrosis and provide a theoretical basis for further
application of emodin-containing drugs in the treatment of
silicosis.
Materials and methods
Preparation of silica particles and
emodin solution
The purity of the silica dust (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) was used in the present study was
>98%, with a particle diameter of 1–5 µm. The silica particles
were sterilized by being boiled at 120°C for 30 min and then washed
with sterilized water and dried. For intratracheal administration,
the particles were resuspened in sterile saline and sonicated for
10 min. The emodin (Sigma-Aldrich; Merck Millipore) was dissolved
in DMSO and adjusted to 0.1% (v/v).
Animals and treatments
Female C57BL/6 mice (SPF class; 6-week old; n=50; 10
mice/group) were provided by the Animal Experimental Center of
Xi'an Jiaotong University (Xi'an, China). The animals were raised
in an environment with an artificial 12/12 h day-night cycle, the
temperature controlled at (25±1)°C and the humidity controlled at
(65±4)%. All animals were maintained with standard food and fresh
water. All animal experimental procedures were approved by the
Experimental Animal Ethics Committee of Xi'an Jiaotong
University.
The mice were anesthetized by intraperitoneal (i.p.)
injection of pentobarbital sodium (2%; 20 mg/kg body weight). The
neck skin was dissected, following which the trachea was exposed by
blunt dissection. A 7-gauge needle was then inserted into the
trachea to initiate intratracheal administration. A 50 µl silica
particle suspension, containing 30 mg silica, was administrated to
induce lung fibrosis, then neck skin of the surgical site was
sutured and cleaned with ethanol and penicillin. The recovery
period was 4 weeks. Control animals received administration of an
equal volume of sterile saline. Following model establishment,
emodin solution was administrated i.p. once per day (20 mg/kg body
weight) for a continuous 7-day period. The Sirt1 inhibitor,
nicotinamide (Beyotime Institute of Biotechnology, Nantong, China),
which is considered to interrupt the deacetylase activity of Sirt1
(17), was administrated once per
day to the animals by i.p. injections (500 mg/kg body weight) for 7
days consecutively following model establishment. For control
animals, equal volumes of saline were administrated i.p.
Assessment of pulmonary function
Prior to sacrifice of the animals, the pulmonary
function was evaluated according to methods described in a previous
study (18). Briefly, the mice
were anesthetized by i.p. injection of pentobarbital sodium (2%; 20
mg/kg body weight), paralyzed via tail vein injection of
pancuronium bromide (5 mg/kg body weight) and then subjected to
mechanical ventilation (FlexiVent; SCIREQ Scientific Respiratory
Equipment, Inc., Montréal, QC, Canada). Respiratory frequency was
set as 100/min and tidal volume was set as 0.2 ml with a flow rate
of 1 ml/sec. The transpulmonary pressure was measured using
pressure transducers. Airway resistance, dynamic compliance and
elastance were calculated according to the occlusion method
described previously (18).
Subsequent to establishment of pulmonary function, the mice were
sacrificed by overdose of anesthesia with pentobarbital sodium.
Fluorescent staining of α-SMA and
collagen I in lung tissues
The lung tissue was harvested, trimmed and embedded
in OCT compound (Sakura Finetek USA, Inc., Torrance, CA, USA) at
−20°C for 20 min. The lung tissue was then cut into slices at a
thickness of 5 µm. The slices were incubated with antibodies
against α-SMA (ab5694; Abcam, Cambridge, MA, USA) and collagen I
(ab34710; Abcam). Following incubation with secondary antibodies
conjugated with Alexa 594 (A-11072; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and Alexa 488 (A27034;
Invitrogen; Thermo Fisher Scientific, Inc.), respectively,
fluorescent images were observed under a fluorescent microscope
(Nikon, Tokyo, Japan).
Determination of bronchoalveolar
lavage fluid (BALF) TGF-β1 levels
Following sacrifice of the mice, BALF was obtained
with the assistance of tracheal cannulation. The BALF was
centrifuged at 200 x g for 10 min at 4°C, following which
the supernatant was collected. The level of TGF-β1 was determined
using an using enzyme-linked immunosorbent assay kit (R&D
Systems, Inc., Minneapolis, MN, USA) according to the
manufacturer's protocol.
Resin preparation
CNBr-activated Sepharose™ 4B (GE Healthcare,
Piscataway, NJ, USA) was used following preparation with HCl (1
mmol/l) and washing in coupling buffer (0.1 mmol/l HaHCO3 and 0.5
mmol/l NaCl). For the emodin- loaded affinity column, emodin was
dissolved in DMSO (5 mmol/l) and then mixed into the resin at a
ratio of 1:10 emodin:resin. The resin was then rotated for 4 h at
room temperature and washed with deionized water. Any possible
remaining active groups were inhibited using capping solution (1
mmol/l ethanolamine) for 2 h at room temperature. Sirt1 protein (15
µl at a concentration of 10 mg/ml) was added into the resin and
then incubated at 4°C for 8 h. Following incubation, the excess
protein was washed, and the resin was added to loading buffer
(Beyotime Insitute of Biotechnology) and boiled. The mixture was
then subjected to SDS-PAGE and assessed using western blot
analysis. For the Sirt1-loaded affinity column, Sirt1 protein (10
mg/ml; Sigma-Aldrich; Merck Millipore) was added to the resin and
then rotated at 4°C for 8 h, followed by washing with capping
solution at room temperature for 2 h. The resin was then treated
with emodin solution for 4 h at room temperature. Following washing
with high pH buffer (0.1 mmol/l Tris-HCl and 0.5 mmol/l NaCl; pH 8)
and low pH buffer (0.1 mmol/l AcOH and 0.5 mmol/l NaCl; pH=4) three
times, a Vivapure C18 spin column (Sartorius AG, Göttingen,
Germany) was used to accomplish the desalt process prior to liquid
chromatography-mass spectrometry (LC/MS) analysis.
LC/MS analysis
A BioBasic Picofrit C18 capillary column (New
Objective, Inc., Woburn, MA, USA) was used to separate the
peptides. Using an acetonitrile gradient (0–100%) with a flow rate
of 1 ml/min for 1 h, the elution was prepared. The 6410B Triple
Quadrupole LC/MS (Agilent Technologies, Inc., Santa Clara, CA, USA)
system was used for LC/MS in the present study according to the
manufacturer's protocol.
Western blot analysis
Activation of the TGF-β1/Smad signaling pathway was
assessed in the present study using western blot analysis. The
pre-cleaned lung tissue was homogenized in RIPA buffer with PMSF.
Following centrifugation at 14,000 × g at 4°C, the
supernatants were used for western blot analysis. The protein
concentration was determined using a BCA protein assay kit (Santa
Cruz Biotechnology, Inc., Dallas, TX, USA). The proteins (50 µg)
were then separated by SDS PAGE (4% stacking gel and 10% separation
gel) vertically and semi-dry transferred onto polyvinylidene
fluoride membranes. Antibodies against TGF-β1 (ab64715; Abcam),
Smad3 (A27034; Cell Signaling Technology, Inc., Danvers, MA, USA),
acetylated (Ac)-Smad3 (#9513; Cell Signaling Technology, Inc,),
Sirt1 (#8469; Cell Signaling Technology, Inc.), α-SMA (Abcam),
collagen I (Abcam) and GAPDH (#MA1-16757; Invitrogen; Thermo Fisher
Scientific, Inc.) were used to incubate the membranes. Rabbit
anti-mouse (sc-358943), bovine anti-goat (sc-2384) and bovine
anti-rabbit (sc-2385) secondary antibodies conjugated to
horseradish peroxidase (Santa Cruz Biotechnology, Inc.) were used
to incubate the membranes. Subsequently, Super Signal West Pico
chemiluminescence reagent (Thermo Fisher Scientific, Inc.) was used
to develop the membranes and the immunoblots were visualized on
X-ray films.
Statistical analysis
The results in the present study are presented as
the mean ± standard deviation and were analyzed using SPSS software
(version 16.0; SPSS, Inc., Chicago, IL, USA). Differences between
values were evaluated using one-way analysis of variance or
Student's-t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Silica exposure induces lung fibrosis
and impairs pulmonary functions
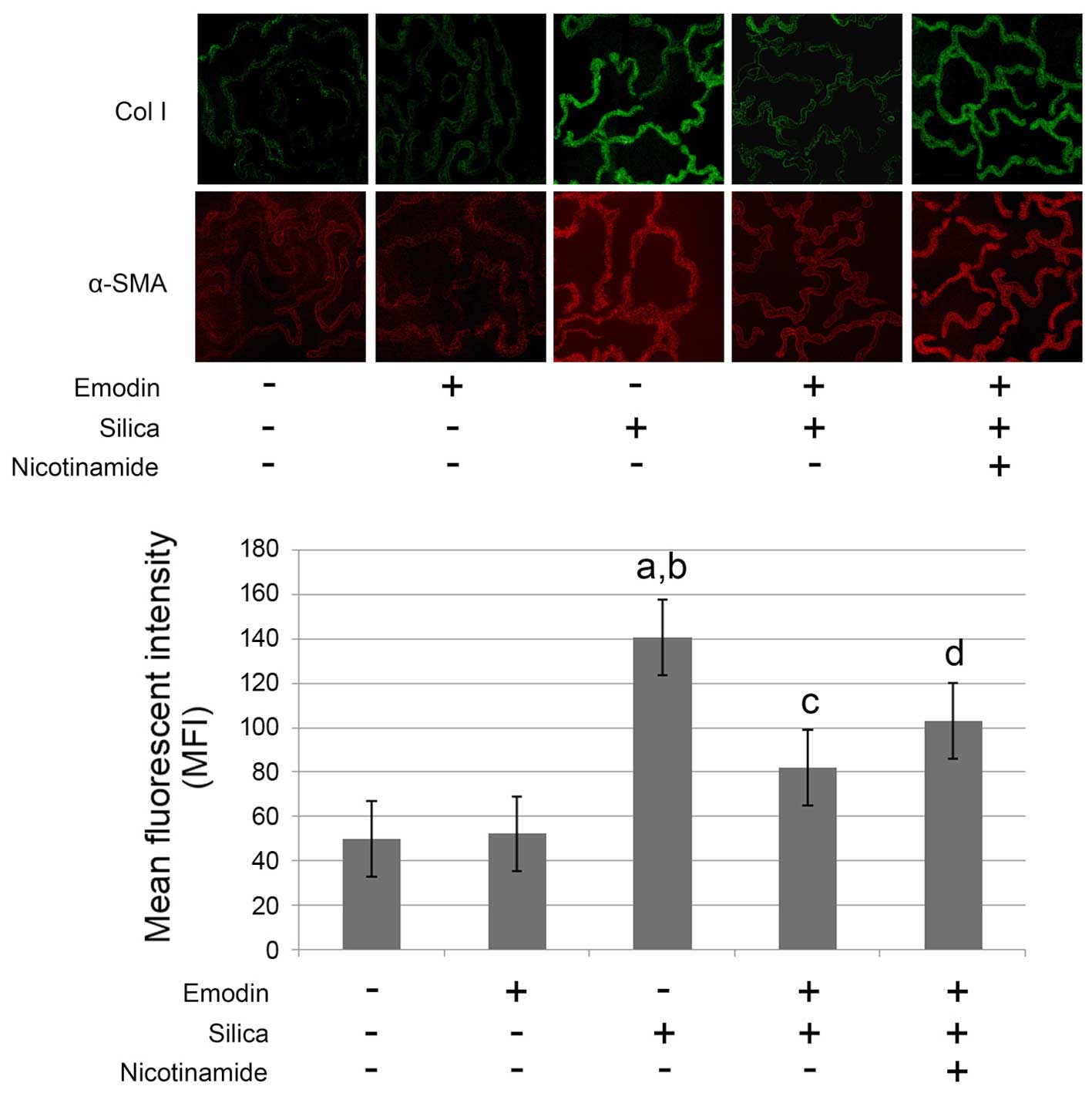
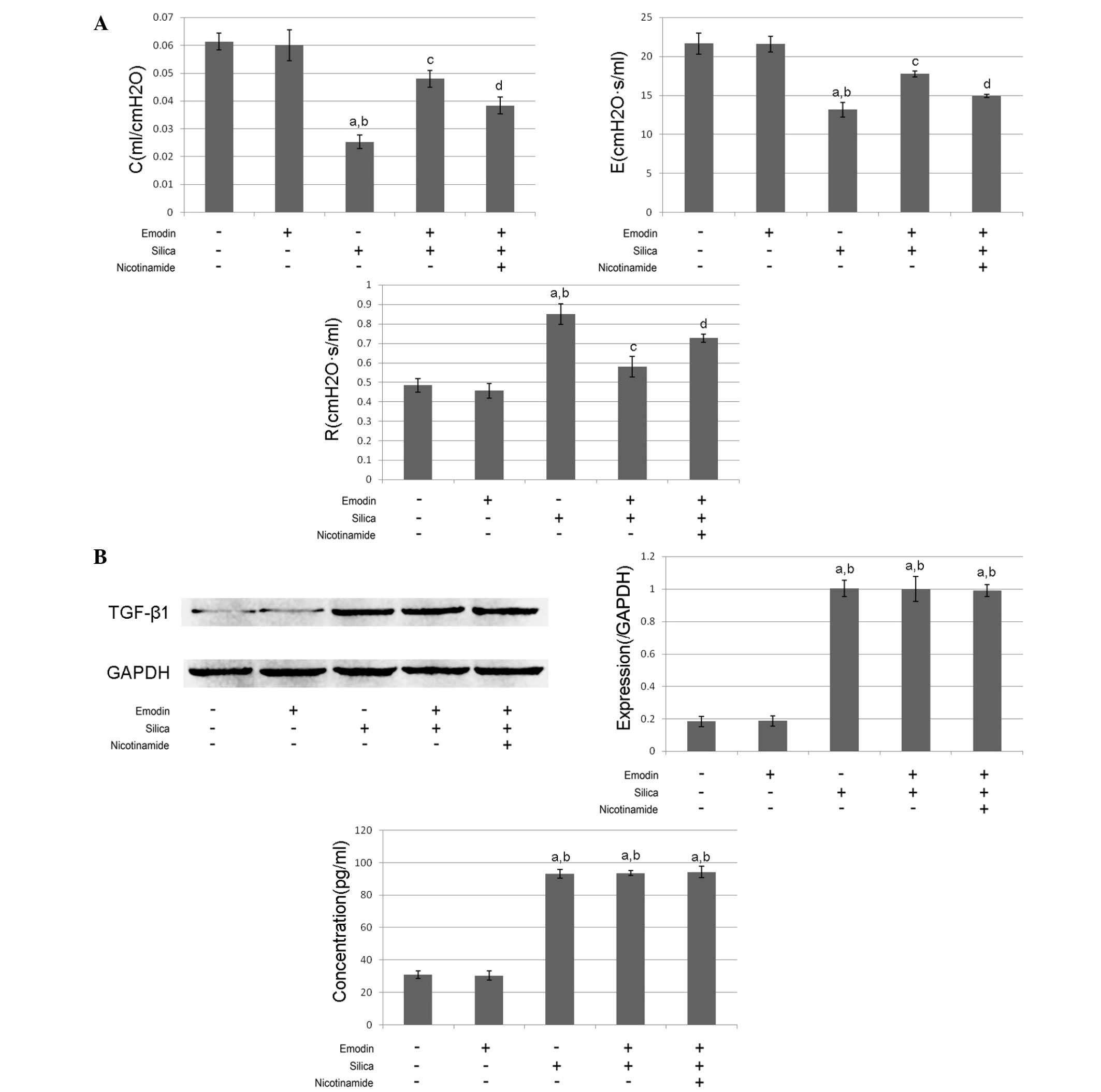
As shown in Figs. 1
and 2, compared with the control
animals, marked lung fibrosis was observed following exposure of
the lungs to silica. The lung fibrosis was evidenced by increased
α-SMA and collagen I deposition in the lung tissues (Fig. 1) Furthermore, the pulmonary
function was impaired following the induction of lung fibrosis. The
airway resistance was increased, whereas the dynamic compliance and
elastance were reduced (Fig. 2A).
The levels of TGF-β1 in the BALF and the lung tissues were markedly
increased, compared with the control group (Fig.2B).
Emodin inhibits silica-induced lung
fibrosis and improves pulmonary functions without affecting levels
of TGF-β1 in the BALF and lung tissue
As shown in Figs. 1
and 2, i.p. treatment with emodin
significantly inhibited lung fibrosis and attenuated the pulmonary
functions of the animal models. Furthermore, the administration of
emodin did not affect the levels of TGF-β1 in the BALF or lung
tissue.
Nicotinamide treatment impairs the
effects of emodin on lung fibrosis and pulmonary function without
affecting levels of TGF-β1
The results of the effects of nicotinamide are also
shown in Figs. 1 and 2. The mice received i.p. treatment with
emodin and nicotinamide following silica inhalation. Compared with
the silica-inhaled mice treated with emodin, nicotinamde
administration significantly impaired the effects of emodin on
inhibiting lung fibrosis and improving pulmonary functions.
However, neither emodin nor nicotinamide affected the levels of
TGF-β1 in the BALF or lung tissue.
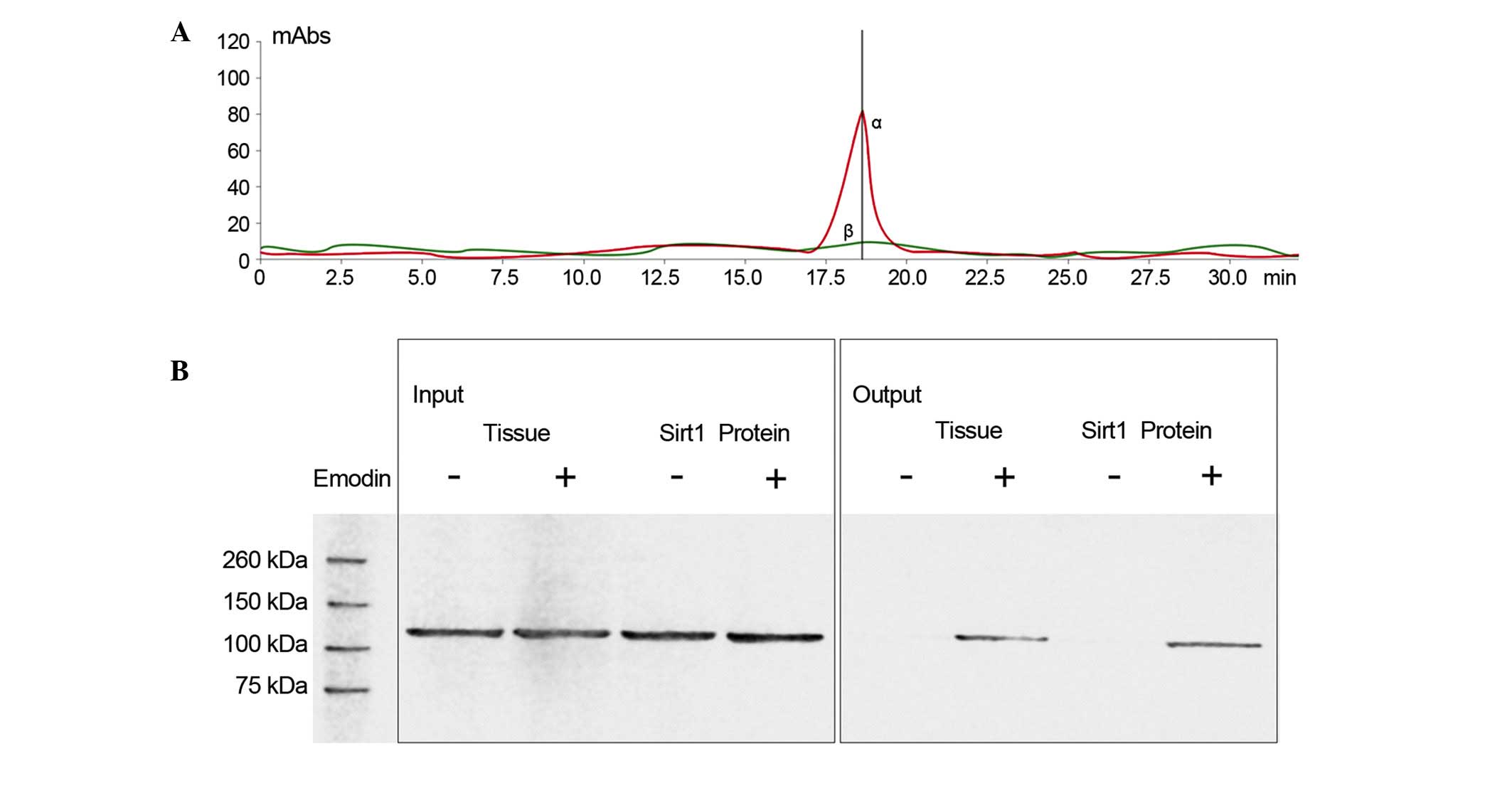
Emodin interacts with Sirt1 by direct
contact
As shown in Fig. 3,
LC/MS analysis confirmed that emodin was able to bind to Sirt1.
Also shown in Fig. 3, following
incubation of the emodin-affinity columns with Sirt1 protein, the
results of the western blot analysis suggested that Sirt1 was in
contact with emodin.
Emodin elevates the acetylation of
Smad3 by increasing the expression of Sirt1
The immunoblots of Sirt1, Smad3, Ac-Smad3, collagen
I and GAPDH are shown in Fig. 4.
Compared with the control animals, silica inhalation markedly
increased Ac-Smad3/Smad3, which subsequently elevated the level of
collagen I in the lung tissues of the model animals. Emodin
administration increased the expression of Sirt1 and decreased
Ac-Smad3/Smad3. However, treatment with the Sirt1 inhibitor,
nicotinamide, impaired the effects of emodin on decreasing
Ac-Smad3/Smad3 and elevating Sirt1. As a result, nicotinamide
undermined the effect of emodin on decreasing the level of collagen
I.
 | Figure 4.Immunoblots of Sirt1, Ac-Smad3, Smad3,
Col I and GAPDH in harvested lungs from mice with silica
inhalation-induced lung fibrosis treated with emodin and/or
nicotinamide. Graphs show the relative expression levels of Sirt1,
Ac-Smad3 and Col I. aP<0.05, compared with untreated
control; bP<0.05, compared with emodin-treated group;
cP<0.05, compared with the silica-exposed group;
dP<0.05, compared with the emodin-treated
silica-exposed group. Sirt1, surtuin 1; Ac-Smad3, acetylated small
mothers against decapentaplegic 3; Col, collagen I. |
Discussion
In the present study, an animal model of pulmonary
silicosis was induced by intratracheal administration of silica
particles. Lung fibrosis was clearly identified in these animals.
As a result, pulmonary functions were impaired in the mice with
silica-induced lung fibrosis, and the present study provided
further insight into the possible mechanism underlying
silica-induced lung fibrosis. It was found that the TGF-β1/Smad3
signaling pathway was activated in silica-induced lung fibrosis.
The present study also examined the antifibrotic activity of emodin
in silica-induced lung fibrosis. Investigation of the mechanism
revealed that, without affecting the expression level of TGF-β1 in
the lungs, emodin downregulated Smad-induced lung fibrosis by
promoting Sirt1 signaling via direct binding, which caused the
deacetylation of R-Smads to inhibit ECM synthesis and
deposition.
Characterized by impaired pulmonary function and
hypoxemia, silicosis is considered to be a common occupational
disease (19). The chronic
inhalation of silicon dioxide particles, which is also referred as
silica, is suggested to be the pathogenic factor of lung silicosis.
Pathologically, chronic lung inflammation and fibrosis are accepted
as the typical characteristics of pulmonary silicosis (20), and the impairment of pulmonary
function is initiated and expedited by lung fibrosis (21). In the present study, lung fibrosis
was identified following exposure of the lungs to silica particles.
Characterized by increased airway resistance, reduced dynamic
compliance and elastance, the pulmonary function was impaired,
accompanied by the lung fibrosis.
During fibrosis, immune cells are activated and
several types of inflammatory cytokines, including interferon-γ,
tumor necrosis factor-α, ILs and TGF-β are released (22). It is generally accepted that TGF-β
is one of the most potent initiators of fibrosis by inducing
fibroblasts to synthesize ECM, including collagen, laminin and
fibronectin (23). On binding to
its receptors, the formed complex recruits and triggers the
phosphorylation of R-Smads, which further target gene-encoding ECM
proteins as transcription factors (24). In the present study, the expression
levels of TGF-β1 were found to be elevated in the BALF and lung
tissue. This result indicated that silica exposure promoted the
synthesis and release of TGF-β1 from immune cells accumulated in
the airway and lung tissues. Previously, it was documented that
Smad3 is one of the underlying mechanisms promoting fibrogenesis in
response to multiple fibrogenic initiators, including angiotensin
II, advanced glycation end products and TGF-β (25,26).
With the exception of the phosphorylation of R-Smads, accumulating
evidence suggests that the acetylation of R-Smads is also a
critical signal, which induces the production and deposition of
ECM, and can be induced by TGF-β1 (15). In the present study, high levels of
R-Smad acetylation were found, which led to lung fibrosis,
characterized by the deposition of ECM in the lung tissue.
Natural agents of herbal origin have attracted
substantial attention in previous decades due to their multiple
biological activities. A number of these agents, including
curcumin, emodin and matrine, have been shown to exert antifibrotic
activity (27,28). Thus, these agents are of potential
therapeutic value in the treatment of fibrosis of pulmonary
silicosis. In the present study, the administration of emodin
resulted in lung fibrosis being relieved, and the pulmonary
function was improved in the animals with lung fibrosis. Further
investigation showed that there was molecular binding between
emodin and Sirt1. These results indicated that emodin may have a
regulatory effect on Sirt1.
In the present study, it was found that, in mice
with silica-induced lung fibrosis, emodin treatment not only
increased the expression level of Sirt1 in lung tissues, but also
significantly promoted the deacetylation of R-Smads in the lung
tissue without affecting the levels of TGF-β1 in the lung tissue or
BALF. Furthermore, treatment with the Sirt1 inhibitor,
nicotinamide, suppressed the emodin-induced deacetylation of
R-Smads. In conclusion, emodin showed significant antifibrotic
activity in inhibiting lung fibrosis in pulmonary silicosis,
improving pulmonary function. Mechanically, the present study
provided evidence confirming that emodin attenuated the
above-mentioned lung fibrosis and improve pulmonary functions by
activating Sirt1 signaling to deacetylate R-Smad-induced ECM
synthesis and deposition.
References
|
1
|
Iossifova Y, Bailey R, Wood J and Kreiss
K: Concurrent silicosis and pulmonary mycosis at death. Emerg
Infect Dis. 16:318–320. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hayes D Jr, Hayes KT, Hayes HC and Tobias
JD: Long-term survival after lung transplantation in patients with
silicosis and other occupational lung disease. Lung. 193:927–931.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leung CC, Yu IT and Chen W: Silicosis.
Lancet. 379:2008–2018. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song J, Rong Y, Cui X and Chen W: Advances
in research on the role of Gas 6/TAMin inflammation response and
silicosis induced by silica dusts. Zhonghua Lao Dong Wei Sheng Zhi
Ye Bing Za Zhi. 32:715–718. 2014.(In Chinese). PubMed/NCBI
|
|
5
|
Gera K, Pilaniya V and Shah A: Silicosis:
Progressive massive fibrosis with eggshell calcification. BMJ Case
Rep. 2014:bcr20142063762014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weng ZP, Zhang JJ, Liu WW, Chen J, Liu YM,
Yu W, Tang LJ, Chen JY, Fang M, Zhang C, et al: The experimental
study of suppressing silicosis fibrosis. Zhonghua Lao Dong Wei
Sheng Zhi Ye Bing Za Zhi. 29:740–745. 2011.(In Chinese). PubMed/NCBI
|
|
7
|
Beamer CA, Migliaccio CT, Jessop F,
Trapkus M, Yuan D and Holian A: Innate immune processes are
sufficient for driving silicosis in mice. J Leukoc Biol.
88:547–557. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davis GS, Pfeiffer LM, Leslie KE and
Hemenway DR: Macrophage-lymphocyte cytokine interactions in
silicosis. Chest. 109(3): Suppl. 49S–50S. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Allison SJ: Fibrosis: Regulation of
fibrotic signalling by TGF-β receptor tyrosine phosphorylation. Nat
Rev Nephrol. 10:4842014. View Article : Google Scholar
|
|
10
|
Gao ZQ and Wang CH: Emodin and organ
fibrosis. Zhongguo Zhong Xi Yi Jie He Za Zhi. 25:1030–1032.
2005.(In Chinese). PubMed/NCBI
|
|
11
|
Chen XH, Sun RS, Hu JM, Mo ZY, Yang ZF,
Jin GY, Guan WD and Zhong NS: Inhibitory effect of emodin on
bleomycin-induced pulmonary fibrosis in mice. Clin Exp Pharmacol
Physiol. 36:146–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu C: Inhibition of mechanical
stress-induced hypertrophic scar inflammation by emodin. Mol Med
Rep. 11:4087–4092. 2015.PubMed/NCBI
|
|
13
|
Pooja T and Karunagaran D: Emodin
suppresses Wnt signaling in human colorectal cancer cells SW480 and
SW620. Eur J Pharmacol. 742:55–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang T, Zhong XG, Li YH, Jia X, Zhang SJ,
Gao YS, Liu M and Wu RH: Protective effect of emodin against airway
inflammation in the ovalbumin-induced mouse model. Chin J Integr
Med. 21:431–437. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang XZ, Wen D, Zhang M, Xie Q, Ma L,
Guan Y, Ren Y, Chen J and Hao CM: Sirt1 activation ameliorates
renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J Cell
Biochem. 115:996–1005. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zerr P, Palumbo-Zerr K, Huang J, Tomcik M,
Sumova B, Distler O, Schett G and Distler JH: Sirt1 regulates
canonical TGF-β signalling to control fibroblast activation and
tissue fibrosis. Ann Rheum Dis. 75:226–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang T, Cui H, Ma N and Jiang Y:
Nicotinamide-mediated inhibition of SIRT1 deacetylase is associated
with the viability of cancer cells exposed to antitumor agents and
apoptosis. Oncol Lett. 6:600–604. 2013.PubMed/NCBI
|
|
18
|
Santos-Silva MA, Pires KM, Trajano ET,
Martins V, Nesi RT, Benjamin CF, Caetano MS, Sternberg C, Machado
MN, Zin WA, et al: Redox imbalance and pulmonary function in
bleomycin-induced fibrosis in C57BL/6, DBA/2 and BALB/c mice.
Toxicol Pathol. 40:731–741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Subra JF, Renier G, Reboul P, Tollis F,
Boivinet R, Schwartz P and Chevailler A: Lymphopenia in
occupational pulmonary silicosis with or without autoimmune
disease. Clin Exp Immunol. 126:540–544. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Connell M and Kennedy M: Progressive
massive fibrosis secondary to pulmonary silicosis appearance on
F-18 fluorodeoxyglucose PET/CT. Clin Nucl Med. 29:754–755. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kitaguchi Y, Fujimoto K, Hanaoka M, Honda
T, Hotta J and Hirayama J: Pulmonary function impairment in
patients with combined pulmonary fibrosis and emphysema with and
without airflow obstruction. Int J Chron Obstruct Pulmon Dis.
9:805–811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Borthwick LA, Wynn TA and Fisher AJ:
Cytokine mediated tissue fibrosis. Biochim Biophys Acta.
1832:1049–1060. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tatler AL and Jenkins G: TGF-β activation
and lung fibrosis. Proc Am Thorac Soc. 9:130–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cutroneo KR: TGF-beta-induced fibrosis and
SMAD signaling: Oligo decoys as natural therapeutics for inhibition
of tissue fibrosis and scarring. Wound Repair Regen. 15:(Suppl 1).
S54–S60. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Roberts AB, Tian F, Byfield SD, Stuelten
C, Ooshima A, Saika S and Flanders KC: Smad3 is key to
TGF-beta-mediated epithelial-to-mesenchymal transition, fibrosis,
tumor suppression and metastasis. Cytokine Growth Factor Rev.
17:19–27. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gauldie J, Bonniaud P, Sime P, Ask K and
Kolb M: TGF-beta, Smad3 and the process of progressive fibrosis.
Biochem Soc Trans. 35:661–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu JL, Li JH, Chengz RG, Ma YM, Wang XJ
and Liu JC: Effect of matrine on transforming growth factor β1 and
hepatocyte growth factor in rat liver fibrosis model. Asian Pac J
Trop Med. 7:390–393. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen N, Geng Q, Zheng J, He S, Huo X and
Sun X: Suppression of the TGF-β/Smad signaling pathway and
inhibition of hepatic stellate cell proliferation play a role in
the hepatoprotective effects of curcumin against alcohol-induced
hepatic fibrosis. Int J Mol Med. 34:1110–1116. 2014.PubMed/NCBI
|