Introduction
Idiopathic pulmonary fibrosis (IPF) is the most
common of the interstitial pneumonias and the most aggressive
interstitial lung disease (1). The
etiology of IPF still remains to be elucidated and thus, a
successful treatment remains to be identified. The disease is more
common in males, particularly those aged between 50 and 70
(2), and the incidence of IPF
rises markedly with age. The prevalence of IPF ranges between 13
cases per 100,000 for women to 20 cases per 100,000 for men and the
figures are increasing (3). The
onset of clinical symptoms is insidious, including shortness of
breath on exertion and a dry cough, and certain patients experience
an initial flu-like malaise (1),
leading to a late diagnosis if ignored.
Usually IPF is confirmed by the histopathological
pattern of usual interstitial pneumonia, and requires an integrated
multidisciplinary approach from pulmonologists, radiologists and
pathologists. The common measurements include high-resolution
computed tomography, surgical lung biopsy and radiologic diagnosis.
However, these diagnoses are performed at a late stage of IPF and
are not useful in proposing a plan of treatment.
A recent genetic study (4) assessed early-stage pulmonary fibrosis
as the majority of these mutations are present at birth, predating
disease development, and thus can provide insights into the early
stages. A study of genetic associations (5) holds promise in exhibiting the
connections between early-stage and advanced disease. Although
progress has been made in the field of IPF genetics in identifying
common variants that are associated with IPF diagnosis, rare
variants remain to be analyzed. The use of genetics in early IPF
detection remains in its infancy.
It has been demonstrated (6) that numerous critical genes and
pathways are deregulated during the initiation and progression of a
cancer, certain studies (7,8) have
identified differential expressed genes in IPF and several studies
(7,9) have analyzed pathways in IPF, however
they were non-uniform. Identifying pathways that are deregulated in
patients with cancer may be useful in identifying cancer from
unknown samples. A number of methods have been proposed to identify
differential pathways, including the attract method (10), personal pathway deregulation score
(11) and individualized pathway
aberrance score (12).
Personalized identification of differential pathways provides
pathway interpretation in a single sample with accumulated normal
data.
Support vector machines (SVM) are among the most
powerful classification and prediction methods, first developed by
Cherkassky (13). They are used in
a wide range of scientific applications (14), including cancer tissue
classification (15), protein
domain classification (16) and
splice site prediction (17), due
to their great accuracy, their ability to deal with
high-dimensional and large datasets, and their flexibility in
modeling diverse sources of data (18).
From this perspective, a pathway aberrance analysis
to identify and determine the extent of IPF using the peripheral
blood transcriptome was performed, with the aim of distinguishing
normal individuals from patients with IPF and, additionally, to
distinguish the extent of the disease when samples were classified
by percent predicted diffusion capacity for carbon monoxide of the
lung, however not forced vital capacity (19). Three methods were employed to
identify differential pathways. To analyze the feasibility of
pathway-based diagnosis in IPF, SVM was introduced.
Materials and methods
Dataset
Gene expression data
Microarray data of E-GEOD-33566 (19), together with the annotation files,
were downloaded from the ArrayExpress database (https://www.ebi.ac.uk/arrayexpress). The data
included 93 patients with IPF and 30 healthy controls. Blood was
collected in PAXgene RNA tubes. The platform in this study was
A-AGIL-28-Agilent Whole Human Genome Microarray 4×44K 014850 G4112F
(85 columnsx532 rows) and the platform was designated. The
Peripheral Blood Transcriptome Predicts the Presence and Extent of
Disease in Idiopathic Pulmonary Fibrosis, by which the gene
expression files were generated. According to the gene ID and
symbol in the annotation file of the platform, the gene ID in the
microarray was changed to its designation.
Pathway data and preprocessing
All the pathways of Homo sapiens were derived
from the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway
Database (http://www.kegg.jp) (20). In total, 300 pathways, including
6,919 genes, were obtained. To simplify pathway data, pathways
containing <5 genes were excluded. Eventually, 284 pathways were
obtained for further analysis. Genes common to pathways and samples
were used in subsequent analysis.
Pathway analysis
The aim of the present study was to analyze the
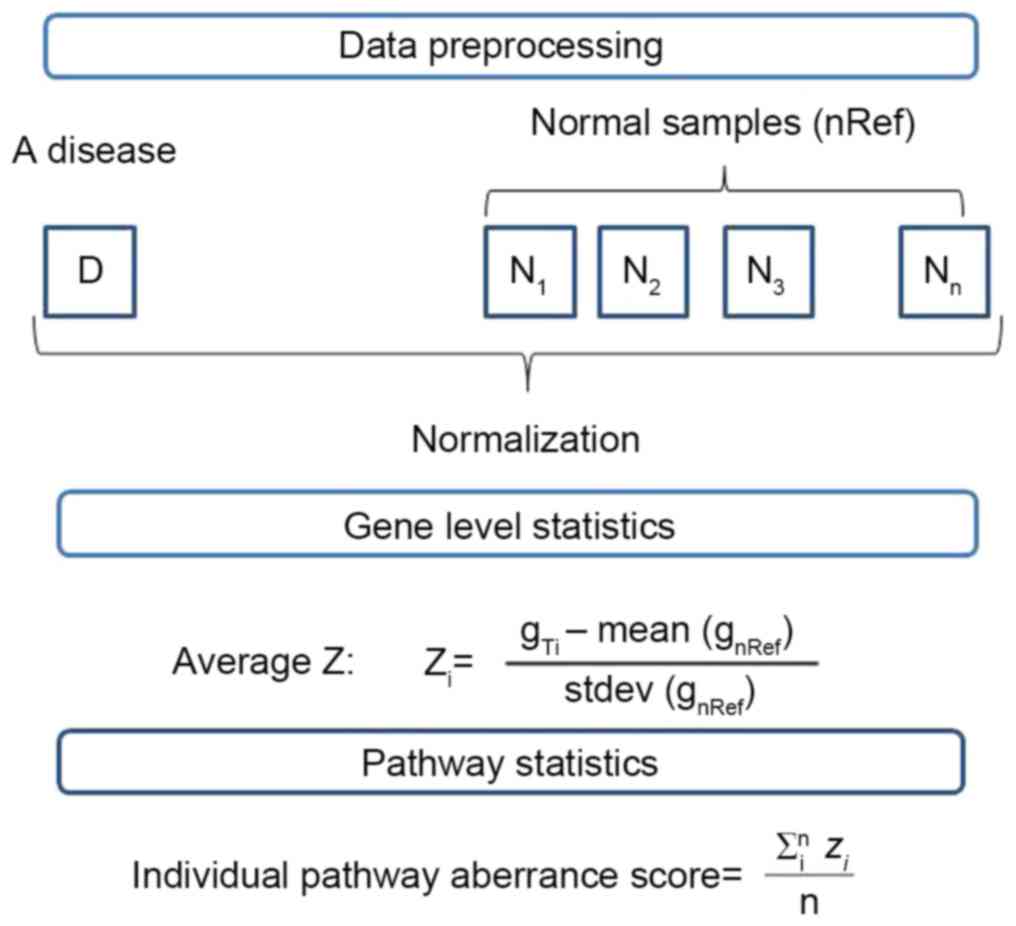
altered pathways in an individual with a disease. The process of
this analysis is presented as Fig.
1.
Gene level statistics
Gene data in the normal group were normalized using
quantile normalization in the preprocessCore package (21), which generated the mean and
standard deviation of gene expression levels. Following the
amalgamation of genes in tumor samples with all the normal samples,
quantile normalization using mean and standard deviation of the
gene expression levels was performed, generating gene level
statistics. The formula was:
Where Zi symbolized the standardized
expression value of the i-th gene, and n represented the
number of genes belonging to the pathway. The results obtained were
gene level statistics.
Zi=gTi–mean(gnRef)stdev(gnRef)
Pathway level statistics
The statistics for each pathway were calculated by
averaging the gene level statistics of all genes belonging to the
pathway, thus:
Pathwaystatistics=∑inzin
Where n represented the number of genes in the
pathway and Zi symbolized the standardized
expression value of i-th gene in the pathway.
Differential pathway screening
A significance test was performed to assess
differential pathways associated with IPF. To identify the best
test protocol to assess differential pathways, three pathway groups
were constructed for comparison.
Wilcoxon-based KEGG Pathway (n>5) group: The
pathway statistics, obtained from the pathways of disease group and
normal group, were tested by Wilcoxon (22) with the function:
E(T)=n(n+1)4
D(T)=n(n+1)(2n+1)24Z=T–E(T)D(T)
Where n is the number of samples.
The significance of the level was corrected by false
discovery rate (FDR) (23).
Subsequently, each pathway was allotted a P-value.
Those pathways with P<0.01 were considered differential
pathways. In total, 106 differential pathways were obtained.
Limma-based KEGG Pathway (n>5) group: The pathway
statistics were performed with Limmae Bayes (24) and top Table functions, generating
P-values. In total, 100 differential pathways were screened out
with P<0.01.
Attract-based KEGG Pathway (n>5) group: Genes in
differential pathways of the Wilcoxon-based KEGG pathway group were
subsequently analyzed using the attract method.
The F-statistics for gene I was calculated by:
F(i)=MSSiRSSi
Where MSSi denotes the mean treatment sum
of squares:
MSSi=1K–1∑k=1Krk[y·k(i)–y··(i)]2
And RSSi denotes the residual sum of
squares:
RSSi=1N–1∑k=1K∑j=1rj[yjk(i)–y··(i)]2
For pathway P consisting of gp genes, the
T-statistic takes the following form:
Tp=[1gp∑i=1gpF(i)]–[1G∑j=1GF(i)](Sp2gp)+(SG2G)
Where G denotes the total number of genes in a
pathway and SP2 and SG2
were defined as sample aberrances.
Following the performance of the t-test and adjusted
with the FDR of Benjamini-Hochberg (25), the pathway statistical value was
transformed into P-values. In total, seven pathways with P<0.05
were identified.
SVM analysis
An SVM method was applied to test the analysis
results of the three pathway groups and 5-fold cross validation was
selected to analyze the SVM model. The pathways statistics of the
normal and disease group were amalgamated and divided into two
sets, the training and the test set, with a ratio of 6:4. These
data were treated with linear SVM, employing the formula:
K(x,xi)=[(x*xi)+1]q
Subsequent to classification, the parameters of the
area under the receiver operator characteristic (ROC) curve (AUC),
accuracy, the Matthews coefficient correlation classification
measure (MCC), the degree of true negative identification
specificity (specificity) and the degree of true positive
identification sensitivity (sensitivity) were ascertained.
Results
Differential pathways
The original KEGG pathway database contains 300
pathways and 6,919 genes. Pathways with <5 genes were deleted,
generating a KEGG Pathway (n>5) group containing 284 pathways
and 4,303 genes. In comparing the healthy (n=30) and diseased
(n=93) lung samples, differential pathways were identified using
three methods.
In the Wilcoxon-based KEGG Pathway (n>5) group,
106 differential pathways were identified, the largest number of
the three groups. By ranking pathways with P-values, five pathways
with the least P-values and gene number are presented in Table I. The P-value can be regarded as an
indicator of the extent of the disease. The first differential
pathway with the least P-value was ‘Amoebiasis’, indicating that it
was among the pathways most susceptible to disease. It is caused by
an extracellular protozoan parasite that invades the intestinal
epithelium and belongs to infectious diseases. The pathway of
‘bladder cancer’ demonstrates that the disease causes urinary
system lesion. The other three pathways are involved in basic
metabolism in the body.
 | Table I.The top five ranked differential
pathways with the least P-values in the Wilcoxon-based KEGG pathway
group (n>5). |
Table I.
The top five ranked differential
pathways with the least P-values in the Wilcoxon-based KEGG pathway
group (n>5).
| Differential
pathway | P-value | Geneno. |
|---|
| Amoebiasis | 0.000151 | 60 |
| Bladder cancer | 0.000186 | 29 |
| Type II diabetes
mellitus | 0.000236 | 30 |
| Primary
immunodeficiency | 0.000386 | 31 |
| Histidine
metabolism | 0.000386 | 9 |
The Limma-based KEGG Pathway (n>5) group
contained 100 differential pathways, six fewer than the
Wilcoxon-based KEGG Pathway (n>5) group. The top five ranked
pathways with the least P-values and gene number are presented in
Table II. Notably, four pathways
were the same as in the Wilcoxon-based KEGG Pathway (n>5) group.
The exception is ‘Notch signaling pathway’, an intercellular
signaling mechanism essential for correct embryonic
development.
| Table II.The top five ranked differential
pathways with P-values in the Limma-based KEGG Pathway group
(n>5). |
Table II.
The top five ranked differential
pathways with P-values in the Limma-based KEGG Pathway group
(n>5).
| Differential
pathway | P-value | Genesno. |
|---|
| Amoebiasis | 0.0000684 | 60 |
| Bladder cancer | 0.0000684 | 9 |
| Type II diabetes
mellitus | 0.00022 | 31 |
| Primary
immunodeficiency | 0.000405 | 30 |
| Histidine
metabolism | 0.000405 | 38 |
The attract-based KEGG Pathway (n>5) group
contained seven differential pathways, and was the smallest group.
These differential pathways were the same as seven of the
differential pathways in Wilcoxon-based KEGG Pathway (n>5)
group, but none of them were in the top five pathways of the latter
group in P-values. The pathways with P-values and gene number are
presented in Table III. The
seven pathways represented the core pathways that reflected the
disease and may aid analysis of the disease. The first ranked
pathway was ‘Ribosome’, which is responsible for genetic
information processing and translation. The ‘Legionellosis’ pathway
is associated with a potentially fatal infectious disease.
‘Pyrimidine metabolism’ is responsible for nucleotide metabolism.
The ‘Renin-angiotensin system’ pathway is a peptidergic system with
endocrine characteristics concerned with the regulation of blood
pressure and hydroelectrolytic balance. The ‘B cell receptor
signaling’ pathway is involved in the immune system. The ‘Oxidative
phosphorylation’ pathway is part of energy metabolism.
| Table III.All the differential pathways with
P-values in the attract-based KEGG Pathway group (n>5). |
Table III.
All the differential pathways with
P-values in the attract-based KEGG Pathway group (n>5).
| Differential
pathways | P-value | Geneno. |
|---|
| Ribosome | 0.000072 | 128 |
| Legionellosis | 0.000072 | 48 |
| Pyrimidine
metabolism | 0.001157 | 79 |
| Renin-angiotensin
system | 0.001157 |
7 |
| B cell receptor
signaling | 0.002139 | 70 |
| Oxidative
phosphorylation | 0.006775 | 115 |
| Osteoclast
differentiation | 0.006775 | 109 |
SVM analysis
To obtain the best performing pathway group, linear
SVM analysis was adopted. In each differential pathway group,
pathways in the normal and disease groups were divided into two
sets, the training and the test set, with a ratio of 6:4. Several
parameters were analyzed to compare the four pathway groups,
including AUC, accuracy, specificity, sensitivity, MCC, true
negative, false positive, true positive and false negative. The
test set of the differential pathway groups with parameters is
presented in Table IV.
| Table IV.Comparison of the test sets of the
three differential pathway groups classified by the method
ofsupport vector machines. |
Table IV.
Comparison of the test sets of the
three differential pathway groups classified by the method
ofsupport vector machines.
| Parameter | Limma-based KEGG
pathway | Wilcoxon-based KEGG
pathway | Attract-based KEGG
pathway |
|---|
| Negative
samples | 14 | 14 | 14 |
| Positive
samples | 36 | 36 | 36 |
| TN | 7 | 8 | 0 |
| FP | 7 | 6 | 14 |
| TP | 31 | 33 | 36 |
| FN | 5 | 3 | 0 |
| AUC |
0.68 |
0.74 |
0.50 |
| Accuracy |
76.00 |
82.00 |
72.00 |
| MCC |
0.38 |
0.53 |
0.00 |
| Specificity |
0.50 |
0.57 |
0.00 |
| Sensitivity |
0.86 |
0.92 |
1.00 |
According to the SVM results, the Wilcoxon-based
KEGG Pathway (n>5) group performed the best, with all the
parameters better than the other two groups.
Discussion
A method to diagnose IPF at an early stage is
required. Since the field of IPF genetics has made significant
progress in identifying common variants that are confidently
associated with IPF diagnosis, a gene-based pathway aberrance
analysis may aid the detection of IPF at an early stage.
In the present study, three pathway groups were
constructed; a Wilcoxon-based KEGG Pathway (n>5) group, a
Limma-based KEGG Pathway (n>5) group and an attract-based KEGG
Pathway (n>5) group. Different groups were obtained due to the
different test methods deployed in pathway statistics and the
quantity of differential pathways in the three groups also
differed; the Wilcoxon-based KEGG Pathway (n>5) group possessed
the greatest number of pathways, the Limma-based KEGG Pathway
(n>5) group possessed fewer pathways and the attract-based KEGG
Pathway (n>5) group the least number of pathways. The
attract-based KEGG Pathway (n>5) group contained only seven
differential pathways, far fewer than the other two groups.
Differential pathways reflected the core metabolisms that were most
influenced by the disease, however the large number of differential
pathways identified suggested further evaluation and study is
required in order to fully elucidate the mechanism.
The SVM method (26), which has been demonstrated to
possess a high identification rate in numerous datasets, was
introduced to perform the comparison. According to the SVM results,
the Wilcoxon-based KEGG Pathway (n>5) group performed the best,
with all parameters better than the other two groups.
To identify which group performed best in
diagnosing IPF with differential pathways, a classifier SVM was
introduced. The results demonstrated that the Wilcoxon-based KEGG
Pathway (n>5) group performed the best, with the parameters of
AUC, accuracy, MCC, specificity and sensitivity. It is therefore
suggested that this pathway group reflected the occurrence of IPF
more exactly. The top five pathways that were most prone to alter
in IPF were ‘Amoebiasis’, ‘Bladder cancer’, ‘Type II diabetes
mellitus’, ‘Primary immunodeficiency’ and ‘Histidine
metabolism’.
The ‘Amoebiasis’ pathway is involved in a type of
infectious disease. The pathogenesis of amoebiasis begins with
parasite attachment and disruption of the intestinal mucus layer,
followed by apoptosis of host epithelial cells. The parasite can
cause extra intestinal infection, including amoebic liver
abscesses, by evading the immune response (27). That the ‘Amoebiasis’ pathway was
inhibited in IPF was identified by Nance et al (28). In the present study, the
‘Amoebiasis’ pathway in the disease group was demonstrated to be
significantly different from the normal group, which was consistent
with the result of Nance et al (28).
The ‘Bladder cancer’ pathway is responsible for
bladder cancer. This pathway was significantly altered in IPF,
which may be the result of the deregulation of a regulator,
caveolin-1, since caveolin-1deregulation has been associated with
several human diseases (29–32).
It has been demonstrated that caveolin-1 mRNA expression is low in
IPF (33), however is high in
bladder cancer (32).
The ‘Type II diabetes mellitus’ pathway was
identified altered in IPF. Among various lifestyle-associated
diseases, diabetes mellitus is a frequent complication in patients
with IPF and may increase the risk of IPF (34).
‘Primary immunodeficienies’ are a heterogeneous
group of disorders, which affect cellular and humoral immunity or
non-specific host defense mechanisms mediated by complement
proteins and cells (35). It has
been previously demonstrated (36)
that in a severe combined immunodeficiency bleomyc in mouse model
of fibrosis, human fibrocytes are also trafficked to the lung, the
primary area of injury.
In summary, differential pathways can be used in
diagnosis of IPF at an early stage, and the best method analyzed by
SVM is by making use of the significant differential pathways
identified in the Wilcoxon-based KEGG Pathway (n>5) group.
Acknowledgements
The authors would like to thank Honghui
Biotechnology Co., Ltd. (Shandong, China) for help in information
analysis.
References
|
1
|
Michaelson JE, Aguayo SM and Roman J:
Idiopathic pulmonary fibrosis: A practical approach for diagnosis
and management. Chest. 118:788–794. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Woodcock HV and Maher TM: The treatment of
idiopathic pulmonary fibrosis. F1000prime Rep. 6:162014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Agabiti N, Porretta MA, Bauleo L, Coppola
A, Sergiacomi G, Fusco A, Cavalli F, Zappa MC, Vignarola R, Carlone
S, et al: Idiopathic pulmonary fibrosis (IPF) incidence and
prevalence in Italy. Sarcoidosis Vasc Diffuse Lung Dis. 31:191–197.
2014.PubMed/NCBI
|
|
4
|
Putman RK, Rosas IO and Hunninghake GM:
Genetics and early detection in idiopathic pulmonary fibrosis. Am J
Respir Crit Care Med. 189:770–778. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Levine DM, Ek WE, Zhang R, Liu X, Onstad
L, Sather C, Lao-Sirieix P, Gammon MD, Corley DA, Shaheen NJ, et
al: A genome-wide association study identifies new susceptibility
loci for esophageal adenocarcinoma and Barrett's esophagus. Nat
Genet. 45:1487–1493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bild AH, Yao G, Chang JT, Wang Q, Potti A,
Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al:
Oncogenic pathway signatures in human cancers as a guide to
targeted therapies. Nature. 439:353–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nance T, Smith KS, Anaya V, Richardson R,
Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G
and Montgomery SB: Transcriptome analysis reveals differential
splicing events in IPF lung tissue. PLoS One. 9:e975502014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deng N, Sanchez CG, Lasky JA and Zhu D:
Detecting splicing variants in idiopathic pulmonary fibrosis from
non-differentially expressed genes. PLoS One. 8:e683522013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Boon K, Bailey NW, Yang J, Steel MP,
Groshong S, Kervitsky D, Brown KK, Schwarz MI and Schwartz DA:
Molecular phenotypes distinguish patients with relatively stable
from progressive idiopathic pulmonary fibrosis (IPF). PLoS One.
4:e51342009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mar JC, Matigian NA, Quackenbush J and
Wells CA: attract: A method for identifying core pathways that
define cellular phenotypes. PLoS One. 6:e254452011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Drier Y, Sheffer M and Domany E:
Pathway-based personalized analysis of cancer. Proc Natl Acad Sci
USA. 110:6388–6393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ahn T, Lee E, Huh N and Park T:
Personalized identification of altered pathways in cancer using
accumulated normal tissue data. Bioinformatics. 30:i422–i429. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cherkassky V: The nature of statistical
learning theory~. IIEEE Trans Neural Netw. 8:15641997. View Article : Google Scholar
|
|
14
|
Ben-Hur A, Ong CS, Sonnenburg S, Schölkopf
B and Rätsch G: Support vector machines and kernels for
computational biology. PLoS Comput Biol. 4:e10001732008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furey TS, Cristianini N, Duffy N,
Bednarski DW, Schummer M and Haussler D: Support vector machine
classification and validation of cancer tissue samples using
microarray expression data. Bioinformatics. 16:906–914. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Karchin R, Karplus K and Haussler D:
Classifying G-protein coupled receptors with support vector
machines. Bioinformatics. 18:147–159. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sonnenburg S, Schweikert G, Philips P,
Behr J and Rätsch G: Accurate splice site prediction using support
vector machines. BMC Bioinformatics. 8 Suppl 10:S72007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Müller KR, Mika S, Rätsch G, Tsuda K and
Schölkopf B: An introduction to kernel-based learning algorithms.
IEEE Trans Neural Net. 12:181–201. 2001. View Article : Google Scholar
|
|
19
|
Yang IV, Luna LG, Cotter J, Talbert J,
Leach SM, Kidd R, Turner J, Kummer N, Kervitsky D, Brown KK, et al:
The peripheral blood transcriptome identifies the presence and
extent of disease in idiopathic pulmonary fibrosis. PLoS One.
7:e377082012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kanehisa M, Goto S, Sato Y, Furumichi M
and Tanabe M: KEGG for integration and interpretation of
large-scale molecular data sets. Nucleic Acids Res. 40(Database
issue): D109–D114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bolstad B: preprocessCore: A collection of
pre-processing functions. Bioconductor. 2013.
|
|
22
|
Gehan EA: A generalized wilcoxon test for
comparing arbitrarily singly-censored samples. Biometrika.
52:203–223. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benjamini Y, Drai D, Elmer G, Kafkafi N
and Golani I: Controlling the false discovery rate in behavior
genetics research. Behav Brain Res. 125:279–284. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McCarthy DJ and Smyth GK: Testing
significance relative to a fold-change threshold is a TREAT.
Bioinformatics. 25:765–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lazar C, Taminau J, Meganck S, Steenhoff
D, Coletta A, Molter C, De Schaetzen V, Duque R, Bersini H and Nowé
A: A survey on filter techniques for feature selection in gene
expression microarray analysis. IEEE/ACM Trans Comput Biol
Bioinform. 9:1106–1119. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Papadonikolakis M and Bouganis CS: Novel
cascade FPGA accelerator for support vector machines
classification. IEEE Trans Neural Netw Learn Syst. 23:1040–1052.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lejeune M, Rybicka JM and Chadee K: Recent
discoveries in the pathogenesis and immune response toward
Entamoeba histolytica. Future Microbiol. 4:105–118. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nance T, Smith KS, Anaya V, Richardson R,
Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G
and Montgomery SB: Transcriptome analysis reveals differential
splicing events in IPF lung tissue. PLoS One. 9:e921112014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang XM, Zhang Y, Kim HP, Zhou Z,
Feghali-Bostwick CA, Liu F, Ifedigbo E, Xu X, Oury TD, Kaminski N
and Choi AM: Caveolin-1: A critical regulator of lung fibrosis in
idiopathic pulmonary fibrosis. J Exp Med. 203:2895–2906. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Williams TM and Lisanti MP: Caveolin-1 in
oncogenic transformation, cancer, and metastasis. Am J Physiol Cell
Physiol. 288:C494–C506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sotgia F, Williams TM, Schubert W, Medina
F, Minetti C, Pestell RG and Lisanti MP: Caveolin-1 deficiency
(−/−) conveys premalignant alterations in mammary epithelia, with
abnormal lumen formation, growth factor independence, and cell
invasiveness. Am J Pathol. 168:292–309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomas S, Overdevest JB, Nitz MD, Williams
PD, Owens CR, Sanchez-Carbayo M, Frierson HF, Schwartz MA and
Theodorescu D: Src and caveolin-1 reciprocally regulate metastasis
via a common downstream signaling pathway in bladder cancer. Cancer
Res. 71:832–841. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nho RS, Peterson M, Hergert P and Henke
CA: FoxO3a (Forkhead Box O3a) deficiency protects idiopathic
pulmonary fibrosis (IPF) fibroblasts from type I polymerized
collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas.
PLoS One. 8:e610172013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Enomoto T, Usuki J, Azuma A, Nakagawa T
and Kudoh S: Diabetes mellitus may increase risk for idiopathic
pulmonary fibrosis. Chest. 123:2007–2011. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Geha RS, Notarangelo LD, Casanova JL,
Chapel H, Conley ME, Fischer A, Hammarström L, Nonoyama S, Ochs HD,
Puck JM, et al: Primary immunodeficiency diseases: An update from
the International union of immunological societies primary
immunodeficiency diseases classification committee. J Allergy Clin
Immunol. 120:776–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Phillips RJ, Burdick MD, Hong K, Lutz MA,
Murray LA, Xue YY, Belperio JA, Keane MP and Strieter RM:
Circulating fibrocytes traffic to the lungs in response to CXCL12
and mediate fibrosis. J Clin Invest. 114:438–446. 2004. View Article : Google Scholar : PubMed/NCBI
|