Introduction
Osteosarcoma (OS) is one of the most common
malignancies in children and young adults worldwide (1). The surgical techniques and
chemotherapeutic treatments for OS have markedly improved; however,
30% of children diagnosed with OS will not survive for >5 years
(1–3). Therefore, a comprehensive
understanding of OS biology is required to optimize the diagnosis,
therapy and prognostic predictions of this disease.
MicroRNAs (miRNAs) are a class of non-coding small
RNAs, 18–22 nucleotides in length, which regulate target gene
expression at the post-transcriptional level, via imperfect
base-pairing with the 3′ untranslated region (3′UTR) of specific
target mRNAs (4). It has
previously been demonstrated that miRNAs may function as oncogenes
or tumor suppressors in cancer development (5). A large number of miRNAs have
previously been identified; however, their roles in OS development
and the underlying molecular mechanisms remain to be fully
elucidated.
miRNA (miR)-34c-3p is one of the mature miRNAs of
the miR-34c family, which are critical modulators of the p53
pathway and potential tumor suppressors in human cancer (6). Downregulation of miR-34c-3p
expression was previously revealed in several types of cancer,
including non-small cell lung cancer (NSCLC) (7,8),
cervical cancer (9) and glioma
(10). It has previously been
demonstrated that miR-34c-3p functions as a tumor suppressor via
the targeting of numerous signaling pathways (7,8).
However, compared with other types of cancer, the role of
miR-34c-3p in the pathogenesis of OS remains to be elucidated.
The aim of present study was to investigate the
expression pattern and the biological effects of miR-34c-3p in OS.
The findings suggested that miR-34c-3p was downregulated and
associated with poor prognosis in OS patients. Forced miR-34c-3p
expression suppressed cell growth in vitro and tumorigenesis
in vivo. In addition, myristoylated alanine-rich protein
kinase C substrate (MARCKS) was identified as a direct target of
miR-34c-3p and its overexpression partly reversed the suppressive
effects of miR-34c-3p. The results support a tumor suppressor role
of miR-34c-3p in OS via inhibition of MARCKS, and may thus be a
promising therapeutic target for the treatment of OS.
Materials and methods
Human tissue samples
A total of 50 paired fresh surgically resected OS
tumor and adjacent non-tumor tissues were collected from the Xi'an
Honghui Hospital (Xi'an, China) between September 2010 and November
2012. Of the 50 OS patients, 32 were male, 28 were female, with a
median age of 32 years old (range 19–51). None of the patients
received pre-operative chemotherapy or radiotherapy prior to
surgery. Specimens were gently washed with normal saline and
flash-frozen in liquid nitrogen immediately following collection
and stored at −80°C until use. Tumor and non-tumor tissue samples
were confirmed by pathological examination. The present study was
approved by the Research Ethics Committee of Xi'an Honghui
Hospital.
Cell culture
Human OS cell lines (MG-63, U2OS, HOS and Saos-2),
and normal human osteoblast (NHOst) cells were purchased from
American Type Culture Collection (Manassas, VA, USA). Cells were
cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (FBS, Gibco; Thermo Fisher Scientific, Inc.),
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C, in a
humidified incubator in an atmosphere containing 5%
CO2.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from tissues and cells using
miRNeasy Mini kit (Qiagen China Co., Ltd., Shanghai, China),
according to the manufacturer's protocol. Total RNA concentration
was assessed by measuring absorbance at a wavelength of 260 nm
using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific,
Inc.). A total of 2 µg total RNA was reverse transcribed using the
PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.,
Otsu, Japan) and miRNA-specific stem-loop RT primer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The stem-loop RT
primer sequence was:
5′-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCTGGC-3′, for
miR-34c-3p. Gene-specific amplification was performed using Applied
Biosystems 7500 fast real-time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) and SYBR-Green PCR Master Mix
(Applied Biosystems, Thermo Fisher Scientific, Inc.) with a first
step at 95°C for 10 min followed by 40 cycles with 95°C for 15 sec
and 60°C for 1 min with a fluorescent reading at the end of this
step. The gene-specific primers used were as follows: MiR-34c-3p,
forward 5′-GGTGGAATCACTAACCACACG-3′ reverse 5′-GTGCAGGGTCCGAGGT-3′;
MARCKS, forward 5′-AGCCCGGTAGAGAAGGAGG-3′ reverse
5′-TTGGGCGAAGAAGTCGAGGA-3′; U6 small nuclear RNA, forward
5′-AGAGCCTGTGGTGTCCG-3′ reverse 5′-CATCTTCAAAGCACTTCCCT-3′; and
GAPDH, forward 5′-CATGTTCGTCATGGGTGTGAACCA-3′ and reverse
5′-AGTGATGGCATGGACTGTGGTCAT-3′. The relative expression levels of
miR-34c-3p and MARCKS were normalized to that of internal control
U6 or GAPDH using the comparative 2-ΔΔCq method
(11). Each sample was analyzed in
triplicate and the mean expression level was calculated.
Cell transfection
The miR-34c-3p mimics, miR-34c-3p inhibitor and
negative control were designed and synthesized by Shanghai
GenePharma Co., Ltd. (Shanghai, China). For transfection, 2×105
MG-63 and Saos-2 cells were seeded into a 6-well plate in growth
medium without serum and antibiotics at a density of 30–40% and
incubated overnight. Subsequently, the cells were transfected with
miR-34c-3p mimics, miR-34c-3p inhibitor or negative control using
HiPerFect Transfection Reagent (Qiagen GmbH, Hilden, Germany)
according to the manufacturer's protocol. The mixture was added to
cells at a final concentration of 100 nM. Following incubation at
37°C in an atmosphere containing 5% CO2 for 4–6 h, the
serum-free medium was removed, and cells were maintained in DMEM
supplemented with 10% FBS.
Cell proliferation assay
A total of 24 h post-transfection, cells were
harvested and seeded into 96-well plates at a density of 5×103
cells/well and cultured at 37°C in an atmosphere containing 5%
CO2 for 1, 2, 3 and 4 days. A total of 10 µl Cell
Counting Kit-8 solution (Dojindo Molecular Technologies, Inc.,
Kumamoto, Japan) was added into the culture medium in each well.
After 1 h incubation, optical density values were measured using a
microplate reader (BioTek Instruments, Inc., Winooski, VT, USA) at
a wavelength of 450 nm. Each time point was repeated in three wells
and the experiment was independently performed three times.
Colony formation assay
A total of 24 h post-transfection, cells were
harvested, seeded into 24-well plates at a density of 5×102
cells/well and cultured at 37°C in an atmosphere containing 5%
CO2. During colony growth, the culture medium was
replaced every 3 days. After 12 days, the plates were stained for
the formation of cell colonies with crystal violet in 70% ethanol
and counted under a microscope (Nikon AZ100; Nikon Corporation,
Tokyo, Japan). The colony was counted only if it contained >50
cells. The experiment was independently performed three times.
Animal experiments
A total of 12 female BALB/c nu/nu mice (6–8 weeks
old and weight 18–20 g; Beijng Vital River Laboratory Animal
Technology Co., Ltd., Beijing, China) were housed together in
specific pathogen-free conditions with a temperature of 25°C,
55–65% relative humidity, and a 12-h light-dark cycle with standard
chow and water ad libitum. A total of 2×106 MG-63 cells transfected
with miR-34c-3p agomir or agomir-negative control (Shanghai
GenePharma Co., Ltd.) were injected subcutaneously into the flank
region of 6–8 week-old female nude mice (n=6/group). Tumor growth
was monitored every 3 days, measured with fine digital calipers.
Tumor volume was calculated using the following formula: Tumor
volume=0.5 × width2 × length. After 4 weeks, the mice were
sacrificed by cervical dislocation and tumor weights were measured.
All animal procedures were performed in accordance with protocols
approved by the Institute Research Ethics Committee of Xi'an
Honghui Hospital.
Prediction of miR-34c-3p target genes
and luciferase reporter assay
Putative miR-34c-3p targets were predicted using
several different algorithms, including TargetScan (www.targetscan.org), Pictar (http://www.pictar.org/) and miRanda (http://www.microrna.org). The 3′-UTR of MARCKS
containing the putative miR-34c-3p binding sequence was amplified
by PCR and cloned downstream of the firefly luciferase gene in the
pMIR-Report vector (Promega Corporation, Madison, WI, USA)
(12). Mutations in the miR-34c-3p
seed regions of the MARCKS 3′UTR were generated using the QuikChang
Multisite-directed mutagenesis kit (Promega Corporation). The
mutated MARCKS 3′-UTR fragment was cloned into the pMIR-Report
vector to develop the pMIR-MARCKS-3′-UTR-mut vector. For the
luciferase assay in the OS cells, MG-63 or Saos-2 cells were
co-transfected with 50 ng/well MARCKS-3′-UTR or MARCKS 3′-UTR-mut,
and 50 nM/well miR-34c-3p mimic or scrambled microRNA negative
control using Lipofectamine® 2000 reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). Cells were collected after 48 h
for analysis using the Dual Luciferase reporter assay system
(Promega Corporation).
Western blotting
MG-63 and Saos-2 cells transfected with miR-34c-3p
mimics, miR-34c-3p inhibitor and negative control were lysed in
radioimmunoprecipitation assay buffer with protease inhibitor at 72
h post-transfection. Lysate protein concentrations were obtained by
BCA assay (Thermo Fisher Scientific, Inc.). A total of 50 µg
protein was separated by 10% SDS-PAGE and transferred onto
polyvinylidene fluoride membranes and blocked for 1 h in 5% bovine
milk diluted in Tris-buffered saline with Tween-20 at room
temperature. The mouse monoclonal antibody against MARCKS (catalog
no. sc-100777; 1:500; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) and mouse monoclonal antibody against β-actin (catalog no.
sc-69879; 1:2,000; Santa Cruz Biotechnology, Inc.) were incubated
with the blot overnight at 4°C. Following extensive washing with
phosphate-buffered saline containing 0.1% Triton X-100, the
membranes were incubated with horseradish peroxidase-conjugated
goat anti-mouse secondary antibody (catalog no. sc-2005; 1:5,000,
Santa Cruz Biotechnology, Inc.) for 30 min at room temperature. The
bands were visualized using an enhanced chemiluminescence system
(EMD Millipore, Billerica, MA, USA).
Statistical analysis
Statistical Package of the Social Sciences version
19.0 for Windows (IBM SPSS, Armonk, NY, USA) was used for
statistical analyses. The survival rate of patients with OS was
calculated using Kaplan-Meier survival analysis. Differences
between experimental groups were assessed using the two-tailed
unpaired Student's t-test. The relationship between miR-34c-3p and
MARCKS expression was assessed using Spearman's correlation
analysis. Data are presented as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
miR-34c-3p is downregulated in human
OS and correlates with poor prognosis
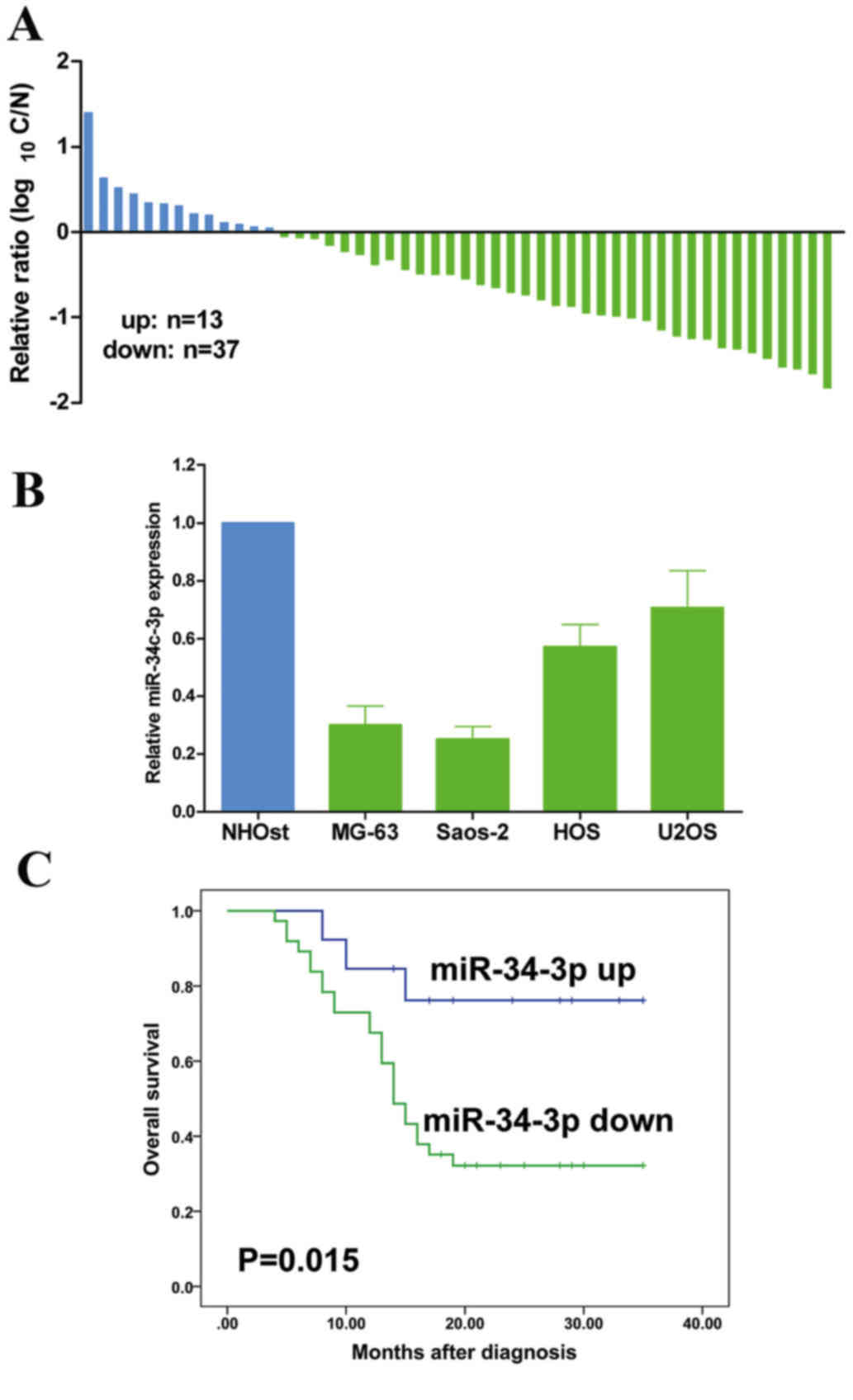
To understand the role of miR-34c-3p in OS,
miR-34c-3p expression was examined by RT-qPCR in 50 OS and
corresponding healthy bone tissues. The results demonstrated that
miR-34c-3p was downregulated in the majority of OS tissues
(Fig. 1A). Furthermore, miR-34c-3p
was downregulated in MG-63, Saos-2, HOS and U2OS human OS cell
lines, compared with the NHOst cell line (Fig. 1B). Furthermore, patients with lower
expression of miR-34c-3p revealed a poor prognosis compared with
those exhibiting a high expression of miR-34c-3p (P=0.015; Fig. 1C). These results suggested that
miR-34c-3p is essential in OS development.
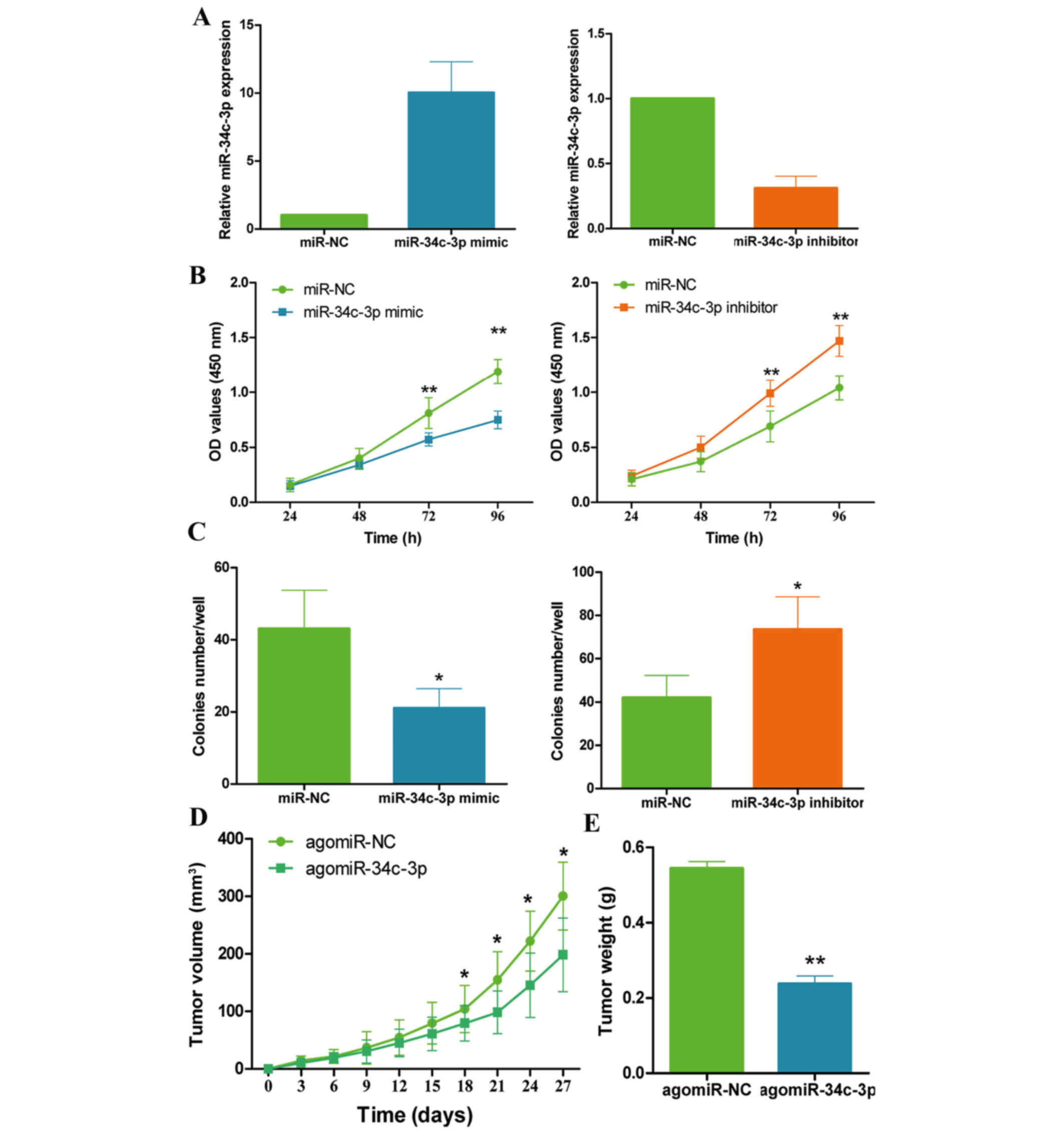
miR-34c-3p suppresses OS growth in
vitro and in vivo
The present study hypothesized that miR-34c-3p was
implicated in OS growth, based on the aforementioned findings. The
effects of miR-34c-3p on OS cell growth were investigated following
overexpression or inhibition of miR-34c-3p in MG-63 cells (Fig. 2A). As presented in Fig. 2B, overexpression of miR-34c-3p
significantly inhibited MG-63 cell growth (P=0.009 at 72 h and
P=0.003 at 96 h. Conversely, inhibition of miR-34c-3p stimulated
MG-63 cell growth (P=0.007 at 72 h and P=0.001 at 96 h). In
addition, colony formation assays revealed that the overexpression
of miR-34c-3p suppressed MG-63 colony formation (P=0.031), whereas
inhibition of miR-34c-3p promoted MG-63 colony formation compared
with the control (P=0.036; Fig.
2C). These results were further confirmed using another OS cell
line Saos-2 (data not shown).
To determine if this effect was also apparent in
vivo, these in vitro results were confirmed using a
xenograft model. MG-63 cells transfected with agomiR-34c-3p or
negative control (agomiR-NC) were subcutaneously injected into the
flank region of nude mice. As presented in Fig. 2D, overexpression of miR-34c-3p
inhibited tumor growth compared with the control group (P=0.024),
and the tumor weights in the agomiR-34c-3p group were significantly
decreased compared with the agomiR-NC group (P=0.008; Fig. 2E). These data revealed that
miR-34c-3p suppresses OS cell growth in vitro and in
vivo.
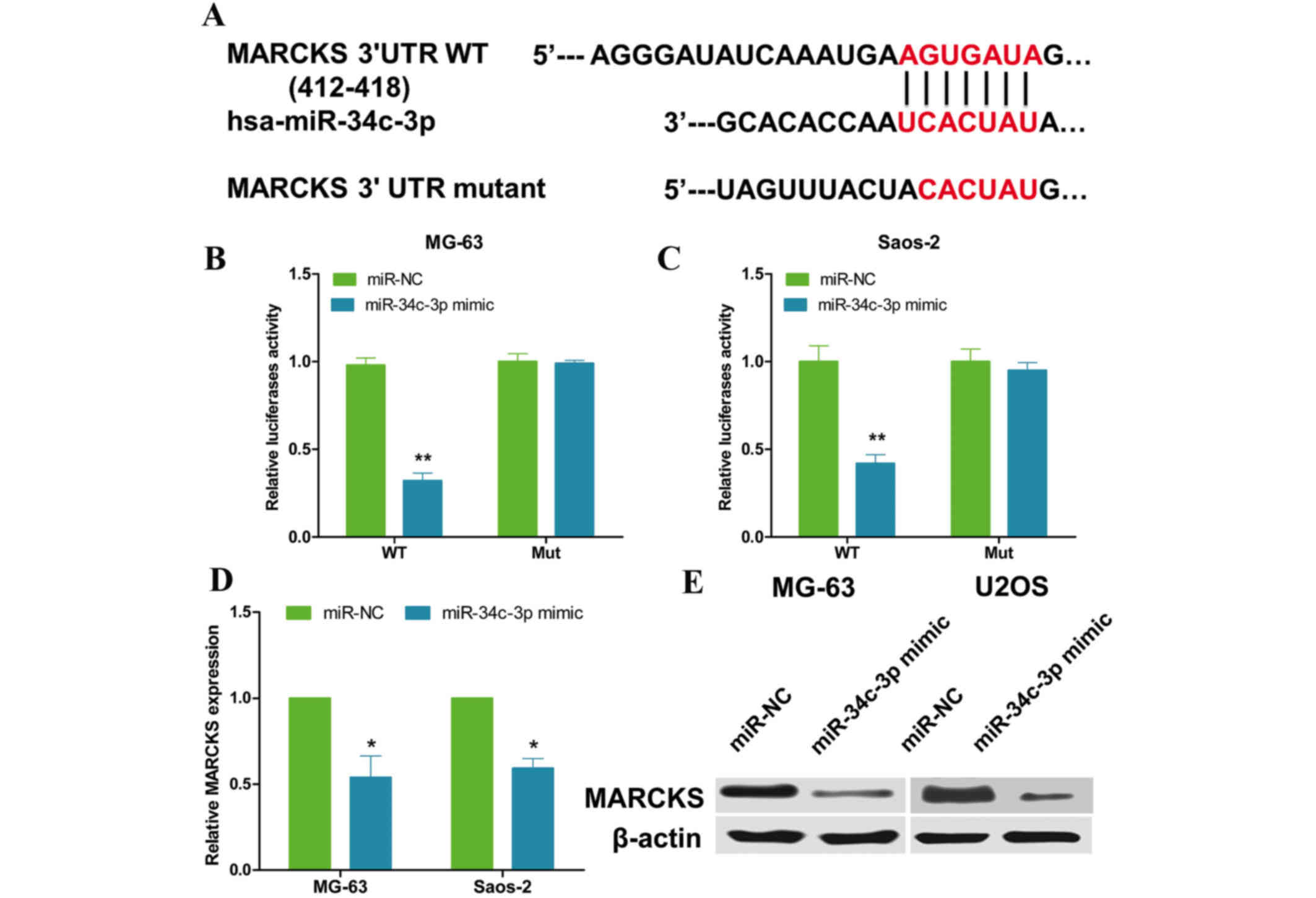
MARCKS is a direct target of
miR-34c-3p in OS cells
Bioinformatic research was conducted to find
potential targets of miR-34c-3p using TargetScan and miRanda. As
presented in Fig. 3A, MARCKS was
identified as a potential target gene of miR-34c-3p, with the
predicted binding site between base positions 412 and 418. To
validate whether the 3′-UTR of MARCKS is a functional target of
miR-34c-3p, a dual-luciferase reporter system was employed. 3′-UTR
sequences were cloned that contained wild type or mutated binding
sites of miR-34c-3p into the pMIR-Report vector and co-transfected
with the miR-34c-3p mimics or NC into OS cells. Data from the
luciferase assay demonstrated that overexpression of miR-34c-3p
significantly suppressed the luciferase activity of the reporter
gene with the wild type construct (P=0.006 and P=0.008;
respectively) but not with the mutant MARCKS 3′-UTR construct in OS
cells (Fig. 3B and C). In
addition, overexpression of miR-34c-3p resulted in a reduction of
MARCKS mRNA and protein expression in OS cells (P=0.042 and
P=0.037, respectively, Fig. 3D and
E). Therefore, these results suggested that MARCKS is a direct
target of miR-34c-3p in OS cells.
Restoration of MARCKS reverses the
suppressive effects of miR-34c-3p in OS cell growth
To further confirm MARCKS as a direct target of
miR-34c-3p, the present study co-transfected MG-63 cells with
miR-34c-3p mimics and MARCKS plasmid with or without 3′-UTR. As
presented in Fig. 4A,
miR-34c-3p-mediated MARCKS downregulation was restored following
co-transfection with a MARCKS plasmid without 3′-UTR. In addition,
the inhibitory role of miR-34c-3p in cell growth and colony
formation was reversed under the condition of overexpression of
MARCKS without 3′-UTR (Fig. 4B and
C). These data suggested that MARCKS is a direct and functional
downstream target of miR-34c-3p in OS cells.
MARCKS is upregulated and inversely
correlated with miR-34c-3p in OS tissues
Finally, it was investigated whether MARCKS was
upregulated in OS tissues and associated with miR-34c-3p
expression. As presented in Fig.
5A, MARCKS was revealed to be upregulated in the majority of OS
tissues compared with matched healthy tissues (P=0.0008). In
addition, correlation analyses revealed that there was a
significant inverse correlation between miR-34c-3p and MARCKS
expression levels in OS tissues (R=−0.43, P=0.0017, Fig. 5B).
Discussion
The present study investigated the biological role
of miR-34c-3p in the progression of OS. It was revealed that
miR-34c-3p was significantly downregulated in OS compared with
matched healthy bone tissue. In addition, functional experiments
demonstrated that miR-34c-3p suppressed cell growth in vitro
and inhibited tumor growth in vivo by targeting MARCKS. The
present study, to the best of our knowledge, has been the first to
reveal the role of miR-34c-3p in OS, as a novel tumor
suppressor.
It has previously been suggested that miR-34c-3p is
downregulated in various tumors, and functions as a tumor
suppressor in numerous types of cancer. Zhou et al (7) and Liu et al (8) demonstrated that miR-34c-3p functions
as a tumor suppressor in NSCLC partially by inhibiting the
pituitary adenylate cyclase 1/mitogen activated protein kinase
pathway and eukaryotic translation initiation factor 4E. Wu et
al (10) demonstrated that the
overexpression of miR-34c-3p suppressed proliferation and invasion
of OS cells by targeting the Notch pathway in glioma. In the
present study, it was demonstrated that miR-34c-3p suppressed OS
cell growth and colony formation in vitro and inhibited OS
growth in vivo, suggesting a tumor suppressive role for
miR-34c-3p in OS.
To determine how miR-34c-3p acts as a tumor
suppressor, the target genes of miR-34c-3p were screened using
bioinformatic analysis. MARCKS was selected as a potential target
gene of miR-34c-3p based on its particular functions and expression
patterns. It had previously remained to be elucidated as to whether
miR-34c-3p directly targeted MARCKS in OS. MARCKS is a ubiquitously
expressed protein kinase C substrate, which binds actin and
calmodulin, and regulates actin dynamics (13). MARCKS is involved in multiple
cellular processes, including cell adhesion, migration, metastasis,
membrane trafficking and motility via regulation of the actin
cytoskeletal structure (14,15).
The expression of MARCKS has been demonstrated to be downregulated
in hepatocellular carcinoma tissues, and downregulation of MARCKS
may increase the migration of human hepatic stellate cells
(16,17). It was additionally involved in
12-O-tetradecanoylphorbol-13-acetate-mediated migration of
neuroblastoma cells (18). It has
previously been suggested that elevated MARCKS phosphorylation
contributes to the unresponsiveness of breast cancer to paclitaxel
treatment (19). The present study
hypothesized that MARCKS was the precise intracellular target of
miR-34c-3p by using the miRanda, TargetScan and Pictar databases.
Furthermore, MARCKS was verified as a direct target of miR-34c-3p
using the luciferase reporter gene assay. Overexpression of
miR-34c-3p suppressed mRNA and protein expression levels of MARCKS.
Furthermore, the inhibitory effects of miR-34c-3p on cell
proliferation and colony formation were reversed under conditions
of MARCKS overexpression. In OS tissues, it was demonstrated that
expression of MARCKS was upregulated, and miR-34c-3p levels were
inversely correlated with MARCKS expression levels. These data
suggested that MARCKS is a direct and functional target of
miR-34c-3p in OS cells. However, the role of MARCKS in OS remains
to be fully elucidated.
In conclusion, the present study combined clinical
and experimental studies to establish a critical role for
miR-34c-3p in OS. The results demonstrated that miR-34c-3p directly
targeted the 3′UTR of MARCKS, and promoted tumor cell growth, which
further contributed to a high mortality rate for patients with OS.
Therefore, miR-34c-3p may represent a novel therapeutic target for
OS treatment in the future.
References
|
1
|
Luetke A, Meyers PA, Lewis I and Juergens
H: Osteosarcoma treatment-where do we stand? A state of the art
review. Cancer Treat Rev. 40:523–532. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Benjamin RS: Osteosarcoma: Better
treatment through better trial design. Lancet Oncol. 16:12–13.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li F, Li S and Cheng T: TGF-β1 promotes
osteosarcoma cell migration and invasion through the
miR-143-versican pathway. Cell Physiol Biochem. 34:2169–2179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berindan-Neagoe I, Pdel Monroig C,
Pasculli B and Calin GA: MicroRNAome genome: A treasure for cancer
diagnosis and therapy. CA Cancer J Clin. 64:311–336. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou YL, Xu YJ and Qiao CW: MiR-34c-3p
suppresses the proliferation and invasion of non-small cell lung
cancer (NSCLC) by inhibiting PAC1/MAPK pathway. Int J Clin Exp
Pathol. 8:6312–6322. 2015.PubMed/NCBI
|
|
8
|
Liu F, Wang X, Li J, Gu K, Lv L, Zhang S,
Che D, Cao J, Jin S and Yu Y: miR-34c-3p functions as a tumour
suppressor by inhibiting eIF4E expression in non-small cell lung
cancer. Cell Prolif. 48:582–592. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lopez JA and Alvarez-Salas LM:
Differential effects of miR-34c-3p and miR-34c-5p on SiHa cells
proliferation apoptosis, migration and invasion. Biochem Biophys
Res Commun. 409:513–519. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu Z, Wu Y, Tian Y, Sun X, Liu J, Ren H,
Liang C, Song L, Hu H, Wang L and Jiao B: Differential effects of
miR-34c-3p and miR-34c-5p on the proliferation, apoptosis and
invasion of glioma cells. Oncol Lett. 6:1447–1452. 2013.PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jiang C, Chen H, Shao L and Wang Q:
MicroRNA-1 functions as a potential tumor suppressor in
osteosarcoma by targeting Med1 and Med31. Oncol Rep. 32:1249–1256.
2014.PubMed/NCBI
|
|
13
|
Swierczynski SL and Blackshear PJ:
Membrane association of the myristoylated alanine-rich C kinase
substrate (MARCKS) protein. Mutational analysis provides evidence
for complex interactions. J Biol Chem. 270:13436–13445. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yarmola EG, Edison AS, Lenox RH and Bubb
MR: Actin filament cross-linking by MARCKS: Characterization of two
actin-binding sites within the phosphorylation site domain. J Biol
Chem. 276:22351–22358. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arbuzova A, Schmitz AA and Vergéres G:
Cross-talk unfolded: MARCKS proteins. Biochem J. 362:1–12. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rombouts K, Lottini B, Caligiuri A, Liotta
F, Mello T, Carloni V, Marra F and Pinzani M: MARCKS is a
downstream effector in platelet-derived growth factor-induced cell
motility in activated human hepatic stellate cells. Exp Cell Res.
314:1444–1454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Masaki T, Tokuda M, Yoshida S, Nakai S,
Morishita A, Uchida N, Funaki T, Kita Y, Funakoshi F, Nonomura T,
et al: Comparison study of the expressions of myristoylated
alanine-rich C kinase substrate in hepatocellular carcinoma, liver
cirrhosis, chronic hepatitis, and normal liver. Int J Oncol.
26:661–671. 2005.PubMed/NCBI
|
|
18
|
Stensman H and Larsson C: Protein kinase
Cepsilon is important for migration of neuroblastoma cells. BMC
cancer. 8:3652008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen CH, Cheng CT, Yuan Y, Zhai J, Arif M,
Fong LW, Wu R and Ann DK: Elevated MARCKS phosphorylation
contributes to unresponsiveness of breast cancer to paclitaxel
treatment. Oncotarget. 6:15194–15208. 2015. View Article : Google Scholar : PubMed/NCBI
|