Introduction
The uterus in mammals serves more than a
reproductive role; it also serves other key physiological functions
and so the maintenance of the normal structure and function of the
endometrium is important. Pathological conditions, including
ischemia, hypoxia, or inflammation, may cause excessive reactive
oxygen species to accumulate in the body, upsetting the
oxidation/antioxidant balance and causing oxidative stress to
trigger endometrial cell damage (1,2).
This is mainly manifested in infertility; endometrial damage is a
difficult clinical problem.
With the development of assisted reproductive
technology, numerous problems in fertility have been solved, but
not endometrial damage. At the time of writing, the mechanism of
endometrial damage remains to be elucidated and there remains a
lack of effective diagnosis and prevention.
The fruits of Lycium barbarum are used in
traditional Chinese medicine and are credited with numerous
biological activities and pharmacological functions. They may serve
a role in antiaging effects (3,4), in
antitumor activity (5,6), immunomodulatory activity (7) and increased metabolism (8). Previous studies have indicated that
of the variety of nutrients and trace elements in Lycium
barbarum, the main effective components are Lycium
barbarum polysaccharides (LBPs) and they have been reported
(9–12) to exhibit a concentration-dependent
antioxidant activity, including antilipid peroxidation, superoxide
anion scavenging and anti-superoxide formation. However, any
specific antioxidant properties of LBPs in the prevention of
endometrium damage remain to be elucidated.
In the present study, selected human endometrial
stromal cells were used to investigate the protective effect of
LBPs on the hydrogen peroxide (H2O2)-induced
injury of human endometrial stroma, and a scientific theoretical
basis was investigated for the application of LBPs in reproductive
health care.
Materials and methods
Endometrial specimen collection
Endometrial tissues were obtained from the
hysterectomies of 20 patients with uterine fibroids in the
Affiliate Hospital of Hebei Engineering University (Handan, China)
between September 2013 and December 2014. The procedure was
approved by the Ethics Committee of Hebei Engineering University,
and informed consent was obtained from the patients with uterine
fibroids prior to the hysterectomies. The patients were aged
between 30 and 45 years old, with menstrual cycles from 24–35 days
(mean, 28 days). No patient received any hormonal treatment for at
least 3 months prior to the hysterectomies. Endometrial tissues
were confirmed as in hyperplasia period and disease-free by
postoperative pathology. Endometrial tissue was scraped immediately
from the uterus under sterile conditions in vitro, and
placed in Dulbecco's modified Eagle's medium/nutrient mixture F-12
(DMEM/F12) (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), including 10% fetal bovine serum, penicillin and streptomycin
(100 mg/ml; Gibco; Thermo Fisher Scientific, Inc.) in an ice bath,
and transported to the laboratory within 2 h.
Preparation of LBPs
LBPs were purchased from Shanghai Organic Chemistry
Institute (Shanghai, China). A stock solution was prepared of 5,000
mg/l in basal medium DMEM/F12 and maintained at −20°C. For the
following experiments, the final concentration of LBPs was 100
µg/ml produced by diluting the stock solution with DMEM/F12.
Isolation, purification and culture of
endometrial stromal cells (ESCs)
Following several washes with phosphate buffered
saline (PBS), the tissue was cut into 1–2 mm3 pieces with sterile
scissors and incubated with 5 ml DMEM/F12 containing 0.2%
collagenase I (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany)
in an incubator with atmosphere of 5% CO2 at 37°C for 60
min. During the incubation, the tissue pieces were pipetted gently
to disperse the cells. The whole cell suspension was centrifuged at
500 × g for 5 min at room temperature. The supernatant fluid, which
contained ESCs, was centrifuged at 1,200 × g for 5 min at room
temperature. The supernatant fluid was discarded, while the
precipitate was resuspended in a culture bottle with 3 ml complete
cell-culture medium [DMEM/F12+10% fetal bovine serum (FBS)+1%
penicillin and streptomycin]. The ESCs attached to the culture
bottle were washed several times with serum-free DMEM/F12 to remove
red blood cells.
A trypan blue exclusion assay was performed to
assess the proportion of living cells. Then, 1 ml cell suspension
(1×105 cells/ml) was placed into a six-well plate
containing coverslips and cultured in atmosphere of 5%
CO2 at 37°C for 5 days for the identification and
assessment of the purity of the cells.
Morphological observation and
identification of ESCs
ESCs were cultured for 0 and 5 days and then stained
with hematoxylin and eosin (H&E) following fixing in 4%
paraformaldehyde for 1–2 h at 4°C and dehydration with 70, 80, 90,
95 and 100% ethanol and 100% xylene. The morphology and structure
of the ESCs were observed with an inverted microscope equipped with
phase-contrast apparatus and with a light microscope.
Immunocytochemical staining was performed to
identify the ESCs and assess their purity. PBS was used instead of
a primary antibody as a negative control. Cells cultured on
coverslips were fixed with 4% paraformaldehyde and treated with
0.25% Triton X-100. After blocking with 5% normal goat serum for 20
min at 37°C, the rabbit anti-human primary antibody vimentin
(1:100; cat. no. MA3-745) and cytokeratin (1:100; cat. no.
PA5-14263; Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China) were incubated with the cells at 4°C overnight. The
cells were subsequently incubated with goat anti-rabbit IgG (1:100;
Boster Corporation, Wuhan, China) for 20 min at 37°C and stained
with 3,3′-diaminobenzidine (5 mg/ml; Sigma Aldrich; Merck
Millipore) for 5 min at room temperature. The specimens were washed
with PBS for 5 min three times and observed using light
microscopy.
Experimental groups
ESCs were divided into four groups according to the
different intervention factors. Control group: Serum-free DMEM/F12;
H2O2 group: H2O2 and
serum-free DMEM/F12; LBP group: LBPs and serum-free DMEM/F12;
H2O2 + LBP group: H2O2,
LBPs and serum-free DMEM/F12. The final concentration of
H2O2 and LBPs was 10–4 M/l and 100 µg/ml
respectively.
Morphological observations
ESCs (4×104/ml) were seeded in 6-well plates and
were divided into the above four groups and cultured for 12 h.
Morphology and structure of the ESCs were observed with an inverted
microscope equipped with phase-contrast apparatus.
Cell proliferation assay
Cell proliferation was determined by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay. ESCs (4×104/ml) were seeded in 96-well plates and cultured
with serum-free DMEM/F12 for 12 h for synchronization. They were
divided into the four groups described previously, each group into
6-well plates and cultured for 24 h. Thereafter, MTT (5 mg/ml) was
added to each well and the plates were incubated at 37°C for 2 h.
The medium was then replaced with 150 µl DMSO and agitated for 10
min at room temperature. The absorbance at 560 nm was measured
using a microplate reader (Packard Instrument Company, Inc.,
Meriden, CT, USA).
Detection of superoxide dismutase
(SOD) activity and malondialdehyde (MDA) content
The four groups of cells were cultured for 12 h,
absorbing the culture supernatant fluid, according to the
manufacturer's protocol of SOD assay kit and MDA assay kit (R&D
Systems, Minneapolis, MN, USA), the activity of SOD was detected by
autoxidation of pyrogallic acid, as previously described (13) and the content of MDA was detected
by the thio-barbituric acid method, as previously described
(14).
Western blot analysis
Cells from the above four groups were grown in 10 cm
dishes and cultured for 12 h, followed by washing with PBS. The
cells were subsequently lysed with lysis buffer (pH 7.4; 1 M
Tris-HCl, 1% Triton X-100, sodium deoxycholate, 10% SDS).
Solubilized proteins were centrifuged at 14,000 × g at 4°C for 30
min. Extracted proteins were quantified by Coomassie Protein assay
reagent (Sigma-Aldrich; Merck Millipore). Western blot analysis of
Bcl-2 and caspase-3 were performed. In brief, 30 mg isolated
protein was electrophoresed on 8% sodium dodecyl
sulphate-polyacrylamide gel (100 V for 1.5 h), and transferred onto
PVDF membranes. The membranes were treated with blocking solution
[Tris-buffered saline (TBS) pH 7.2, 0.1% Tween, 5% milk] for 1 h at
room temperature and incubated for 12 h at 4°C with rabbit
anti-human monoclonal Bcl-2 (cat. no. 13-8800) and caspase-3 (cat.
no. 700182) antibodies (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), diluted 1:1,000 in TBS (pH 7.2) 0.1% Tween.
After four washings for 15 min with TBS (pH 7.2) 0.1% Tween, the
membranes were incubated with the goat anti-rabbit IgG antibody
horseradish peroxidase conjugate (cat. no. 35552) (Invitrogen;
Thermo Fisher Scientific, Inc.), diluted 1:1,000 in TBS pH 7.2,
0.1% Tween for 1 h at room temperature. They were then washed four
times for 15 min with TBS (pH 7.2), treated with an enhanced
chemiluminescent method according to the protocols of an enhanced
chemiluminesence detection kit (eECL western blot detection kit)
(Invitrogen; Thermo Fisher Scientific, Inc.) and exposed to Kodak
X-ray film for 0.5–20 min as necessary to detect the signals. The
relative intensity of the immunoreactive bands on the films was
quantified by a computer-assisted densitometry program (SmartView;
Major Science, Saratoga, CA, USA). Proteins expression was
quantified by comparison with internal control β-actin.
RNA extraction
Total RNA was extracted from the cells in the four
groups by culturing for 12 h with the TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol, and the RNA was dissolved in RNase-free water. The
integrity of the RNA was assessed by ethidium bromide agarose gel
electrophoresis and the quantity of RNA was determined by the
relative absorbance at 260 vs. 280 nm. cDNA (2 µg) was synthesized
in a volume of 10 µl with a cDNA synthesis kit (Takara Bio, Inc.,
Otsu, Japan) according to the manufacturer's protocol. The cDNA was
stored at −20°C.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR, primers (Invitrogen; Thermo Fisher
Scientific, Inc.) were derived from the GeneBank database. β-actin
was used as the housekeeping gene. The primers used were as
follows: Caspase-3 forward, 5′-TATCCTGAGATGGGTTTA-3′ and reverse,
5′-TGTTTCCCTGAGGTTTGC-3′; Bcl-2 forward,
5′-CTGGGAGAACAGGGTACGATAA-3′ and reverse,
5′-CCCACCGAACTCAAAGAAGG-3′; β-actin forward,
5′-CGGGAAATCGTGCGTGAC-3′ and reverse,
5′-CAGGAAGGAAGGCTGGAAG-3′.
The RT-qPCR reactions were performed using Brilliant
SYBR-Green QRT-PCR Master Mixture according to the manufacturer's
protocol (Invitrogen; Thermo Fisher Scientific, Inc.). RNA for
caspase-3 and Bcl-2 was amplified using an ABI Prism 7500 Sequence
Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.). For all RT-qPCR studies, synthesizing by PCR procedure was
performed with the following time courses: 94°C for 10 min, 40
cycles at 94°C for 15 sec, 59°C for 30 sec, and 72°C for 32 min for
amplification. The amplified products were subjected to a stepwise
increase in temperature from 59–94°C and dissociation curves were
constructed.
Target mRNA was quantified by measuring the
threshold cycle and reading against a calibration curve. The
relative amount of each mRNA was normalized to the housekeeping
gene, β-actin mRNA. Results were analyzed using the relative
standard curve method of analysis/2−ΔΔCq method of
analysis (15).
Statistical analysis
All data are presented as bar graphs with the mean ±
standard deviation of six independent experiments with samples from
different subjects. Data were analyzed using SPSS for windows,
version 15.0 (SPSS Inc., Chicago, IL, USA). Statistical analysis
was performed by one-way analysis of variance, Student-Newman-Keuls
was used to compare individual groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Morphological observation of ESCs
The ESCs were observed using an inverted microscope
equipped with phase-contrast apparatus. Differential centrifugation
identified the purity of the isolated ESCs as 92–95%. Trypan blue
staining demonstrated that the viability of the EECs was 96–98%.
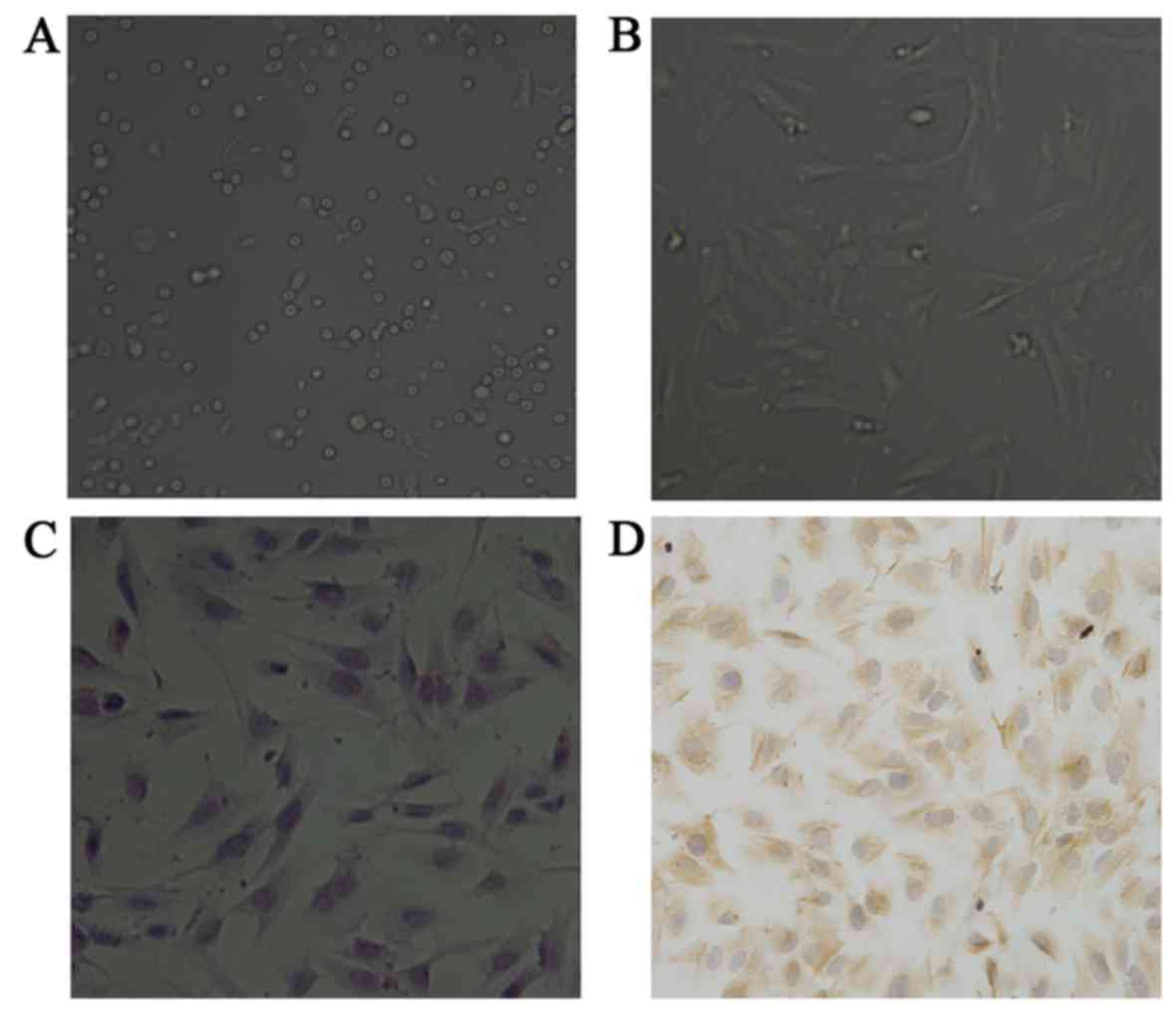
Recently isolated ESCs exhibited mostly round morphology (Fig. 1A), later growing as spindle and
polygon-shaped cells with long cytoplasmic processes, usually
reaching confluence after 5 days culture, exhibiting a single-cell
monolayer growth pattern (Fig.
1B). The ESCs were stained by H&E staining after being
cultured for 5 days (Fig. 1C) to
indicate the cell morphology.
Identification of ESCs
The ESCs were detected by immunocytochemical
staining. The cytoplasm of positive ESCs was stained as
brownish/yellow granules. The ESCs were positive for vimentin
(Fig. 1D).
LBP inhibits
H2O2-induced cell death
After the four groups of ESCs had been cultured for
12 h, the changes in the ESCs morphology and structure were
observed using an inverted microscope equipped with phase-contrast
apparatus. No significant changes were observed in the morphology
and structure of ESCs in the LBPs group compared with the control
group, while those in the H2O2 group became
irregular in shape and declined in number. The
H2O2 + LBP group demonstrated significantly
less deterioration compared with the H2O2
group (Fig. 2A).
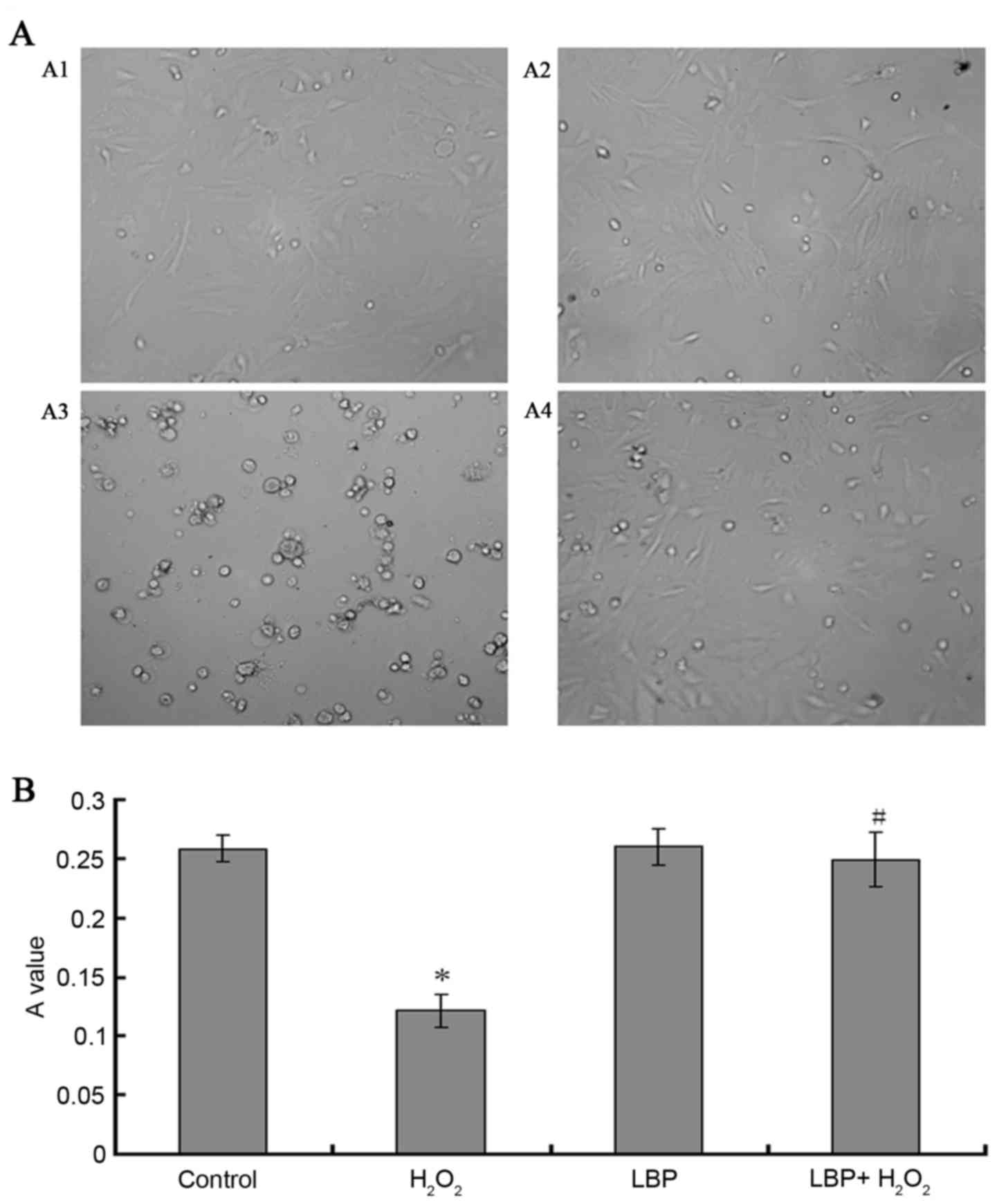 | Figure 2.Morphological observations and
proliferation assays of ESCs, ESCs were incubated with different
intervention factors for 12 h. (A) Morphological observation of
ESCs under an inverted microscope equipped with phase-contrast
apparatus. A1, Control; A2 H2O2; A3, LBPs;
A4, H2O2 + LBPs. (B) The proliferation assay
of ESCs was evaluated by MTT. Control, serum-free DMEM/F12;
H2O2, H2O2 and
serum-free DMEM/F12; LBP, LBPs and serum-free DMEM/F12;
H2O2 + LBP, H2O2, LBPs
and serum-free DMEM/F12, the final concentration of
H2O2 and LBPs was 10−4 M/l and 100
µg/ml respectively. The data are presented as the mean ± standard
deviation (*P<0.05 vs. control group, #P<0.05 vs.
the H2O2 group). ESCs, endometrial stromal
cells; H2O2, hydrogen peroxide; LBPs,
Lycium barbarum polysaccharides; MTT,
3-(4,5-dimethylthiazol-2-yl) −2,5-diphenyltetrazolium bromide;
DMEM/F12, Dulbecco's modified Eagle's medium/nutrient mixture
F-12. |
MTT assay
After the cells had been cultured for 12 h, the
change in the number of the ESCs was observed by detecting the
purple crystals in each culture well at an absorbance value of 550
nm. The result demonstrated that compared with the control group,
the number of ESCs in the LBP group had no significant change
(P=0.7976), while H2O2 significantly
decreased the proliferation of ESCs in a time dependent manner
(P<0.0001), the H2O2 + LBP group
demonstrated significantly less deterioration compared with the
H2O2 group (Fig.
2B).
LBP inhibits
H2O2-induced content of MDA and activity of
SOD in ESCs
The result of colorimetric analysis demonstrated
that compared with the control group, no significant change was
observed in the content of MDA and the activity of SOD in the LBP
group, while the content of MDA increased and the activity of SOD
decreased significantly in H2O2 group
(P<0.0001 and P=0.0002, respectively). Compared with the
H2O2 group, the content of MDA decreased and
the activity of SOD increased significantly in the
H2O2 + LBP group (P<0.0001 and
P<0.0001; Fig. 3).
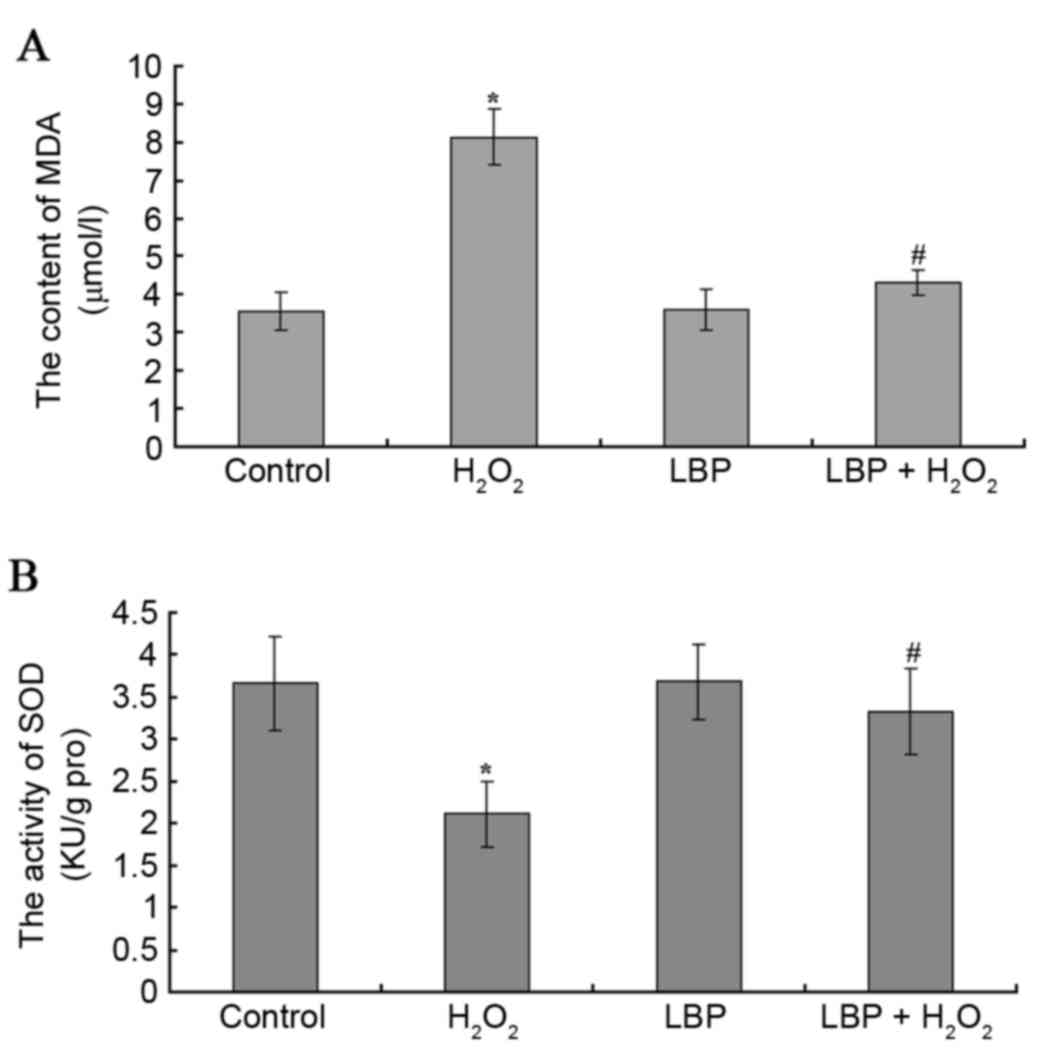 | Figure 3.MDA content and SOD activity. The
content of (A) MDA and activity of (B) SOD were assessed. Control,
serum-free DMEM/F12; H2O2,
H2O2 and serum-free DMEM/F12; LBP, LBP and
serum-free DMEM/F12; H2O2 + LBP,
H2O2, LBP and serum-free DMEM/F12, the final
concentration of H2O2 and LBPs was
10−4 M/l and 100 µg/ml respectively. The data are
presented as the mean ± standard deviation (*P<0.05 vs. control
group, #P<0.05 vs. the H2O2
group). MDA, malondialdehyde; SOD, superoxide dismutase; DMEM/F12,
Dulbecco's modified Eagle's medium/nutrient mixture F-12;
H2O2, hydrogen peroxide; LBPs, Lycium
barbarum polysaccharides. |
Effect of LBP on
H2O2-induced protein expression levels of
caspase-3 and Bcl-2 in ESCs
After cells were cultured for 12 h, the results of
western blot analysis demonstrated that the control group cells had
positive expression levels of caspase-3 and Bcl-2, and that the LBP
group had similar expression levels of caspase-3 and Bcl-2 to the
control group (P>0.05). The expression levels of caspase-3 in
the H2O2 group increased significantly
compared with that of the control group (P<0.0001), and
decreased significantly in the LBP + H2O2
group compared with that of the H2O2 group
(P<0.05); while the expression levels of Bcl-2 increased
significantly in the H2O2 group compared with
that of the control group (P<0.0001), and decreased in the LBP +
H2O2 group compared with that of the
H2O2 group (P<0.0001; Fig. 4A and B).
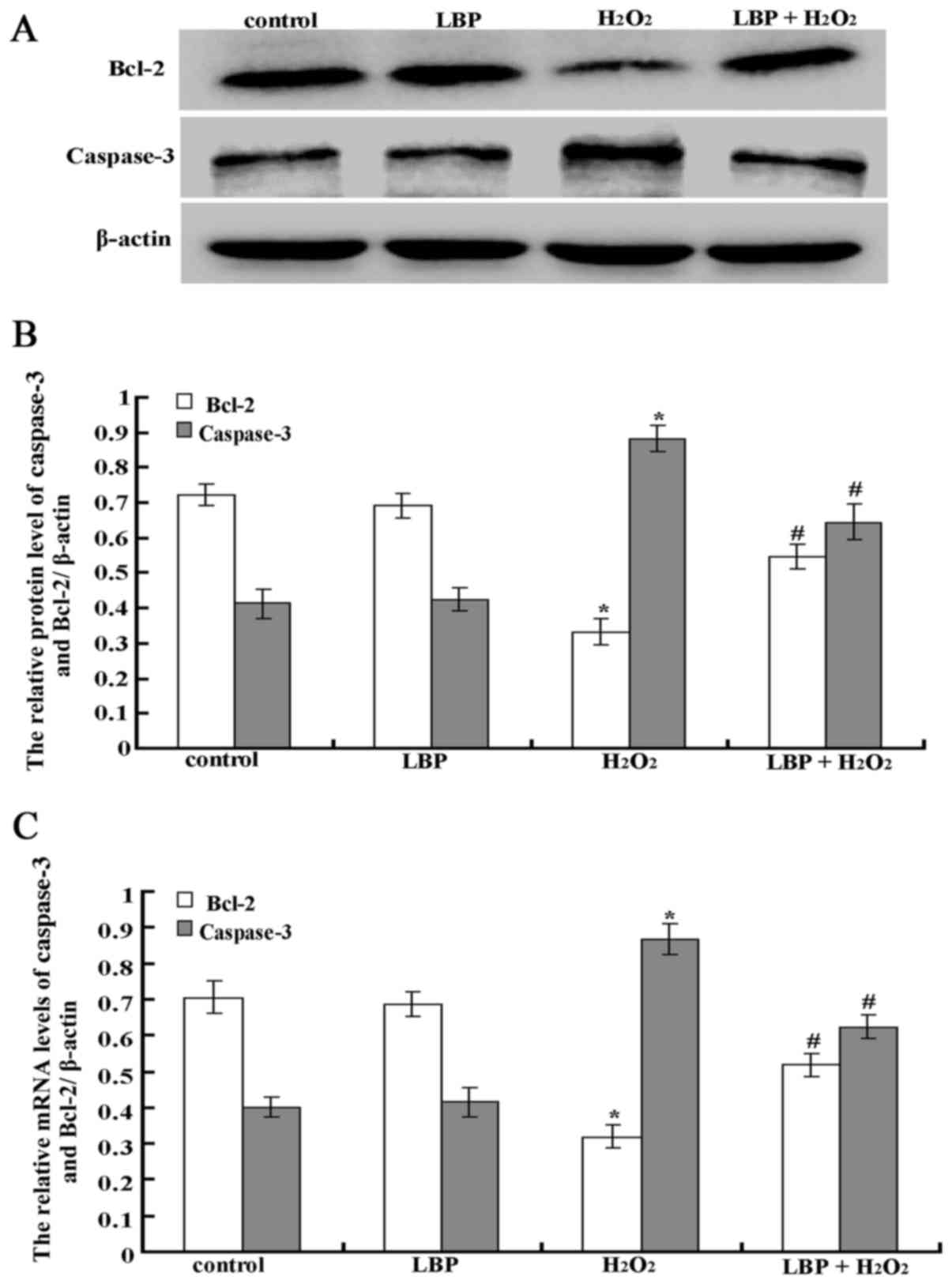 | Figure 4.LBPs inhibit
H2O2-induced expression of caspase-3 and
Bcl-2 in ESCs. (A) The protein expression of caspase-3 and Bcl-2
was analyzed by western blotting. (B) The protein expression levels
of caspase-3 and Bcl-2 were quantified by densitometry. (C) The
mRNA expression levels of caspase-3 and Bcl-2 was analyzed by
reverse transcription-quantitative polymerase chain reaction.
Control, serum-free DMEM/F12; H2O2,
H2O2 and serum-free DMEM/F12; LBP, LBPs and
serum-free DMEM/F12; H2O2 + LBP,
H2O2, LBPs and serum-free DMEM/F12, the final
concentration of H2O2 and LBPs was
10−4 M/l and 100 µg/ml. The data are presented as the
mean ± standard deviation (*P<0.05 vs. control group,
#P<0.05 vs. the H2O2 group).
LBPs, Lycium barbarum polysaccharides;
H2O2, hydrogen peroxide; ESCs, endometrial
stromal cells; DMEM/F12, Dulbecco's modified Eagle's
medium/nutrient mixture F-12; H2O2, hydrogen
peroxide. |
LBP inhibits
H2O2-induced mRNA expression levels of
caspase-3 and Bcl-2 in ESCs
After cells were cultured for 12 h, the result of
RT-qPCR demonstrated that the control group cells had positive
expression of caspase-3 and Bcl-2, as did the LBP group. Compared
with the control group, the expression level of caspase-3 of the
H2O2 group was increased significantly
(P<0.0001) and the expression level of Bcl-2 was decreased
significantly (P<0.05). Compared with the
H2O2 group, the expression levels of
caspase-3 in the LBP + H2O2 group was
decreased significantly (P<0.0001), although the expression of
Bcl-2 increased significantly (P<0.0001; Fig. 4C).
Discussion
An increasing number of studies suggest that LBPs
may have powerful antioxidant activity in vitro and in
vivo, be able to remove free radicals and prevent lipoprotein
lipid peroxidation, and to protect the biological macromolecules,
including DNA, membrane lipids and cytoplasmic proteins, from
oxidative damage (16,17). Wu et al (18) demonstrated that LBPs (10 mg/kg)
significantly reduced blood glucose, nitric oxide and MDA in
streptozotocin-induced diabetic rats. Li (19) also demonstrated that LBPs (20–50
mg/kg) protected liver and kidney tissue from oxidative damage in
streptozotocin-induced diabetic rats. Luo et al (20) demonstrated that LBPs alleviated
heat-induced damage of rat testes, as well as
H2O2-induced DNA damage in mouse testicular
cells, by increasing their resistance to oxidative stress-induced
injury.
Oxidative stress causes membrane lipid peroxidation
(21), directly damaging proteins
and nucleic acids. H2O2 is an important
intermediate in the body's redox reaction (22) and using exogenous
H2O2 to treat a cell is one of the most
common methods of establishing an oxidative stress damage model,
which can effectively induce intracellular reactive oxygen species
generation and cell damage (23).
MDA is the product of the lipid peroxidation of free radicals with
biomembrane polyunsaturated fatty acids and its content reflects
the level of oxygen free radicals and the degree of lipid
peroxidation. In addition, MDA crosslinks with nucleic acids and
proteins and damages them. MDA is thus not only the one of the
metabolites of cellular damage but also one of the substances
leading to cell damage. SOD is the main antioxidant enzyme that can
degrade and remove H2O2 and other free
radicals. The present study identified that
H2O2 can promote ESCs apoptosis, increase the
content of MDA and decrease the activity of SOD, while LBPs
suppress H2O2-induced ESCs apoptosis, reduce
the content of MDA and increase the activity of SOD. This suggests
that H2O2 has a damaging effect on ESCs,
while LBPs serve a protective role in ESCs threatened by
H2O2.
Apoptosis in mammalian cells has two pathways,
intracellular and extracellular, which can activate caspase family
proteins and cause cell lysis (24). The intracellular pathway is also
called the mitochondrial pathway and is mainly regulated by the
Bcl-2 family proteins (25),
including apoptosis promoting proteins (Bax, Bak, Bid and Bik) and
antiapoptotic proteins (Bcl-2 and Bcl-xl). In normal physiological
conditions, apoptosis-promoting proteins and anti-apoptotic
proteins form different dimers to counteract the activity of
apoptosis promoting proteins and to achieve a balance between
promoting and suppressing apoptosis (26). The Bcl-2 protein, located on the
outer membrane of the mitochondria, has an antiapoptosis function
and high expression levels can protect the integrity of the
mitochondrial membrane, avoiding mitochondrial apoptosis factors,
including the release of cytochrome c to prevent cell
apoptosis (cytochrome c is a key factor in the formation of
apoptotic bodies). The caspase family is divided into two subsets,
the apoptosis initiator proteins (caspase-2, 8, 9, 10 and 15) and
apoptosis effector proteins [caspase-3, 6 and 7, and poly
(ADP-ribose) polymerase] (27).
Mitochondria are activated and releases apoptosis factors,
including cytochrome c, the latter activating apoptosis
initiating caspase proteins, thus activating the apoptosis effect
and finally destroying the cells (28,29).
The results from the present study suggested that
H2O2 reduces the protein and mRNA expression
levels of Bcl-2 in ESCs, and increases the protein and mRNA
expression level of caspase-3. LBPs suppress
H2O2-induced ESCs damage, decreases the
protein and mRNA expression levels of Bcl-2, and increases the
protein and mRNA expression level of caspase-3. It is therefore
speculated that ESCs oxidative stress induced apoptosis is mainly
performed through the mitochondrial pathway, as this process is
chiefly regulated by the Bcl-2 protein family. This suggests that
the LBPs can improve the antiapoptosis protein expression levels of
Bcl-2, protect the integrity of the mitochondrial membrane, prevent
the release of cytochrome c from mitochondria into the
cytoplasm and induce the activation of caspase-3 to trigger cell
apoptosis.
In conclusion, LBPs are capable of inhibiting
H2O2-induced ESCs apoptosis, reducing the
content of MDA and increasing the activity of SOD. LBPs are also
capable of inhibiting H2O2-induced ESCs
damage, decreasing the protein and mRNA expression levels of Bcl-2,
and increasing the protein and mRNA expression levels of caspase-3.
This has significance for the clinical prevention and treatment of
endometrial damage.
Acknowledgements
The authors would like to thank Dr Bing-Jun Zhang at
the Affiliated Hospital of Hebei University of Engineering (Handan,
Heibei, China) for the collection of endometrial tissue.
References
|
1
|
Xie JH, Dong CJ, Nie SP, Li F, Wang ZJ,
Shen MY and Xie MY: Extraction, chemical composition and
antioxidant activity of flavonoids from Cyclocarya paliurus
(Batal.) Iljinskaja leaves. Food Chem. 186:97–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang ZJ, Xie JH, Kan LJ, Wang JQ, Shen MY,
Li WJ, Nie SP and Xie MY: Sulfated polysaccharides from Cyclocarya
paliurus reduce H2O2-induced oxidative stress in RAW264.7 cells.
Int J Biol Macromol:. 80:410–417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang RC and So KF: Use of anti-aging
herbal medicine, Lycium barbarum, against aging-associated
diseases. What do we know so far? Cell Mol Neurobiol. 28:643–652.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li XM, Ma YL and Liu XJ: Effect of the
Lycium barbarum polysaccharides on agerelated oxidative stress in
aged mice. J Ethnopharmacol. 111:504–511. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mao F, Xiao B, Jiang Z, Zhao J, Huang X
and Guo J: Anticancer effect of Lycium barbarum polysaccharides on
colon cancer cells involves G0/G1 phase arrest. Med Oncol.
28:121–126. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo Q, Li Z, Yan J, Zhu F, Xu RJ and Cai
YZ: Lycium barbarum polysaccharides induce apoptosis in human
prostate cancer cells and inhibits prostate cancer growth in a
xenograft mouse model of human prostate cancer. J Med Food.
12:695–703. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Du G, Liu L and Fang J: Experimental study
on the enhancement of murine splenic lymphocyte proliferation by
Lycium barbarum glycopeptide. J Huazhong Univ Sci Technolog Med
Sci. 24:518–520. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Amagase H and Nance DM: A randomized,
double-blind, placebo-controlled, clinical study of the general
effects of a standardized Lycium barbarum (Goji) juice, GoChi. J
Altern Complement Med. 14:403–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Potterat O: Goji (Lycium barbarum and L.
chinense): Phytochemistry, pharmacology and safety in the
perspective of traditional uses and recent popularity. Planta Med.
76:7–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu SJ, Ng LT and Lin CC: Antioxidant
activities of some common ingredients of traditional Chinese
medicine, Angelica sinensisLycium barbarum and Poria cocos.
Phytother Res. 18:1008–1012. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Luo Q, Cai Y, Yan J, Sun M and Corke H:
Hypoglycemic and hypolipidemic effects and antioxidant activity of
fruit extracts from Lycium barbarum. Life Sci. 76:137–149. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao J, Liong EC, Ching YP, Chang RC, So
KF, Fung ML and Tipoe GL: Lycium barbarum polysaccharides protect
mice liver from carbon tetrachloride induced oxidative stress and
necroinflammation. J Ethnopharmacol. 139:462–470. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Marland S and Marklund G: Involvement of
the superoxide anion radical in the autoxidation of pyrogollol and
a convenient assay for super oxide dismutase. Eur J Biochem.
47:469–474. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yagi K: A simple fluorometric assay for
lipoperoxide in blood plasma. Biochem Med. 15:212–215. 1976.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li GR: Study on isolation Lycium barbarum
polysaccharides and its effect on anti-active oxygen free radicals.
Chin J Mod Appl Pharm. 12:94–96. 2002.(In Chinese).
|
|
17
|
Wang JH, Wang HZ, Zhang M and Zhang SH:
Anti-aging function of poly saccharides from fructus lycii. J Acta
Nutrimenta Sinica. 12:189–4191. 2002.(In Chinese).
|
|
18
|
Wu H, Guo H and Zhao R: Effect of Lycium
barbarum polysaccharide on the improvement of antioxidant ability
and DNA damage in NIDDM rats. Yakugaku Zasshi. 126:365–371. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li XM: Protective effect of Lycium
barbarum polysaccharides on streptozotocin-induced oxidative stress
in rats. Int J Biol Macromol. 40:461–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Luo Q, Li Z, Huang X, Yan J, Zhang S and
Cai YZ: Lycium barbarum polysaccharides: Protective effects against
heat-induced damage of rat testes and H2O2-induced DNA damage in
mouse testicular cells and beneficial effect on sexual behavior and
reproductive function of hemicastrated rats. Life Sci. 79:613–621.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin MT and Beal MF: Mitoehondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jelie S, Padeletti M, Kawtt SM, Higgins C,
Canfield SM, Onat D, Colombo PC, Basner RC, Factor P and LeJemtel
TH: Inflammation, oxidative stress, and repair capacity of the
vascular endothelium in obstructive sleep apnea. Circulation.
117:2270–2278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Starke PE and Farber JL: Endogenous
defenses against the cytotoxicity of hydrogen peroxide in cultured
rat hepatocytes. J Biol Chem. 260:86–92. 1985.PubMed/NCBI
|
|
24
|
Suen DF, Norris KL and Youle RJ:
Mitoehondrial dynamies and apoptosis. Genes Dev. 22:1577–1590.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15andmiR-16 induce apoptosis by targeting Bcl-2. Proc Natl
Acad Sci U S A. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Heath-Engel H, Chang N and Shore G: The
endoplasmic reticulum in apoptosis and autophagy: Role of the Bcl-2
protein family. Oncogene. 27:6419–6433. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan TJ, Han LH, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dirks AJ and Lecuwenburgh C: Aging and
lifelong calorie restriction result in adaptations of skeletal
muscle apoptosis repressor, apoptosis-inducing factor, X-linked
inhibitor of apoptosis, caspase-3, and caspase-12. Free Radic Biol
Med. 36:27–39. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mazumder S, Plesca D and Almasan A:
Caspase-3 activation is a critical determinant of genotoxie
stress-induced apoptosis. Methods Mol Biol. 414:13–21.
2008.PubMed/NCBI
|