Introduction
Inflammation results in the initiation and
progression of atherosclerosis. Atherosclerosis is the primary
cause of cardiovascular disease and conditions that affect the
cerebral, coronary and peripheral vasculature, and is the most
common cause of morbidity and mortality worldwide (1). Endothelial dysfunction is
characterized by reduced endothelial nitric oxide (NO) synthase
(eNOS)-derived NO bioactivity, and the impairment of
endothelium-dependent vascular dysfunction is the progenitor of
atherogenesis (2). Approaches
designed to improve endothelial function are expected to have
therapeutic value in the prevention or treatment of atherosclerosis
(3). Perivascular adipose tissue
(PVAT) directly surrounds vessels and influences their function via
a paracrine effect.
Adenosine monophosphate-activated protein kinase
(AMPK) is an important regulator of energy metabolic homeostasis
and emerging evidence demonstrates its anti-inflammatory action in
vessel and adipose tissue (4,5).
AMPK may modulate a number of signaling cascades that are expected
to have anti-endothelial cell dysfunction, including the
attenuation of free radicals (6).
In a preliminary experiment, the authors demonstrated that
pharmacological activation of AMPK beneficially regulated
adipocytokine expression in PVAT against inflammatory insult and
ameliorated endothelial dysfunction. These findings demonstrated
the role of AMPK activation in the regulation of PVAT and
endothelial function (7).
Methotrexate (MTX), a non-specific anti-inflammatory
therapy, may be an ideal agent to directly test the inflammatory
hypothesis of atherosclerosis, as it inhibits inflammation with
only minimal impact on other components of the atherosclerotic
process and exhibits an acceptable safety profile (8,9). A
recent pre-clinical study with cholesterol-fed rabbits revealed
that MTX markedly reduced atherosclerosis, without effects on
plasma lipid and lipoprotein levels (10). The Cardiovascular Inflammation
Reduction Trial evaluates the use of very low-dose MTX on
cardiovascular events and plasma lipid levels in coronary artery
disease patients with elevated C-reactive protein levels and is
expected to offer more conclusive results in the future (9).
The present study investigated the effect of MTX on
AMPK activation and adipocytokine expression in PVAT with emphasis
on the regulation of endothelial function. It was observed that MTX
ameliorated endothelial dysfunction by inhibiting inflammation in
PVAT. These findings provide novel information regarding the
underlying mechanism of MTX in the management of cardiovascular
diseases.
Materials and methods
Animals
Sprague-Dawley male rats (n=24; 8-weeks old; weight,
200–250 g) were supplied by the Laboratory Animal Center of Nanjing
Qinglongshan (Nanjing, China). The care and treatment of these rats
was performed in accordance with the Provisions and General
Recommendation of Chinese Experimental Animals Administration
Legislation. All animals were housed in a room with a constant
temperature (22±1°C) and a humidity of 50±%, allowed access to a
standard diet and water ad libitum, and exposed to a 12-h
light/dark cycle. The present study was approved by the Ethics
Committee of Ninth Hospital of Xi'an (Xi'an, China).
Preparation of PVAT-derived
conditioned medium (CM)
Sprague-Dawley rats were anesthetized with diethyl
ether and sacrificed by cervical dislocation. PVAT (located around
the thoracic aorta) was isolated, sectioned into small pieces and
rinsed in phosphate buffered saline (PBS). Equal quantities (40 mg)
of PVAT were individually pretreated with MTX (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) (0.1, 0.25 and 0.5 µM) or salicylate
(5 mM; Tianjin Kemiou Chemical Reagent Co., Ltd., Tianjin, China)
or aminoimidazole-4-carboxamide ribonucleotide (AICAR; 500 µM;
Sigma-Aldrich, Merck Millipore) in the presence or absence of 25 µM
AMPK inhibitor compound C (Sigma-Aldrich; Merck Millipore) and
stimulated with 100 µM palmitic acid (PA; Sinopharm Chemical
Reagent Co., Ltd., Shanghai, China) for 2 h. Following treatment,
the PVAT was washed twice with PBS to remove the reagents, and
cultured in fresh Dulbecco's modified Eagle's medium (DMEM, Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% (v/v) fetal bovine serum (FBS) for 22 h at 37°C. The medium was
collected as CM. The supernatant was then collected as conditioned
CM and stored at −70°C.
Endothelium-dependent relaxation
assessment
Endothelium-dependent relaxation was assayed as
previously described (7). Briefly,
the prepared aortic ring was suspended in an organ bath containing
10 ml K-H solution (118.3 mM NaCl, 4.7 mM KCl, 1.2 mM
MgSO4, 1.2 mM KH2PO4, 2.5 mM
CaCl2, 25 mM NaHCO3, 0.026 mM calcium
disodium EDTA and 5.0 mM glucose, pH 7.4) maintained at 37°C pH
7.4, and continuously aerated with 95% O2 and 5%
CO2. Following a 60 min stabilization period, the
contractive ability of the vessel was examined by contractive
response to 60 mM KCl, and the functionality of vascular
endothelium was confirmed by relaxation to 10 µM acetylcholine
(ACh; Sigma-Aldrich; Merck Millipore). The aortic ring (relaxation,
≥80%) was treated with CM for 30 min. Following washing, the aortic
ring was pre-contracted with phenylephrine and the
endothelium-dependent relaxation was induced by cumulative addition
of ACh (0.001–10 µM). Relaxation was expressed as a percentage of
the phenylephrine-induced contraction.
Cell culture
3T3-L1 pre-adipocytes were purchased from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China). A total
of 2×105/ml cells were cultured in 6-well plates in DMEM containing
10% FBS and 25 mM glucose at 37°C in a humidified atmosphere
containing 5% CO2.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
PVAT from rats were pretreated with MTX in the
presence of PA (100 µM) for 2 h. Following washing with PBS, PVAT
was cultured in fresh DMEM supplemented with 10% (v/v) FBS for 22
h. PVAT was then collected and homogenized with 500 µl
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc.) on
ice. Total RNA was obtained using Total RNA Extraction Reagent
(Nanjing Sunshine Biotechnology Co., Ltd., Nanjing, China). cDNA
was synthesized with the TransScript First-strand cDNA Synthesis
SuperMix kit (Beijing Transgen Biotech Co., Ltd., Beijing, China).
qPCR was performed using SsoFast™ EvaGreen Supermix (Bio-Rad
Laboratories Inc., Hercules, CA, USA). Amplification was performed
with the Bio-Rad iQ5 sequence detection system (Bio-Rad
Laboratories Inc.) with the following conditions: 95°C for 10 min,
followed by 40 cycles at 95°C for 30 sec, and 52°C for 15 sec, and
a final extension at 72°C for 5 min. The following primers were
used: Adiponectin forward, 5′-AAGGGGACAACAATGGACTCTA-3′, and
reverse, 5′-CTACGGGCTGCTCTGAATTAGT-3′; β-actin forward,
5′-GACGTTGACATCCGTAAAGACC-3′, and reverse,
5′-TGCTAGGAGCCAGGGCAGTA-3′. The mRNA expression level of individual
genes was normalized and presented as a ratio to β-actin, and
calculated using the 2-ΔΔCq method (11).
Western blot analysis
PVAT from Sprague-Dawley rats was pretreated with
MTX for 30 min and then incubated with PA for a further 30 min. The
aorta was incubated with CM for 30 min and then exposed to ACh for
another 30 min. PVAT was isolated directly following the sacrifice
of the rats. For protein analysis, PVAT or the aorta was
homogenized in radio immunoprecipitation assay lysis buffer with
phenylmethane sulfonyl fluoride. Protein concentration in the
supernatants was assayed using Bicinchoninic Acid Protein Assay kit
(Nanjing Baxi Technology Co., Ltd. Nanjing, China). Equal amounts
of protein (30 µg) were separated by 10% SDS-PAGE and transferred
to 0.45 µM polyvinylidene difluoride (PVDF) membranes (EMD
Millipore, Billerica, MA, USA) by semi-dry electrophoretic transfer
(Bio-Rad Laboratories, Inc.). The PVDF membranes were blocked with
5% non-fat milk in Tris-buffered saline with Tween-20 (TBST) and
then incubated with the appropriate primary antibodies overnight at
4°C: Rabbit monoclonal anti-phospho-AMPK (cat. no. 2531) and
anti-AMPK (cat. no. 2532), rabbit monoclonal anti-eNOS (cat. no.
9586) and anti-p-eNOS (cat. no. 9571), anti-NF-κB p65 (cat. no.
4764) and anti-p-NF-kB p65 (cat. no. 3033; all at 1:1,000 and
obtained from Cell Signaling Technology, Inc., Beverly, MA, USA).
The PVDF membrane was washed three times with TBST buffer and then
incubated with the HRP-conjugated secondary antibodies (cat. no.
BS13278; 1:10,000, MyBioSource, Inc., San Diego, CA, USA) at room
temperature for 2 h. The membranes were developed with an enhanced
chemiluminescence detection reagent (Thermo Fisher Scientific,
Inc.) and quantitated by densitometry with Image-Pro Plus 6.0
software (Media Cybernetics, Inc., Rockville, MD, USA).
ELISA assay
The levels of TNF-α and IL-6 in the supernatant were
assayed with commercial enzyme-linked immunosorbent assay (ELISA)
kits (R&D, Minneapolis, MN, USA).
AMPK small interfering (si)RNA
transfection
3T3-L1 pre-adipocytes were transfected with siRNA
for AMPKα (cat. no. sc-45313) to knock down levels of endogenous
AMPKα, and negative control siRNA (cat. no. sc-37007), using siRNA
transfection reagent (cat. no. sc-29528) in transfection medium
(cat. no. sc-36868) for 6 h, according to the manufacturer's
protocol. All siRNAs were obtained from Santa Cruz Biotechnology,
Inc., Dallas, TX, USA. Following an additional 48 h, the efficiency
of siRNA-mediated AMPK knockdown was confirmed by western blot
analysis as aforementioned.
Statistical analysis
Data were expressed as the mean ± standard deviation
from at least three independent experiments. SPSS version 13.0
software (SPSS, Inc., Chicago, IL, USA) was used for the
statistical analysis. Individual group statistical comparisons were
analyzed using an unpaired Student's t-test with Bonferroni
correction and multiple-group comparisons were evaluated by one-way
analysis of variance followed by Tukey's post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
MTX regulates AMPK activation in
PVAT
AMPK is important in the regulation of lipid
metabolism, therefore the present study first investigated the
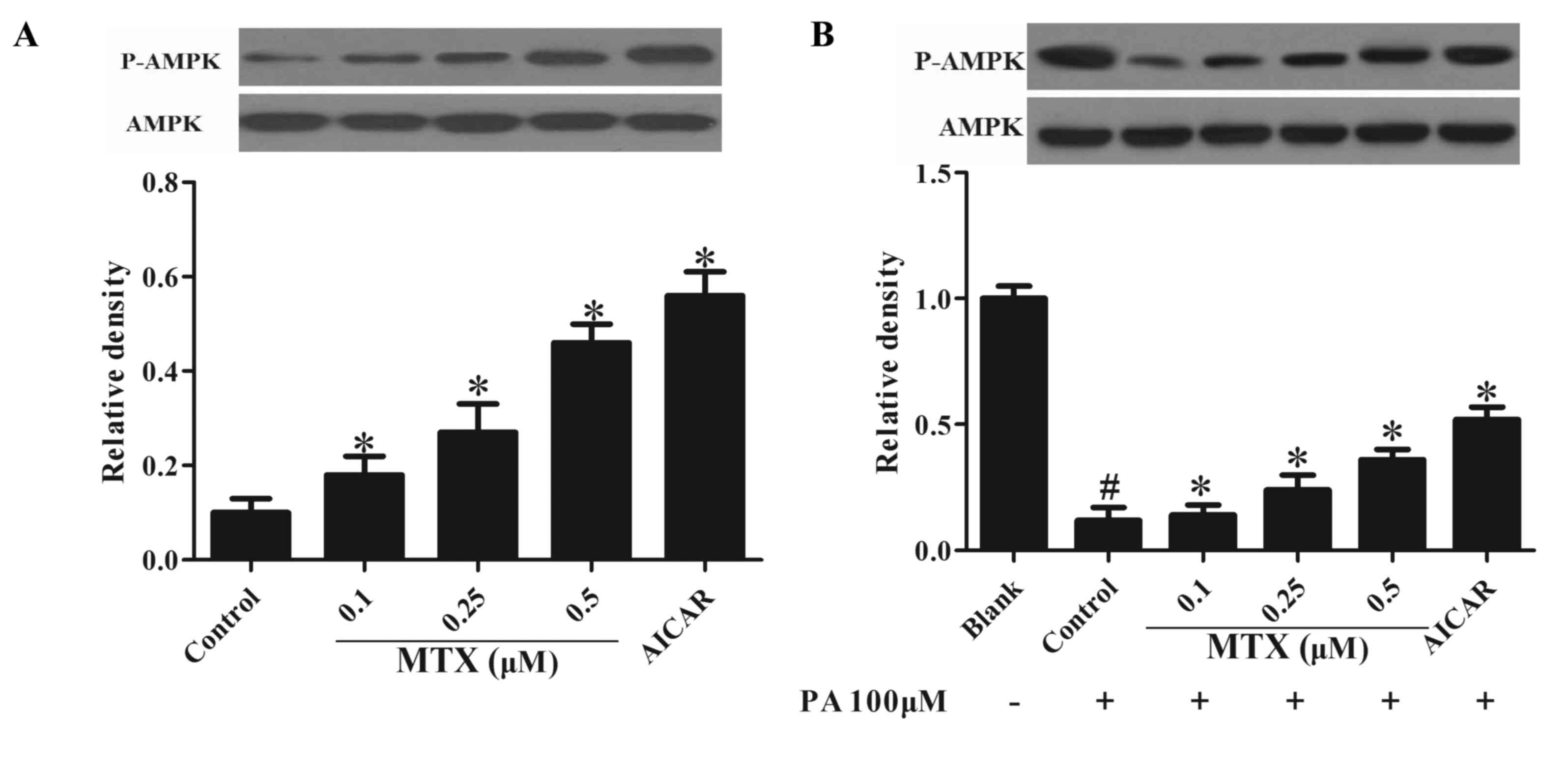
effect of MTX on AMPK activity in PVAT. As presented in Fig. 1A, MTX treatment increased basal
AMPK activity at concentrations ranging from 0.1 to 0.5 µM,
indicated by enhanced AMPK phosphorylation. In addition, it was
observed that when PVAT was exposed to PA, a reduction of AMPK
phosphorylation was observed, and this alteration was prevented by
pretreatment with MTX (P<0.05, Fig.
1B). Furthermore, treatment with AICIR which is an AMPK
agonist, exhibited similar effects (P<0.05). These results
indicated that MTX enhanced AMPK activation under basal and
inflammatory conditions.
MTX inhibits inflammation and
modulates adipocytokine expression in PVAT
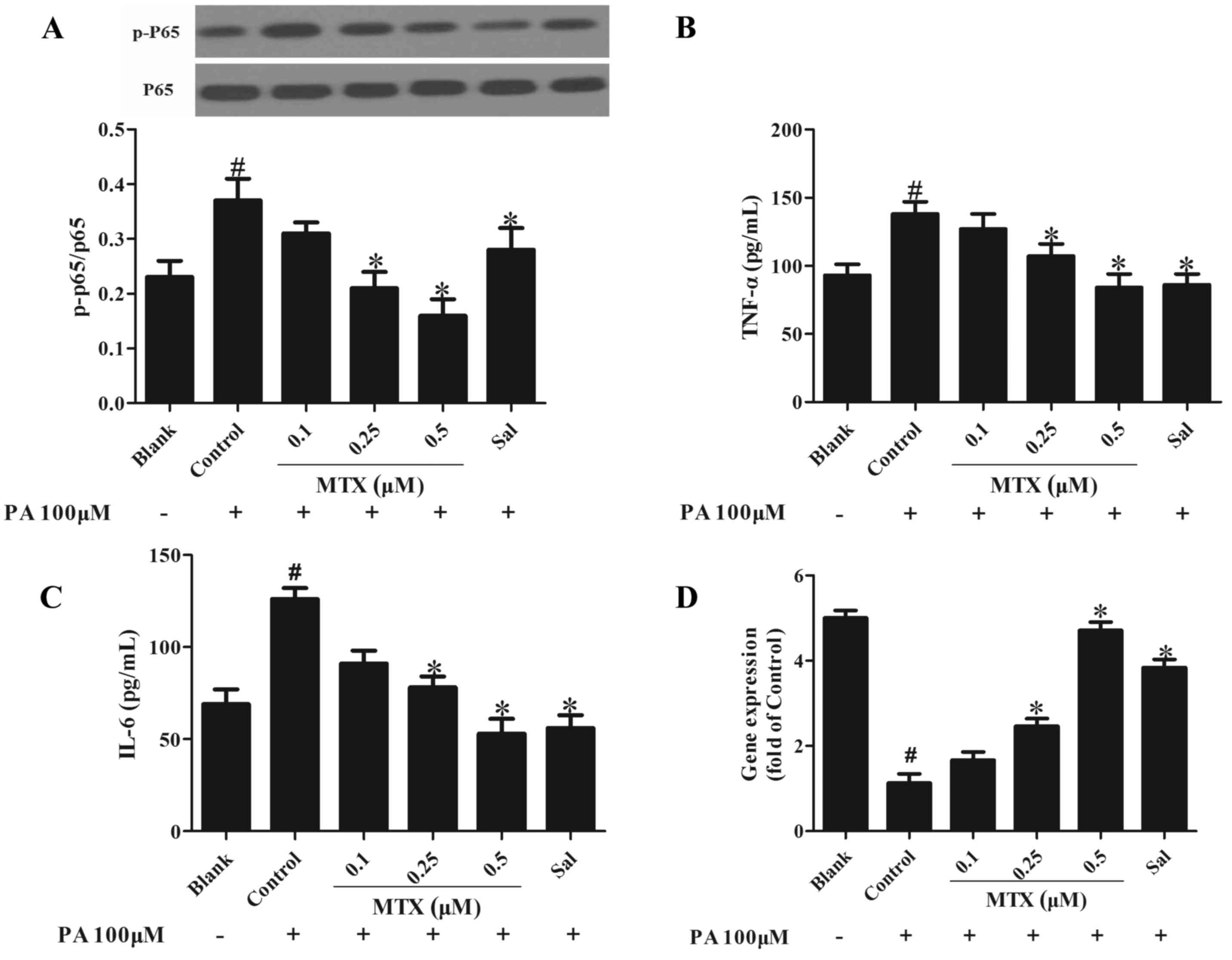
It has previously been demonstrated that free fatty
acids induce inflammation in the endothelium via the nuclear factor
(NF)-κB pathway and stimulation with PA enhanced phosphorylation of
NF-κB p65 in PVAT (12). As
presented in Fig. 2A, MTX
effectively reduced NF-κB p65 phosphorylation, indicative of its
anti-inflammatory activity (P<0.05). In addition, the effect of
MTX on the expression of adipocytokines implicated in inflammation
was examined. As presented in Fig. 2B
and C, the expression levels of pro-inflammatory cytokines,
including tumor necrosis factor (TNF)-α and interleukin (IL)-6,
were increased in cells treated with PA for 24 h and this was
reversed by MTX pretreatment (P<0.05), demonstrating its
anti-inflammatory activity. Furthermore, the expression of
adiponectin was downregulated, however these alterations were
reversed by treatment with MTX (P<0.05, Fig. 2D).
AMPK silencing blocks the
anti-inflammatory effect of MTX
The AMPK inhibitor to confirm the role of AMPK in
the anti-inflammatory effect of MTX, 3T3-L1 cells were transfected
with AMPKa1/2-specific siRNA to knockdown AMPK expression. As
presented in Fig. 3A, silencing
AMPK significantly decreased the inhibitory effect of MTX on
PA-mediated NF-κB p65 phosphorylation (P<0.05). MTX treatment
altered the production of TNF-α (Fig.
3B), IL-6 (Fig. 3C) and
adiponectin (Fig. 3D) upon PA
challenge, however, this action was also blocked by knockdown of
AMPK (P<0.05, Fig. 3B-D). These
results indicated that MTX inhibited inflammation in a
AMPK-dependent manner.
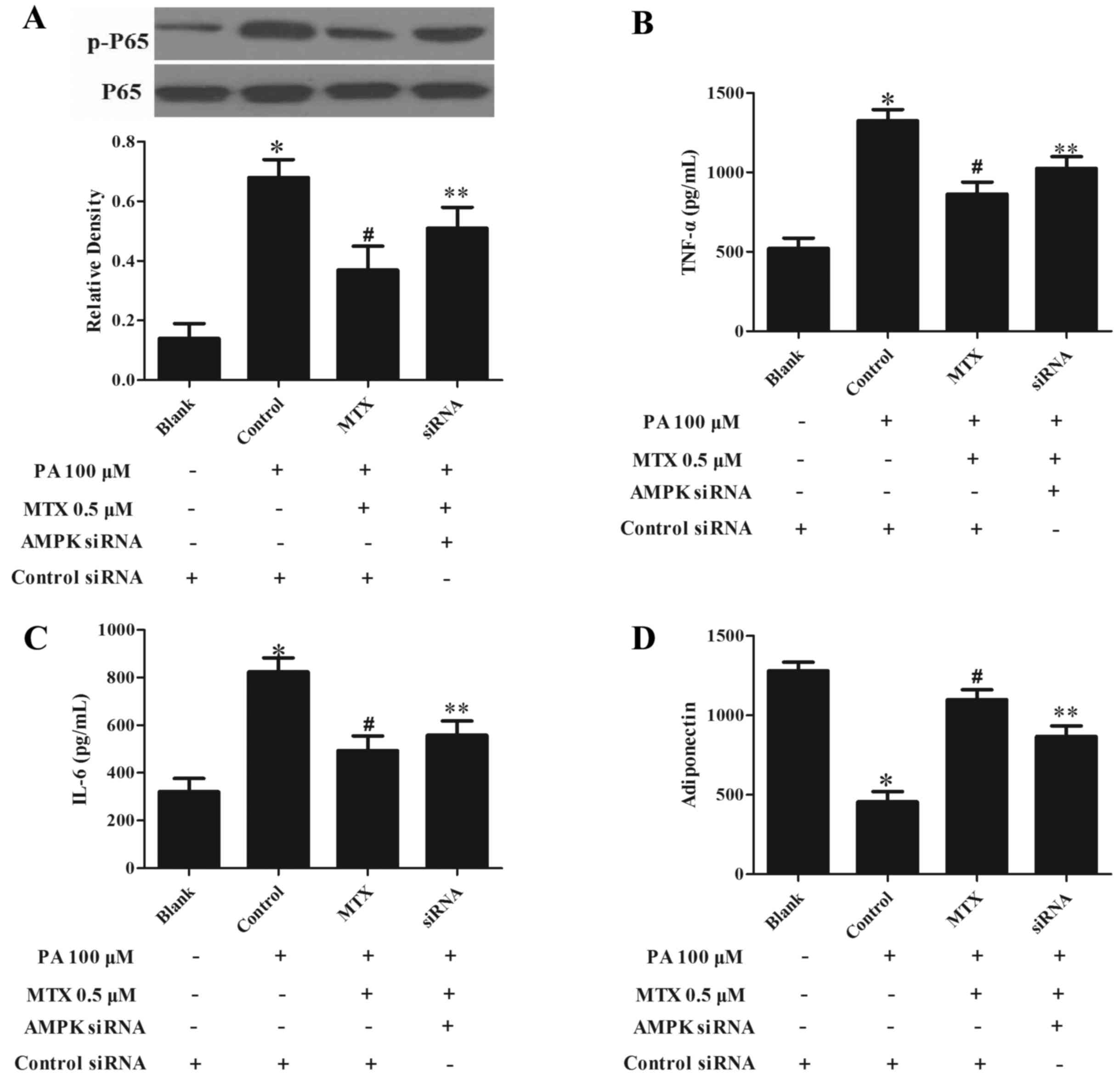 | Figure 3.AMPK siRNA transfection impairs the
ability of MTX to suppress inflammation in 3T3-L1 pre-adipocytes.
(A) 3T3-L1 pre-adipocytes were transfected with AMPKα1/2 siRNA or
control siRNA. siRNA-transfected adipocytes were pretreated with
MTX for 30 min, then stimulated with 100 µM PA for a further 30
min. Nuclear factor-κB p65 phosphorylation was assayed by western
blot analysis. Following transfection, cells were incubated with
MTX in the presence of 100 µM PA for 2 h, then cultured in fresh
medium for a further 22 h, and the concentrations of (B) TNF-α, (C)
IL-6 and (D) adiponectin in the supernatant were measured with
ELISA kits. Data are expressed as the mean ± standard deviation.
*P<0.05 vs. Blank; #P<0.05 vs. control;
**P<0.05 vs. AMPK siRNA treatment. MTX, methotrexate; PA,
palmitic acid; p, phosphorylated; TNF-α, tumor necrosis factor-α;
IL-6, interleukin-6; AMPK, adenosine monophosphate-activated
kinase; siRNA, small interfering RNA. |
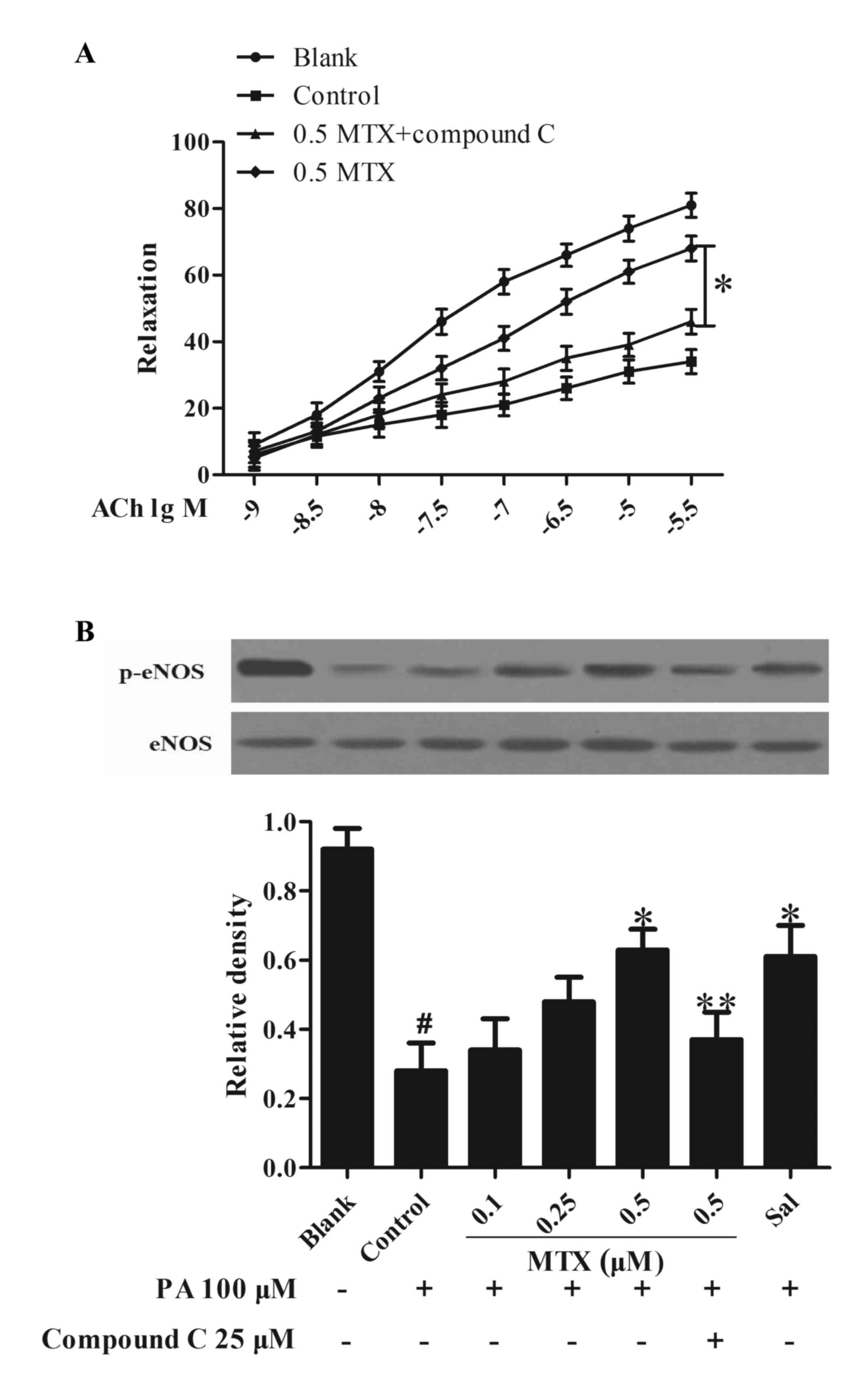
Treatment of PVAT with MTX restores
the loss of ACh-induced vasodilation
Endothelial dysfunction is characterized by the loss
of endothelium-dependent vasodilation. To investigate the influence
of PVAT on vessel function, PVAT was incubated with PA and
collected the medium as CM to stimulate the rat aorta. Data
presented in Fig. 4A indicated
that ACh induced vasodilation, whereas stimulation with PA led to a
significant loss in vessel relaxation (P<0.05). Pretreatment of
PVAT with MTX prevented the alterations and effectively restored
the loss of ACh-mediated vessel relaxation in a
concentration-dependent manner, but this action was attenuated by
co-treatment with AMPK inhibitor compound C (P<0.05, Fig. 4A). Furthermore, it was observed
that treating PVAT with MTX effectively normalized eNOS
phosphorylation in the aorta subjected to CM challenge (P<0.05,
Fig. 4B). AMPK inhibitor compound
C blocked the actions of MTX in the regulation of vasodilation and
eNOS phosphorylation, suggesting the involvement of AMPK in the
mechanism of MTX action. These results suggested that MTX improved
vessel function via regulation of AMPK activity.
Discussion
Endothelial homeostasis is important in the
regulation of vessel function and may predict the development of
cardiovascular diseases independently of other known risk factors
(13). PVAT is a functional
component of the vasculature, exerting paracrine influences on
endothelial homeostasis. A previous study has investigated the
regulation of vessel tone by PVAT, attempting to identify
adipocyte-derived relaxation and constriction factors, which are
endothelium-independent (14).
Inflammation in PVAT induces dysregulation of adipocytokine
expression and subsequently impairs the integrity of the
endothelium, which results in its dysfunction. The present study
prepared an ex vivo model of PVAT/endothelial dysfunction by
treating rat aorta with PVAT-derived CM and successfully observed
the beneficial effects of MTX on adipocytokine expression and
further implications in endothelial function.
In addition to regulating energy metabolism, AMPK
exerts anti-inflammatory activity, and this action has been
implicated in the normalization of adipose and endothelial
functions. The present study first investigated the role of MTX in
the regulation of AMPK activation in PVAT, and observed that AMPK
activity was increased by enhancing phosphorylation. PA stimulation
was demonstrated to reduce AMPK phosphorylation in PVAT, consistent
with results from a previous study (15).
It has previously been demonstrated that nanomolar
concentrations of NO have anti-inflammatory and protective
properties that are mediated by the inhibition of the activation of
NF-κB (16). PA stimulation evoked
inflammation in PVAT, which induced adipose dysfunction as
demonstrated by enhanced NF-κB p65 phosphorylation. MTX attenuated
NF-κB p65 phosphorylation, suggesting the inhibition of NF-κB
inflammatory signaling. This finding is consistent with a previous
study in which PA induced NF-κB-dependent inflammation by binding
to toll-like receptor 4 in the endothelium (17). The expression levels of
pro-inflammatory adipocytokines, including TNF-α and IL-6, were
increased, whereas gene expression levels of adiponectin were
downregulated. MTX reversed the alteration of adipocytokine
expression, exhibiting a similar effect to that induced by
salicylate, demonstrating its anti-inflammatory potency in the
endothelium. To further elucidate the role of AMPK in the
anti-inflammatory activity of MTX, the activity of MTX in
adipocytes was observed. AMPKα knockdown using siRNA diminished the
inhibitory effect of MTX on NF-κB activation and blocked its
beneficial regulation of TNF-α, IL-6 and adiponectin production,
further verifying the role of AMPK in the anti-inflammatory
activity of MTX.
Endothelial dysfunction is characterized by the loss
of endothelium-dependent vasodilation (18,19).
eNOS-derived NO, which is a gaseous signaling molecule, is
important in the maintenance of vascular homeostasis by promoting
vasodilation and inhibiting inflammation (20). Therefore, in order to observe the
effects of AMPK agents on endothelial homeostasis via regulation of
PVAT function, the present study stimulated PVAT with MTX and
collected the medium as CM to treat rat aorta. Furthermore,
ACh-induced endothelium-dependent vasodilation is mediated via eNOS
activation and subsequent NO production. Pretreatment of PVAT with
PA reduced eNOS phosphorylation and impaired ACh-mediated
vasodilation, indicating the association between dysregulation of
adipocytokine expression and endothelial dysfunction. Co-treatment
with the AMPK inhibitor compound C blocked the action of MTX on
vasodilation, further demonstrating the involvement of AMPK in its
regulation.
In conclusion, the present study established an
ex vivo model of PVAT/endothelial dysfunction by stimulating
rat aorta with CM derived from PVAT. It was demonstrated that
pharmacological activators of AMPK regulated adipocytokine
expression by inhibiting PVAT inflammation, and thereby ameliorated
endothelial dysfunction in an AMPK interdependent manner. These
findings provide a novel insight into the potential underlying
mechanism by which MTX protects endothelial function against
inflammatory insult.
References
|
1
|
World Health Statistics, . 2012, World
Health Organization. Geneva: 2012
|
|
2
|
Vita JA and Keaney JF: Endothelial
function: A barometer for cardiovascular risk? Circulation.
106:640–642. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li AC and Glass CK: The macrophage foam
cell as a target for therapeutic intervention. Nat Med.
8:1235–1242. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bijland S, Mancini SJ and Salt IP: Role of
AMP-activated protein kinase in adipose tissue metabolism and
inflammation. Clin Sci (Lond). 124:491–507. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gauthier MS, O'Brien EL, Bigornia S, Mott
M, Cacicedo JM, Xu XJ, Gokce N, Apovian C and Ruderman N: Decreased
AMP-activated protein kinase activity is associated with increased
inflammation in visceral adipose tissue and with whole-body insulin
resistance in morbidly obese humans. Biochem Bioph Res Commun.
404:382–387. 2011. View Article : Google Scholar
|
|
6
|
Tsai KL, Chen LH, Chiou SH, Chiou GY, Chen
YC, Chou HY, Chen LK, Chen HY, Chiu TH, Tsai CS, et al: Coenzyme
Q10 suppresses oxLDL-induced endothelial oxidative injuries by the
modulation of LOX-1-mediated ROS generation via the AMPK/PKC/NADPH
oxidase signaling pathway. Mol Nutr Food Res. 55 Suppl 2:S227–S240.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun Y, Li J, Xiao N, Wang M, Kou J, Qi L,
Huang F, Liu B and Liu K: Pharmacological activation of AMPK
ameliorates perivascular adipose/endothelial dysfunction in a
manner interdependent on AMPK and SIRT1. Pharmacol Res. 89:19–28.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Saag KG, Teng GG, Patkar NM, Anuntiyo J,
Finney C, Curtis JR, Paulus HE, Mudano A, Pisu M, Elkins-Melton M,
et al: American College of Rheumatology 2008 recommendations for
the use of nonbiologic and biologic disease-modifying antirheumatic
drugs in rheumatoid arthritis. Arthritis Rheum. 59:762–784. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ridker PM: Testing the inflammatory
hypothesis of atherothrombosis: Scientific rationale for the
cardiovascular inflammation reduction trial (CIRT). J Thromb
Haemost. 7 Suppl 1:S332–S339. 2009. View Article : Google Scholar
|
|
10
|
Bulgarelli A, Dias AA Martins, Caramelli B
and Maranhão RC: Treatment with methotrexate inhibits atherogenesis
in cholesterol-fed rabbits. J Cardiovasc Pharmacol. 59:308–314.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mugabo Y, Mukaneza Y and Renier G:
Palmitate induces C-reactive protein expression in human aortic
endothelial cells. Relevance to fatty acid-induced endothelial
dysfunction. Metabolism. 60:640–648. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calles-Escandon J and Cipolla M: Diabetes
and endothelial dysfunction: A clinical perspective. Endocr Rev.
22:36–52. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gollasch M: Vasodilator signals from
perivascular adipose tissue. Br J Pharmacol. 165:633–642. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Steinberg GR, Michell BJ, van Denderen BJ,
Watt MJ, Carey AL, Fam BC, Andrikopoulos S, Proietto J, Görgün CZ,
Carling D, et al: Tumor necrosis factor alpha-induced skeletal
muscle insulin resistance involves suppression of AMP-kinase
signaling. Cell Metab. 4:465–474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng HB, Libby P and Liao JK: Induction
and stabilization of I kappa B alpha by nitric oxide mediates
inhibition of NF-kappa B. J Biol Chem. 270:14214–14219. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Maloney E, Sweet IR, Hockenbery DM, Pham
M, Rizzo NO, Tateya S, Handa P, Schwartz MW and Kim F: Activation
of NF-kappaB by palmitate in endothelial cells: A key role for
NADPH oxidase-derived superoxide in response to TLR4 activation.
Arterioscl Throm Vas Biol. 29:1370–1375. 2009. View Article : Google Scholar
|
|
18
|
Félétou M and Vanhoutte PM: Endothelial
dysfunction: A multifaceted disorder (The Wiggers Award Lecture).
Am J Physiol Heart Circ Physiol. 291:H985–H1002. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cersosimo E and DeFronzo RA: Insulin
resistance and endothelial dysfunction: The road map to
cardiovascular diseases. Diabetes Metab Res Rev. 22:423–436. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Moncada S: Nitric oxide in the
vasculature: Physiology and pathophysiology. Ann NY Acad Sci.
811:60–69. 1997. View Article : Google Scholar : PubMed/NCBI
|