Introduction
Monocytes and macrophages are critical mediators of
rheumatoid arthritis (RA) (1,2).
High numbers of macrophages are present in inflamed tissues,
particularly at the cartilage-pannus interface in human RA, and
have been reported to be correlated with RA severity (3,4). In
addition, inhibiting macrophage migration to sites of inflammation
may markedly reduce inflammation and tissue destruction (5). Therefore, inhibiting the migration of
macrophages is considered a potential key strategy for the
treatment of RA. However, macrophage migration is a continuous and
complex process, which includes rolling of monocytes along the
endothelium, firm adhesion to the endothelium, and transendothelial
cell migration. Within macrophage migration, chemotaxis and
adhesion are considered the most important processes. Monocytes
traverse the endothelium in order to enter developing and
established lesions; this trafficking is directed by chemokines,
such as macrophage inflammatory protein (MIP)-1α, monocyte
chemoattractant protein (MCP)-1 and interleukin (IL)-8. C-C
chemokine receptor (CCR)1, CCR2 and CXC chemokine receptor 2 are
considered classical chemokine receptors, which have been proposed
to affect monocyte recruitment in mouse models of arthritis
(6–8). In addition, the firm adhesion of
monocytes is mediated via the endothelial expression of members of
the immunoglobulin superfamily, such as intercellular adhesion
molecule (ICAM)-1, and their counterligands, which are expressed by
leukocytes, including cluster of differentiation (CD)11b/CD18.
The cholinergic anti-inflammatory pathway (CAP),
which transmits information from the brain to the peripheral immune
system via the parasympathetic nervous system, was initially
described in 2000 (9). Cholinergic
stimulation, by vagus nerve electrical stimulation or treatment
with selective cholinergic agonists, suppresses the production of
cytokines in preclinical models of systemic inflammation, including
endotoxemia, hemorrhagic shock, ischemia-reperfusion injury and
acute lung injury (9–11). Macrophages express alpha-7
nicotinic acetylcholine receptor, pharmacological stimulation of
which reduces the production of inflammatory cytokines by
macrophages (12,13). In previous experiments by authors
of the present study, and others, the CAP has been revealed to
exert a protective effect on RA (14–16);
the underlying mechanism may be associated with the inhibition of T
helper (Th)17 cell responses and may improve the Th1/Th2 imbalance
in collagen-induced arthritis (CIA). However, the specific
mechanisms remain unclear.
The present study established that reduced numbers
of macrophages are present in synovial tissues following activation
of cholinergic signaling, since synovial tissues are not innervated
by the vagus nerve. The present study aimed to elaborate the
protective effects of the CAP on RA and to elucidate its cellular
molecular mechanisms, thus providing an experimental basis
regarding how the CAP regulates inflammatory and immune responses
in RA.
Materials and methods
Mice
A total of 56 male DBA/1 mice (weight, 20 g; age,
6–8 weeks; 24 mice died prematurely whilst the model was
established) were purchased from the SJA Laboratory Animal Co.
(Shanghai, China) for use in the CIA studies. The mice were housed
under specific-pathogen-free conditions at 24–28°C, 40–70%
humidity, 12 h light/dark cycle and free access to food and water.
The animal experiments were performed in accordance with the
institutional guidelines for animal care, and were approved by the
committee for the use and care of animals at Central South
University (Changsha, China).
Experimental groups
The mice were randomly divided into four groups
(n=8): Control group [sham vagotomy + phosphate-buffered saline
(PBS)], model group (sham vagotomy + CIA + PBS), vagotomy group
(vagotomy + CIA + PBS), and nicotine group (sham vagotomy + CIA +
nicotine). To inhibit the CAP, mice in the vagotomy group were
subjected to left-side cervical vagotomy 4 days prior to CIA
induction (14). In sham-operated
mice, the left vagus nerve was exposed and isolated from the
surrounding tissue but was not transected. In the nicotine group,
the peripheral segment of the CAP was stimulated by intraperitoneal
pretreatment with nicotine daily (250 µg/kg; Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany), beginning 4 days prior to CIA
induction. The other groups were injected with PBS as a
control.
Induction and evaluation of CIA
CIA was induced in the DBA/1 mice as previously
described (14). Bovine type II
collagen (2 mg/ml; Chondrex, Inc., Redmond, WA, USA) was emulsified
in an equal volume of Freund's complete adjuvant (containing 4
mg/ml Mycobacterium tuberculosis; Chondrex, Inc.) at 4°C by
magnetic stirring until fully emulsified. The final concentration
of collagen was 1 mg/ml. All male DBA/1 mice, with the exception of
the control mice, were initially immunized intracutaneously at the
base of the tail with 0.1 ml of this emulsion (100 µg collagen). On
day 21, the mice were administered booster injections of 0.1 ml
emulsion in Freund's incomplete adjuvant (Chondrex, Inc.) in the
same manner.
Clinical signs of arthritis in the joints were
visually assessed every 3 days by two observers. All animals were
regularly examined for signs of arthritis, and the disease severity
for each paw was graded on a scale between 0 and 4, according to
the levels of erythema, swelling and induration. Arthritis index
score criteria were as follows: 0, no redness or swelling of the
joints; 1, red or slight swelling; 2, moderate swelling; 3, severe
swelling; and 4, severe swelling and unloadable. Assessment of
incidence and macroscopic score were carried out by two independent
observers, without knowledge of the experimental groups. The total
arthritic score per mouse was derived from the sum of the
individual scores of four paws. Two independent observers without
knowledge of the experimental groups assessed the incidence and
macroscopic score.
Immunohistochemical evaluation of
CIA
The mice were sacrificed by cervical dislocation on
day 42, and their hind paws were collected. The joint tissues were
fixed in 4% paraformaldehyde overnight at room temperature,
decalcified in 13% ethylenediamine tetra-acetic acid for 2–3 weeks
and embedded in paraffin and cut into 5 µm sections. The sections
(2.5×2.5 cm) were then dewaxed using xylene, dehydrated using an
alcohol gradient, and were stained with hematoxylin and eosin
(H&E) for 1 min at room temperature for histological
assessment. Arthritis severity in histological samples was
determined by cumulative assessment of synovial inflammation. The
scoring was: 0, normal; 1, minimal; 2, mild; 3, moderate; 4,
marked; and 5, severe. This was completed by two independent
observed, without knowledge of the experimental groups.
Histopathological sections of the hind paws were examined for
hyperplasia of the synovial membrane, infiltration with mononuclear
cells, and cartilage and bone damage (14).
For immunofluorescence detection of macrophage
infiltration, sections were dewaxed and dehydrated as
aforementioned. Slides were retrieved using a heat-mediated
retrieval method, and endogenous peroxidase activity was quenched
with 3% H2O2 for 5 min at room temperature.
The sections were then blocked with 3% bovine serum albumin
(Sijiqing, Hanzhou, China) for 1 h at room temperature and were
incubated overnight at 4°C with an anti-CD11b primary antibody
(1:200; cat. no. 101213; BioLegend, San Diego, CA, USA).
Subsequently, the sections were incubated with Alexa Fluor
555-conjugated goat anti-rat immunoglobulin G (IgG; cat. no.
A-21434; 1:150; Molecular Probes; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) for 1 h at room temperature. The number of Alexa
Fluor 555-labeled CD11b-positive macrophages was counted under a
confocal microscope (LSM780; Carl Zeiss Jena GmbH, Jena, Germany)
using a ×63 oil immersion objective by a blinded observer.
For staining of CCR2 and ICAM-1, the sections were
pretreated with proteinase K for CCR2 retrieval for 20 min at 37°C,
and were then treated with citric buffer (pH 6) for heat-mediated
antigen retrieval of ICAM-1 at 80°C for 20 min. Sections were
incubated with rabbit anti-mouse CCR2 (1:200; cat. no. ab32144;
Abcam, Cambridge, MA, USA), rabbit anti-mouse ICAM-1 monoclonal
antibody (1:400; cat. no. ab124759; Abcam) or normal rabbit IgG
(1:200; cat. no. A7016; Biyuntian, Shanghai, China) at 4°C
overnight. Subsequently, the samples were washed three times for 5
min in PBS and were incubated with biotin-conjugated goat
anti-rabbit IgG (ABC kit; cat. no. PK-6100; Vector Laboratories,
Burlingame, CA, USA) for 2 h at room temperature. After three 5-min
washes in PBS, the sections were incubated with an
avidin-biotinylated enzyme complex (ABC kit; Vector Laboratories)
for 2 h at room temperature. Positive signals were visualized using
a diaminobenzidine kit (Vector Laboratories), the sections were
counterstained with hematoxylin, images were captured using an
Olympus microscope (IMT-2, Olympus Corporation, Tokyo, Japan) and
were analyzed using Image-Pro Plus 6.0 software (Media Cybernetics,
Inc., Rockville, MD, USA).
Chemokine quantification by ELISA
On experimental day 42, the mice were sacrificed,
and serum samples were harvested. Whole blood samples (0.8–1 ml
each) were centrifuged at 12,000 × g for 20 min at 4°C. The
serum levels of MIP-1α (cat. no. MMA00; R&D Systems, Inc.,
Minneapolis, MN, USA), MCP-1 (cat. no. 81-BMS6005; eBioscience,
Inc., San Diego, CA, USA) and MIP-2 (cat. no 70-EK21422;
Multiscience Biotech, Co., Ltd., Hangzhou, China) were measured
using ELISA kits, according to the manufacturers' protocols.
Isolation of mononuclear cells and
flow cytometry
Spleen samples were obtained from the sacrificed
mice. Splenocytes were separated from the spleen using
lymphocyte-separation medium (Histopaque®-1119;
Sigma-Aldrich; Merck Millipore) after filtering the samples with a
cell strainer (BD Biosciences, Franklin Lakes, NJ, USA). Briefly,
the cells were incubated for 1 h at 4°C with
phycoerythrin-conjugated anti-CD11b (eBioscience, Inc.) or the
respective isotype control (cat. no. 12-4031; eBioscience, Inc.).
The cells were then washed with PBS and resuspended in 100 µl PBS
for flow cytometric analysis (BD FACS Canto II; BD Biosciences).
The data were analyzed using FlowJo software version 9.9.4 (FlowJo,
LLC, Ashland, OR, USA).
Statistical analysis
Data were analyzed using GraphPad Prism software
version 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) and are
presented as the mean ± standard deviation. The experiments were
repeated twice. The significance of differences between groups was
determined by one-way analysis of variance followed by a multiple
comparisons test (Student-Newman-Keuls). The Kruskal-Wallis test
was used to determine the heterogeneity of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
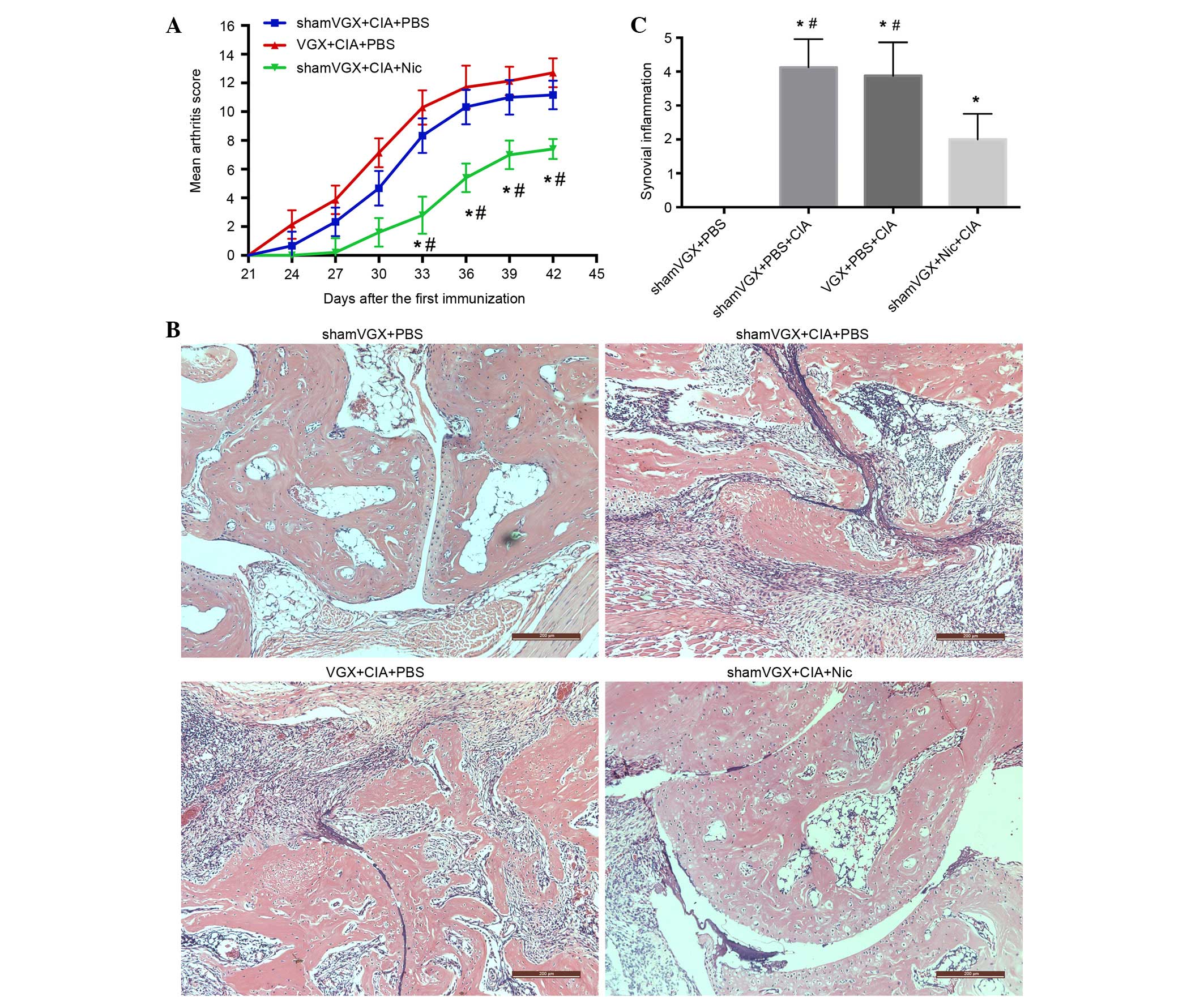
Nicotine attenuates the clinical and
histopathological changes associated with CIA
The CIA model was induced as aforementioned. The
arthritis index score was recorded by two researchers every 3 days
after the second immunization until the mice were sacrificed.
Slight swelling could be observed in the vagotomy group beginning
at day 24, and reaching a peak at day 42. The model group began to
present with arthritic symptoms at day 27, with symptoms peaking
after 42 days. Arthritis developed slowly and was milder in the
nicotine group (Fig. 1A). The
histopathological features of the four groups were assessed by
H&E staining. The results were consistent with the
aforementioned findings, in that the histopathological changes in
the nicotine group were reduced, as detected by decreased synovial
proliferation, inflammatory cell infiltration and bone destruction
(Fig. 1B). Semi-quantitative
analysis of the histopathological features is presented in Fig. 1C.
Nicotine reduces macrophage
infiltration in the synovium and spleen of CIA mice
Since the present study suggested that nicotine
attenuates clinical arthritis and restrains cytokine production,
such as TNF-α in our previous study (14), it was determined whether nicotine
can modulate macrophage infiltration in the synovium and spleen.
Macrophage infiltration serves a critical role in modulating the
immune responses. In mice, the CD11b antigen, as a marker of
monocytes/macrophages, is highly expressed on monocytes,
macrophages, and to a lesser extent, on granulocytes and a subset
of dendritic cells. The present study detected the distribution of
CD11b-positive macrophages in the synovium and spleen.
Immunofluorescence analysis detected an increase in infiltrated
CD11b-positive macrophages in the synovium of the model, vagotomy
and nicotine groups, as compared with in the control group.
However, among the model, vagotomy and nicotine groups, the
percentage of CD11b-positive macrophages was lowest in the nicotine
group (Fig. 2A and B). In
addition, the percentage of CD11b-positive cells was detected among
the splenic mononuclear cells in the four groups, using flow
cytometry (Fig. 2C and D). In the
model group, the percentage of CD11b-positive cells was increased
compared with in the control group. Treatment with nicotine
significantly reduced the percentage of CD11b-positive cells in the
spleen, as compared with in the model and vagotomy groups.
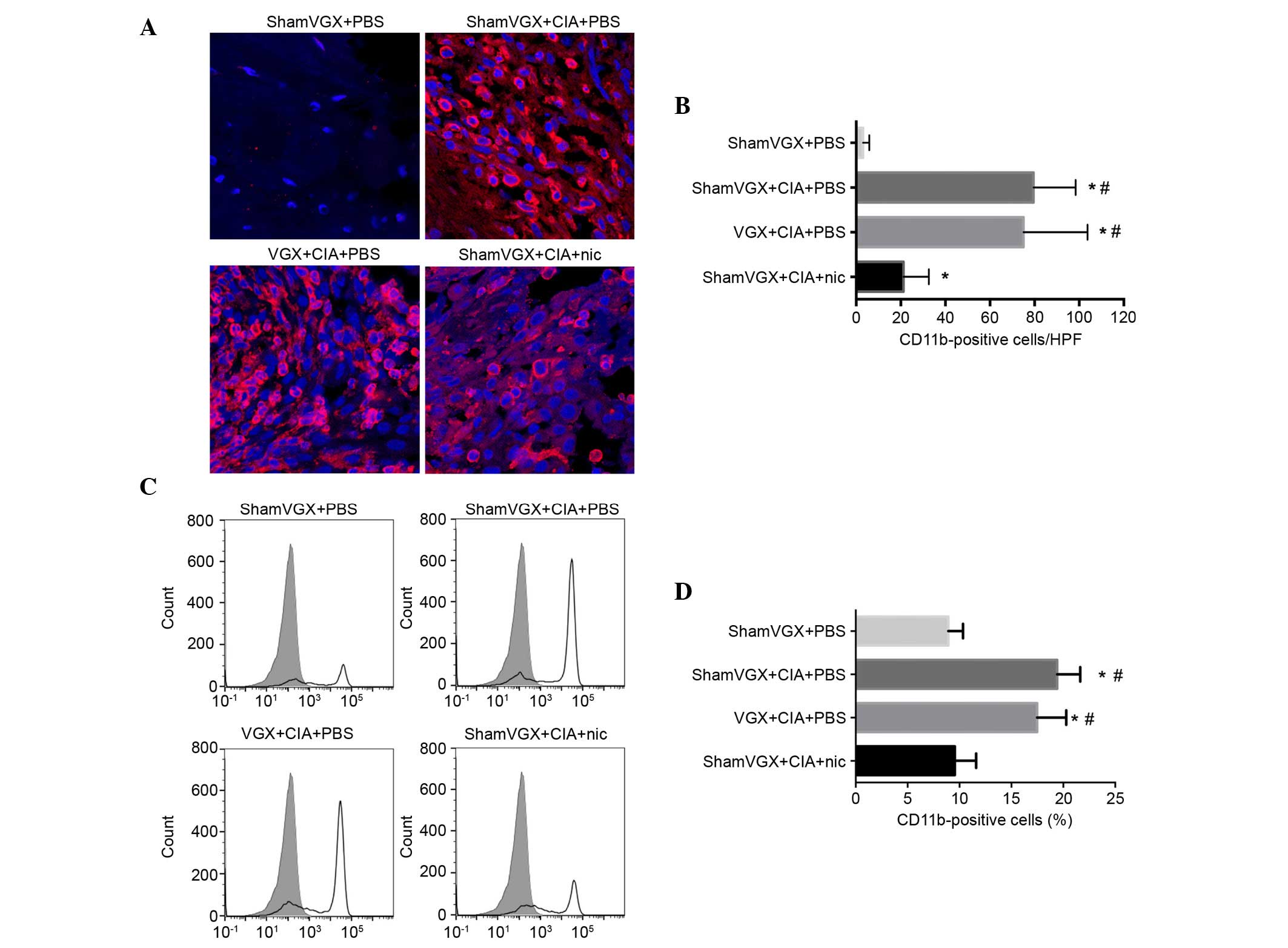 | Figure 2.Nic reduced the migration of
CD11b-positive macrophages into the synovium and spleen. After 42
days, the mice were sacrificed, and their ankle joints and spleen
tissues were harvested and examined for migrated macrophages. (A)
Immunofluorescence staining was used to detect macrophages in the
ankle joints of the mice. CD11b-positive macrophages were labeled
with Alexa Fluor 555. (B) Average number of CD11b-positive cells in
the synovial sections from the four groups. Data are presented as
the mean ± standard deviation (n=8 mice/group). (C) Single cells
were separated from the spleen cells of the mice, labeled with PE,
and detected by flow cytometry. Solid lines indicate staining with
PE-labeled anti-CD11b antibodies; shaded histograms indicate
staining with respective isotype antibodies. (D) Percentage of
CD11b-positive cells. Data are presented as the mean ± standard
deviation (n=8 mice/group). *P<0.05 vs. the shamVGX + PBS group;
#P<0.05 vs. the shamVGX + CIA + Nic group. CIA,
collagen-induced arthritis; VGX, vagotomy; Nic, nicotine; PBS,
phosphate-buffered saline; CD11b, cluster of differentiation 11b;
PE, phycoerythrin. |
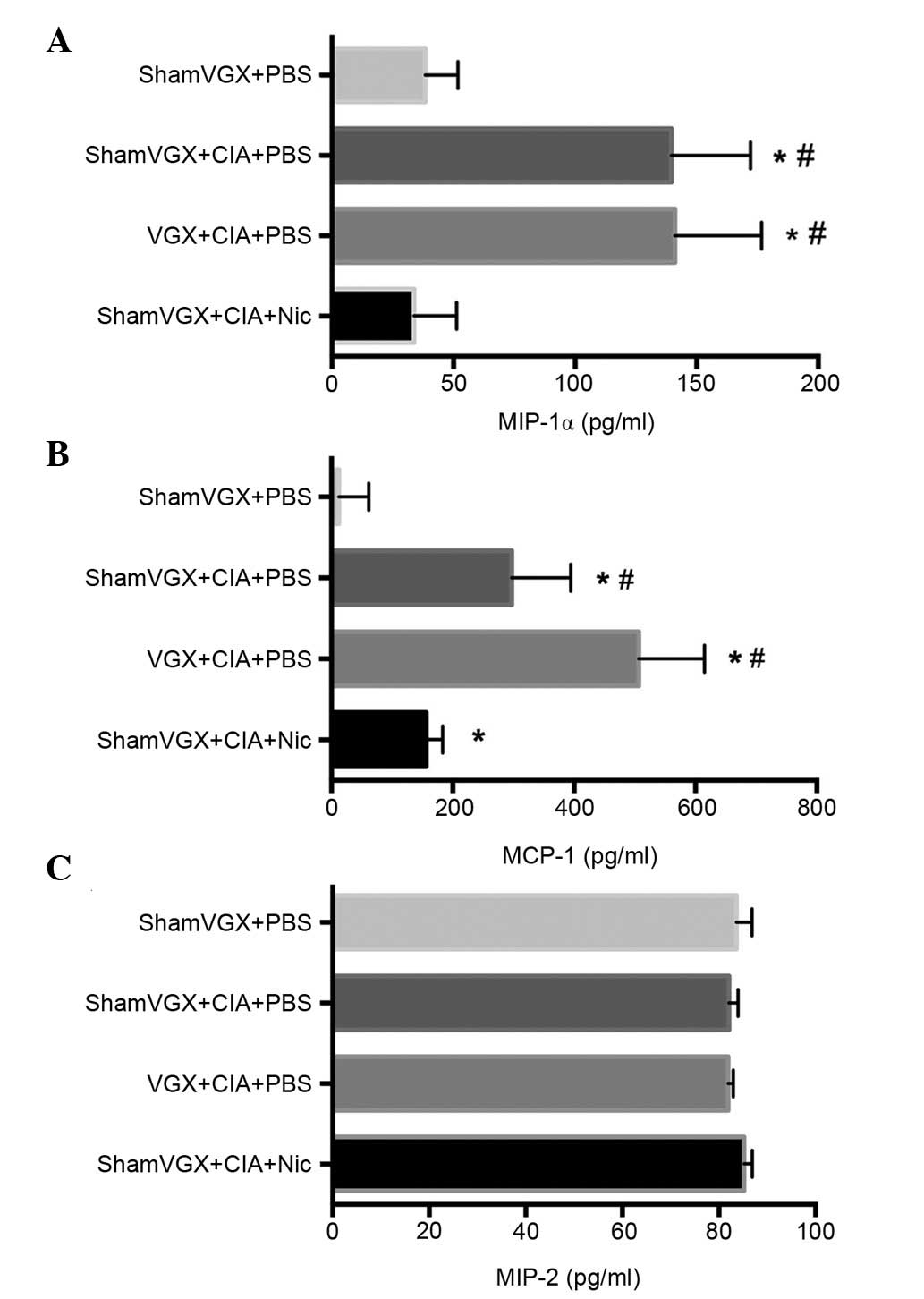
Nicotine suppresses the serum levels
of MIP-1α and MCP-1, but not MIP-2
Chemokines serve a key role in the process of
leukocyte extravasation. To determine whether the CAP affects
chemotaxis of macrophages in the CIA model, the present study
detected the expression levels of the macrophage-associated
chemokines MIP-1α, MCP-1 and MIP-2, which have been reported to
have major roles in the trafficking of monocytes toward synovial
tissue, in the serum of the four groups by ELISA (7,17).
Compared with its release in the model and vagotomy groups,
nicotine, a classical cholinergic agonist, significantly attenuated
release of the chemokines MIP-1α and MCP-1 (Fig. 3A and B), but not MIP-2 (Fig. 3C).
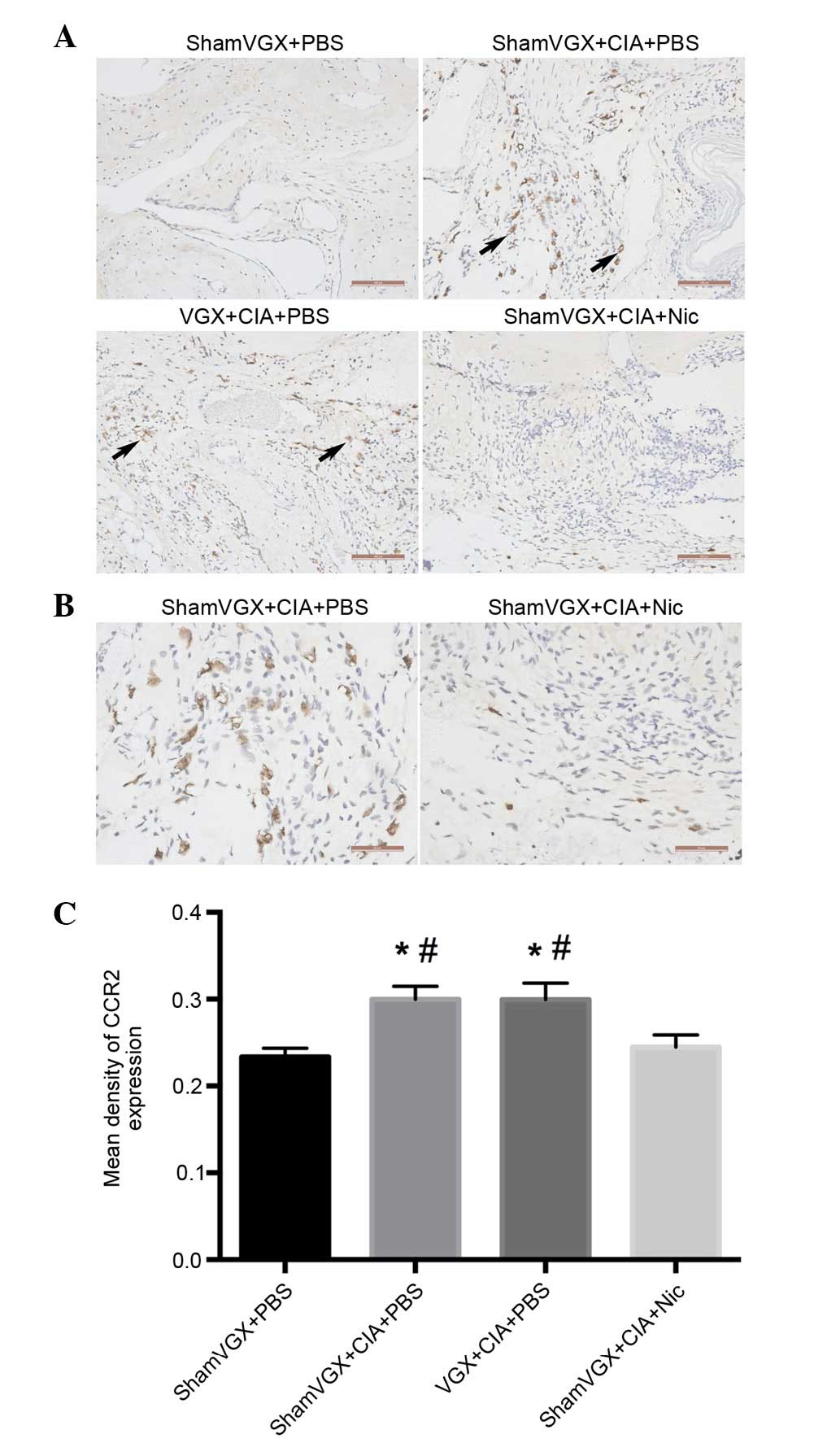
Nicotine treatment results in reduced
CCR2 expression in the synovium of CIA mice
Chemokines control the directed movement of cells
expressing their respective receptors. The chemokine receptor CCR2
is bound by MCP-1, thus contributing to the migration of monocytes
from the bloodstream into the tissue (18). The present study examined CCR2
expression in the synovial tissue by immunohistochemistry. The
results of a semi-quantitative analysis indicated that CCR2 was
highly expressed on synoviocytes in mice with CIA but not in normal
mice, and nicotine treatment reduced CCR2 expression in the CIA
mice (Fig. 4A-C).
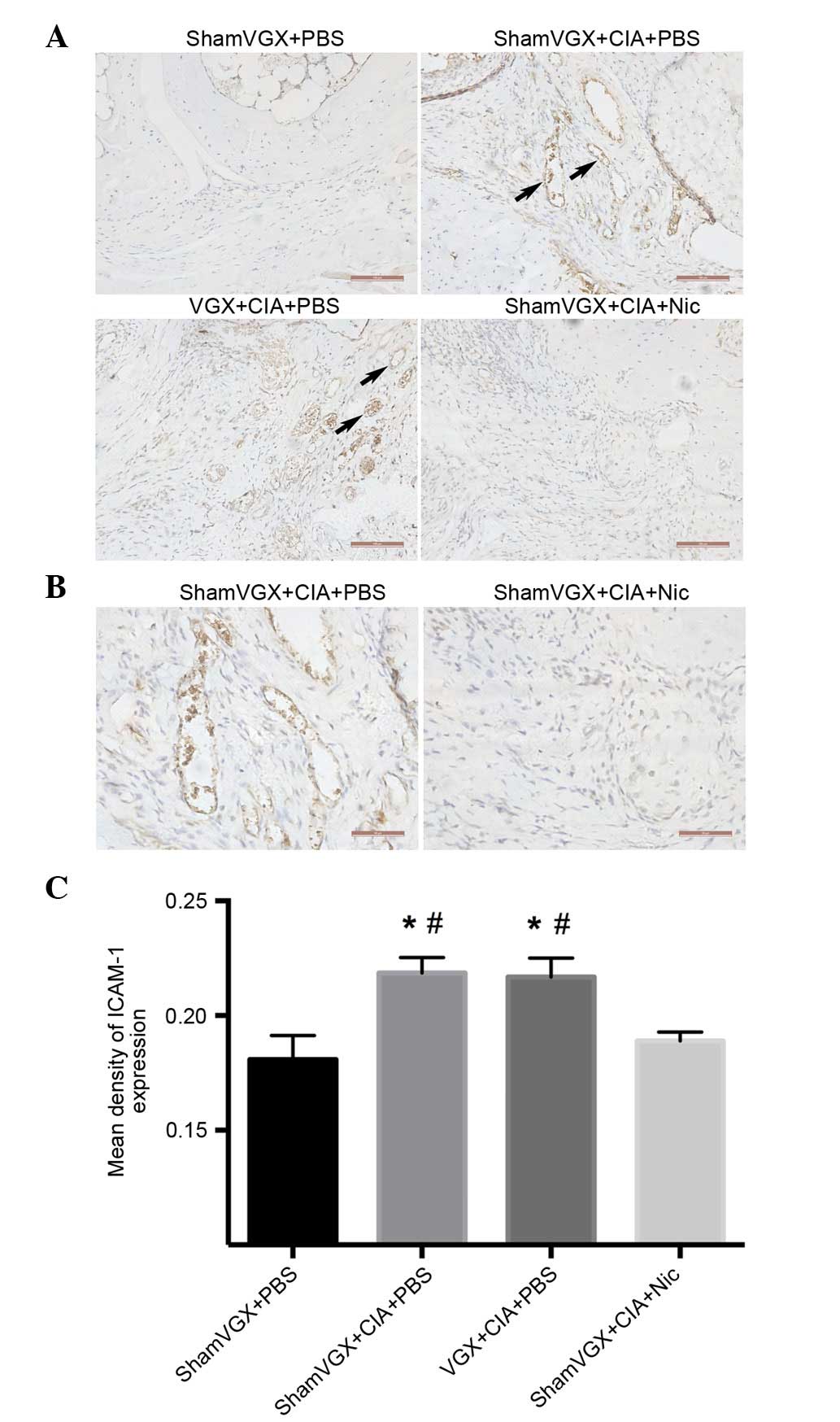
Nicotine decreases endothelial cell
surface levels of ICAM-1 in CIA mice
ICAM-1 has classically been considered to serve a
role in intercellular adhesion. To evaluate the effects of nicotine
on the expression of adhesion molecules on vascular endothelial
cells, ICAM-1 production was measured in the synovium.
Immunohistochemical analysis of mouse synovial sections revealed
that ICAM-1 was highly expressed in the model and vagotomy groups.
Conversely, ICAM-1 expression was significantly suppressed in
nicotine-treated mice (Fig.
5A-C).
Discussion
Vagally mediated anti-inflammatory action, also
known as the CAP, was clearly described by Tracey (19). In previous years, several
researches have reported that activation of the CAP suppresses the
clinical and histological manifestations of CIA in mice with
established disease (14–16,20,21).
The underlying mechanisms may be associated with downregulation of
the expression of tumor necrosis factor-α and IL-6, inhibition of
Th17 cell responses and improvement in the Th1/Th2 imbalance in
CIA; however, the specific mechanism underlying the regulatory
effects of nicotine on RA remains complex and only partially
understood.
Notably, the present results revealed that nicotine
treatment reduced the number of CD11b-positive macrophages in the
synovium and spleen samples of CIA mice. Synovial macrophages
participate in several of the events that direct inflammation,
including angiogenic stimulation, leukocyte and lymphocyte
recruitment, fibroblast proliferation, and protease secretion, thus
resulting in joint destruction (3,22,23).
Synovial macrophages are currently considered the most reliable
biomarker for disease severity and therapeutic response in RA
(24). Therefore, the present
findings suggested that the reduced infiltration of macrophages
mediated by nicotine may serve an important role in protection
against autoimmune arthritis. However, in the present study, the
mice in the vagotomy group were subjected to left-side cervical
vagotomy 4 days prior to CIA induction, the vagotomy did not
exacerbate CIA-associated inflammation, which may be attributed to
a compensatory role for the other side of the vagus nerve. This
finding was consistent with the results of previous studies
(15,16).
Macrophage infiltration is a process that involves
complex and consecutive changes. Monocyte transmigration through
endothelial monolayers is directed by chemotaxis, which is a
crucial step in the process of complete monocyte recruitment to the
vascular wall. The present study demonstrated that nicotine
treatment in a CIA model reduced the production of MIP-1α and
MCP-1. MIP-1α, which belongs to the CC chemokine family, is
chemotactic for monocytes and lymphocytes in RA. MIP-1α may be
considered one of the major cytokines that contributes to the
chemoattraction and retention of RA macrophages in inflamed joints
(25). MCP-1, another CC
chemokine, was initially identified as a monocyte-specific
chemoattractant, and has also been reported to serve a role in
T-cell differentiation and angiogenesis (26), which may be important in RA
pathogenesis. Treatment with an MCP-1 antagonist has been reported
to ameliorate disease severity in adjuvant-induced arthritis by
decreasing macrophage infiltration (27). However, in the present study, there
were no significant differences in MIP-2 levels among the sera.
MIP-2, which is a CXC chemokine (and is known as the murine
equivalent of CXC chemokine ligand 8/IL-8), was among the first
chemokines reported to be involved in leukocyte chemotaxis. MIP-2
has also been demonstrated to serve a major role in neutrophil and
monocyte trafficking toward the synovial tissue (7,28).
The results of the present study suggested that the action of
nicotine may not depend on the production of MIP-2 in the CIA
model.
CCR2 is the major chemokine receptor used to
classify monocyte subsets (29),
and is the predominant functional receptor for MCP-1. MCP-1-CCR2
signaling may activate p42/44 mitogen-activated protein kinases
(MAPK) and protein kinase C (PKC) through G proteins, in order to
regulate cellular adhesion and motility in macrophages (30). Based on previous data, ~50% of
infiltrating cells in the synovial tissue are CCR2-positive
monocytes (31). While
investigating whether activation of the CAP also influences CCR2
expression in the synovium, the present study demonstrated that
nicotine treatment reduced CCR2 expression, as determined by
immunohistochemical semi-quantitative analysis. These results
indicated that the interaction between MCP-1/C-C chemokine ligand
(CCL)2 and CCR2 may promote macrophage migration into the synovium
during CIA. A previous study demonstrated that CCL2-CCR2 signaling
may activate p42/44 MAPK and PKC through G proteins to regulate
cellular adhesion and motility in macrophages (30). Our studies, however, do not rule
out the possibility of more direct evidence of macrophage migration
in synovial tissues. Further investigations are required to clarify
this point. Other interactions between chemokines and their
receptors, such as between MIP-1α/CCL3 and CCR1, may also
contribute to macrophage migration (32). In addition, a previous study
revealed that a CCR1 antagonist, which reduces macrophage
accumulation in RA synovium, is effective for treating RA (33).
ICAM-1 is an adhesion molecule expressed by
activated endothelial cells, which is believed to mediate leukocyte
migration across the endothelium. However, the potential effects of
nicotine on adhesion molecule expression remain controversial.
Takahashi et al reported that nicotine inhibits
IL-18-enhanced expression of ICAM-1 on monocytes (34). Conversely, Cirillo et al and
others demonstrated that nicotine promotes ICAM expression on
endothelial cells (35,36). The present study revealed that
stimulation of the CAP reduced production of ICAM-1, not only on
endothelial cells but also on leukocytes in the peripheral blood
(Fig. 5A). Therefore, based on
this finding, it may be hypothesized that the CAP has a dominant
influence on monocyte adhesion to endothelial cells.
In conclusion, the results of the present study
indicated that activation of the CAP via nicotine stimulation
reduces CIA-associated inflammation by decreasing the number of
macrophages in synovial tissues, which is mediated by effects not
only on the chemotaxis of macrophages but also on macrophage
adhesion to endothelial cells. These observations suggest that
stimulation of cholinergic signaling potentially serves a key role
in initiating and maintaining joint inflammation in patients with
RA. Therefore, further investigation regarding the role of CAP in
this context is required.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81571602).
References
|
1
|
Davignon JL, Hayder M, Baron M, Boyer JF,
Constantin A, Apparailly F, Poupot R and Cantagrel A: Targeting
monocytes/macrophages in the treatment of rheumatoid arthritis.
Rheumatology (Oxford). 52:590–598. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kinne RW, Stuhlmüller B and Burmester GR:
Cells of the synovium in rheumatoid arthritis. Macrophages.
Arthritis Res Ther. 9:2242007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mulherin D, Fitzgerald O and Bresnihan B:
Synovial tissue macrophage populations and articular damage in
rheumatoid arthritis. Arthritis Rheum. 39:115–124. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tak PP and Bresnihan B: The pathogenesis
and prevention of joint damage in rheumatoid arthritis: Advances
from synovial biopsy and tissue analysis. Arthritis Rheum.
43:2619–2633. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Richards PJ, Williams AS, Goodfellow RM
and Williams BD: Liposomal clodronate eliminates synovial
macrophages, reduces inflammation and ameliorates joint destruction
in antigen-induced arthritis. Rheumatology (Oxford). 38:818–825.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haringman JJ, Smeets TJ, Reinders-Blankert
P and Tak PP: Chemokine and chemokine receptor expression in paired
peripheral blood mononuclear cells and synovial tissue of patients
with rheumatoid arthritis, osteoarthritis, and reactive arthritis.
Ann Rheum Dis. 65:294–300. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hayashida K, Nanki T, Girschick H, Yavuz
S, Ochi T and Lipsky PE: Synovial stromal cells from rheumatoid
arthritis patients attract monocytes by producing MCP-1 and IL-8.
Arthritis Res. 3:118–126. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brühl H, Cihak J, Plachý J, Kunz-Schughart
L, Niedermeier M, Denzel A, Gomez M Rodriguez, Talke Y, Luckow B,
Stangassinger M and Mack M: Targeting of Gr-1+,
CCR2+ monocytes in collagen-induced arthritis. Arthritis
Rheum. 56:2975–2985. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Borovikova LV, Ivanova S, Zhang M, Yang H,
Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW and Tracey
KJ: Vagus nerve stimulation attenuates the systemic inflammatory
response to endotoxin. Nature. 405:458–462. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boland C, Collet V, Laterre E, Lecuivre C,
Wittebole X and Laterre PF: Electrical vagus nerve stimulation and
nicotine effects in peritonitis-induced acute lung injury in rats.
Inflammation. 34:29–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao M, He X, Bi XY, Yu XJ, Gil Wier W and
Zang WJ: Vagal stimulation triggers peripheral vascular protection
through the cholinergic anti-inflammatory pathway in a rat model of
myocardial ischemia/reperfusion. Basic Res Cardiol. 108:3452013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wilund KR, Rosenblat M, Chung HR, Volkova
N, Kaplan M, Woods JA and Aviram M: Macrophages from alpha 7
nicotinic acetylcholine receptor knockout mice demonstrate
increased cholesterol accumulation and decreased cellular
paraoxonase expression: A possible link between the nervous system
and atherosclerosis development. Biochem Biophys Res Commun.
390:148–154. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rosas-Ballina M, Goldstein RS,
Gallowitsch-Puerta M, Yang L, Valdés-Ferrer SI, Patel NB, Chavan S,
Al-Abed Y, Yang H and Tracey KJ: The selective alpha7 agonist
GTS-21 attenuates cytokine production in human whole blood and
human monocytes activated by ligands for TLR2, TLR3, TLR4, TLR9,
and RAGE. Mol Med. 15:195–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li T, Zuo X, Zhou Y, Wang Y, Zhuang H,
Zhang L, Zhang H and Xiao X: The vagus nerve and nicotinic
receptors involve inhibition of HMGB1 release and early
pro-inflammatory cytokines function in collagen-induced arthritis.
J Clin Immunol. 30:213–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu S, Luo H, Xiao X, Zhang H, Li T and Zuo
X: Attenuation of collagen induced arthritis via suppression on
Th17 response by activating cholinergic anti-inflammatory pathway
with nicotine. Eur J Pharmacol. 735:97–104. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
van Maanen MA, Lebre MC, van der Poll T,
LaRosa GJ, Elbaum D, Vervoordeldonk MJ and Tak PP: Stimulation of
nicotinic acetylcholine receptors attenuates collagen-induced
arthritis in mice. Arthritis Rheum. 60:114–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szekanecz Z, Koch AE and Tak PP: Chemokine
and chemokine receptor blockade in arthritis, a prototype of
immune-mediated inflammatory diseases. Neth J Med. 69:356–366.
2011.PubMed/NCBI
|
|
18
|
Katschke KJ Jr, Rottman JB, Ruth JH, Qin
S, Wu L, LaRosa G, Ponath P, Park CC, Pope RM and Koch AE:
Differential expression of chemokine receptors on peripheral blood,
synovial fluid, and synovial tissue monocytes/macrophages in
rheumatoid arthritis. Arthritis Rheum. 44:1022–1032. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tracey KJ: The inflammatory reflex.
Nature. 420:853–859. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Maanen MA, Stoof SP, Larosa GJ,
Vervoordeldonk MJ and Tak PP: Role of the cholinergic nervous
system in rheumatoid arthritis: Aggravation of arthritis in
nicotinic acetylcholine receptor α7 subunit gene knockout mice. Ann
Rheum Dis. 69:1717–1723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu S, Zhao H, Luo H, Xiao X, Zhang H, Li T
and Zuo X: GTS-21, an α7-nicotinic acetylcholine receptor agonist,
modulates Th1 differentiation in CD4+T cells from
patients with rheumatoid arthritis. Exp Ther Med. 8:557–562.
2014.PubMed/NCBI
|
|
22
|
Haringman JJ, Gerlag DM, Zwinderman AH,
Smeets TJ, Kraan MC, Baeten D, McInnes IB, Bresnihan B and Tak PP:
Synovial tissue macrophages: A sensitive biomarker for response to
treatment in patients with rheumatoid arthritis. Ann Rheum Dis.
64:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Abeles AM and Pillinger MH: The role of
the synovial fibroblast in rheumatoid arthritis: Cartilage
destruction and the regulation of matrix metalloproteinases. Bull
NYU Hosp Jt Dis. 64:20–24. 2006.PubMed/NCBI
|
|
24
|
Kennedy A, Fearon U, Veale DJ and Godson
C: Macrophages in synovial inflammation. Front Immunol. 2:522011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koch AE, Kunkel SL, Harlow LA, Mazarakis
DD, Haines GK, Burdick MD, Pope RM and Strieter RM: Macrophage
inflammatory protein-1 alpha. A novel chemotactic cytokine for
macrophages in rheumatoid arthritis. J Clin Invest. 93:921–928.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luther SA and Cyster JG: Chemokines as
regulators of T cell differentiation. Nat Immunol. 2:102–107. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shahrara S, Proudfoot AE, Park CC, Volin
MV, Haines GK, Woods JM, Aikens CH, Handel TM and Pope RM:
Inhibition of monocyte chemoattractant protein-1 ameliorates rat
adjuvant-induced arthritis. J Immunol. 180:3447–3456. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Haringman JJ, Ludikhuize J and Tak PP:
Chemokines in joint disease: The key to inflammation? Ann Rheum
Dis. 63:1186–1194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Geissmann F, Jung S and Littman DR: Blood
monocytes consist of two principal subsets with distinct migratory
properties. Immunity. 19:71–82. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiménez-Sainz MC, Fast B, Mayor F Jr and
Aragay AM: Signaling pathways for monocyte chemoattractant protein
1-mediated extracellular signal-regulated kinase activation. Mol
Pharmacol. 64:773–782. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brühl H, Cihak J, Schneider MA, Plachý J,
Rupp T, Wenzel I, Shakarami M, Milz S, Ellwart JW, Stangassinger M,
et al: Dual role of CCR2 during initiation and progression of
collagen-induced arthritis: Evidence for regulatory activity of
CCR2+ T cells. J Immunol. 172:890–898. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Haringman JJ, Kraan MC, Smeets TJ,
Zwinderman KH and Tak PP: Chemokine blockade and chronic
inflammatory disease: Proof of concept in patients with rheumatoid
arthritis. Ann Rheum Dis. 62:715–721. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amat M, Benjamim CF, Williams LM, Prats N,
Terricabras E, Beleta J, Kunkel SL and Godessart N: Pharmacological
blockade of CCR1 ameliorates murine arthritis and alters cytokine
networks in vivo. Br J Pharmacol. 149:666–675. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takahashi HK, Iwagaki H, Hamano R, Yoshino
T, Tanaka N and Nishibori M: Effect of nicotine on IL-18-initiated
immune response in human monocytes. J Leukoc Biol. 80:1388–1394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cirillo P, Pacileo M, De Rosa S, Calabrò
P, Gargiulo A, Angri V, Prevete N, Fiorentino I, Ucci G, Sasso L,
et al: HMG-CoA reductase inhibitors reduce nicotine-induced
expression of cellular adhesion molecules in cultured human
coronary endothelial cells. J Vasc Res. 44:460–470. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Breen LT, McHugh PE and Murphy BP: HUVEC
ICAM-1 and VCAM-1 synthesis in response to potentially athero-prone
and athero-protective mechanical and nicotine chemical stimuli. Ann
Biomed Eng. 38:1880–1892. 2010. View Article : Google Scholar : PubMed/NCBI
|