Introduction
The liver is the primary organ involved in drug
metabolism and disposition. Most drugs are excreted following
biotransformation in the liver, in which some toxic metabolites,
free radicals and reactive intermediates are produced, resulting in
hepatic toxicity (1–3). As a result, the liver is a crucial
target of drug toxicity.
Drug-induced liver injury (DILI) is a serious public
health concern, often leading to a decline in drug efficacy,
discontinuation of therapy, and even instigates liver failure and
death (4,5). These are the most significant reasons
for withdrawal of drugs from the market (6–8).
Therefore, novel strategies for the prevention and treatment of
DILI are critical to future drug safety and efficacy.
Many types of drugs have reported DILI occurrences
throughout treatment. Among them, antineoplastic agents are one of
the most common types of drugs to cause DILI (9). Previous studies have demonstrated
that most chemotherapeutic drugs or their metabolites may cause
liver dysfunction and liver failure (10,11).
Docetaxel belongs to the groups of taxane antitumor agents, and has
been widely used in the treatment of various forms of cancer alone
or combination with other chemotherapeutic drugs (12–15).
However, docetaxel-induced liver injury has, thus far, limited its
clinical success (16,17). Therefore, methods of reducing the
hepatotoxicity of docetaxel are required.
Gap junctions (GJs) are plasma membrane channels
composed of connexin (Cx). Six Cx monomers are assembled into a
hemichannel (connexon), which docks to a counterpart from the
adjacent cell to form a GJ channel. Small molecules, metabolites
and second messengers can spread and transfer between neighboring
cells via GJs. Gap junction intercellular communication (GJIC) is
important in cell proliferation, differentiation and homeostasis
(18,19).
Recent studies have demonstrated that the
hepatotoxicities of some drugs are associated with GJ function.
Blocking GJ channels composed of Cx32 (a major Cx isoform in
hepatocytes) can resist liver damage caused by acetaminophen in
vitro and in vivo (20,21).
The protective effect may be linked to preventing the intercellular
transmission of an ‘injury signal’ (such as free radicals) via GJs
to adjacent cells. This action has been reported in D-galactosamine
and carbon tetrachloride, where loss of GJIC reduced their
hepatotoxicities (22). These
evidences illustrate that GJs may become an important mediator for
the treatment of DILI. However, there is little investigation into
the action of GJs on docetaxel-induced liver injury. The purpose of
this study was to determine the effect and possible mechanisms that
underlie Cx32 GJ activity in docetaxel hepatotoxicity.
Materials and methods
Materials
Docetaxel was purchased from the National Institutes
for Food and Drug Control (Beijing, China). 2-aminoethoxydiphenyl
borate (2-APB), Hoechst 33258, all primary and secondary antibodies
for western blot analysis were obtained from Sigma-Aldrich;
Merck-Millipore (Darmstadt, Germany). Lipofectamine™ 2,000,
calcein-AM and cell culture reagents were purchased from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). Cell Counting Kit-8
(CCK-8) was obtained from Dojindo Molecular Technologies, Inc.
(Kumamoto, Japan). Caspase colorimetric activity assay kits were
purchased from Beyotime Institute of Biotechnology (Haimen,
China).
Cell culture
The rat liver-derived BRL-3A cell line used in this
study was obtained from American Type Culture Collection (Manassas,
VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin at 37°C in a 5%
CO2 incubator (Thermo Fisher Scientific, Inc.).
Drug treatment
Docetaxel and 2-APB were dissolved in
dimethylsulfoxide (DMSO) and diluted in culture medium, in which
the final concentration of DMSO was less than 0.1% (v/v). 2-APB was
added to the cells at 50 µM for 1 h prior to incubation with
docetaxel and remained during the docetaxel treatment.
RNA interference
Cells were seeded at a density of 5,000
cells/cm2 into plates and achieved 30–50% confluency by
the time of Cx32 small interfering (si)RNA transfection. The
negative control siRNA (NCsiRNA) or targeted Cx32 siRNA (Guangzhou
Ribobio Co., Ltd., Guangzhou, China) were transfected into BRL-3A
cells using Lipofectamine™ 2000. The synthetic sequences of siRNA
for targeting Cx32 (si-Cx32) were as follows: si-Cx32-1,
5′-CACCAACAACACATAGAAA-3′; and si-Cx32-2,
5′-GCATCTGCATTATCCTCAA-3′. Knockdown of Cx32 expression and
inhibition of GJIC were confirmed by western blot and parachute
assays.
Assay of ‘parachute’ dye-coupling
The GJ function was determined by the ‘parachute’
dye-coupling assay as described previously (23,24).
Donor cells were labeled with 5 µM calcein-AM, which is transformed
intracellularly into calcein that exhibits GJ permeability. The
donor cells were seeded on receiver cells at a 1:150 ratio
(donor/receiver). The cells were cultured for 4 h at 37°C to form
GJs, and then were monitored with a fluorescence microscope (IX71;
Olympus Corporation, Tokyo, Japan). For each condition, 12
different visual fields were assessed in triplicate. To evaluate
the GJ function, the average number of receiver cells containing
calcein dye/donor cell were counted and normalized against the
vehicle control, containing no siRNA.
Western blot analysis
Western blot analysis was performed as described
previously (25). Following
sonication and centrifugation at 14,167 × g for 30 min at 4°C, 20
µg whole-cell lysate was loaded per well, fractionated by 10%
SDS-PAGE, and transferred onto a nitrocellulose membrane. The
membranes were blocked with 5% milk for 1 h at room temperature.
The antibodies against Cx32 (catalog no. C6344; 1:1,000) and
β-actin, which was used as a loading control (catalog no. A2228;
1:2,000) were applied overnight at 4°C. The blots were then
incubated with a horseradish peroxidase-conjugated goat anti-mouse
IgG secondary antibody (catalog no. A4416) at a 1:4,000 dilution
for 1 h at room temperature. Immunopositive bands were detected by
an Amersham ECL™ Plus Western Blotting Detection kit (GE Healthcare
Bio-Sciences, Pittsburgh, PA, USA). The bands intensities were
quantified by the Quantity One software version 4.6.2 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Assay of cell viability
Cell viability was evaluated using the CCK-8 kit.
Briefly, BRL-3A cells were exposed to docetaxel for 24 h followed
by treatment with CCK-8 for another 3 h at 37°C. The absorbance of
each well was detected at 450 nm by a microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA). Four independent experiments
were performed. The cell viability was assessed by normalizing the
surviving fraction of the drug-treated group to the vehicle
control.
Hoechst 33258 staining
BRL-3A cells were plated in 12-well dishes and
cultured until they reached 80–100% confluency. The cells were
exposed to agents, followed by washing with PBS and incubation with
serum-free culture medium for another 24 h. Cells were rinsed with
PBS and fixed in 4% paraformaldehyde solution for 20 min. Hoechst
33258 (10 µg/ml) was applied for 5 min in the dark to stain the
nuclei of the cells. Apoptotic cells exhibited nucleus shrinkage
with chromatin condensation. Following three washes with PBS, the
cells were visualized and photographed by an Olympus IX71
fluorescence microscope. The cell apoptosis was assessed using the
percentage of apoptotic cells/total cells under five randomly
selected fields.
Caspase activities measurement
BRL-3A cells at 70–80% confluency were incubated
with docetaxel for 24 h. Cells were harvested, lysed and
centrifuged at 16,000 × g for 15 min at 4°C. The supernatant of
each sample was collected for caspase analysis. Caspase
colorimetric assay kits evaluated activities of caspases-3, −8 and
−9. Absorbance detection was measured at 405 nm using a microplate
reader (BioTek Instruments, Inc.).
Statistical analysis
Data were analyzed by the Sigma Plot software
version 10.0 (Jandel Scientific, San Rafael, CA, USA) and expressed
as the mean ± standard error of the mean. The unpaired Student's
t-test was used. P<0.05 was considered to indicate statistical
significance.
Results
Effect of cell density on
docetaxel-induced hepatotoxicity
For initially determining the effect of GJs on
docetaxel hepatotoxicity, BRL-3A cells were cultured under low- and
high-density conditions. GJ channels did not form as the cells were
dispersed into single cells at low-density culture, while GJ
formation was permitted at high-density culture for the cells that
could contact each other (data not shown). Following exposure to
docetaxel for 24 h in the two density conditions, cell survival was
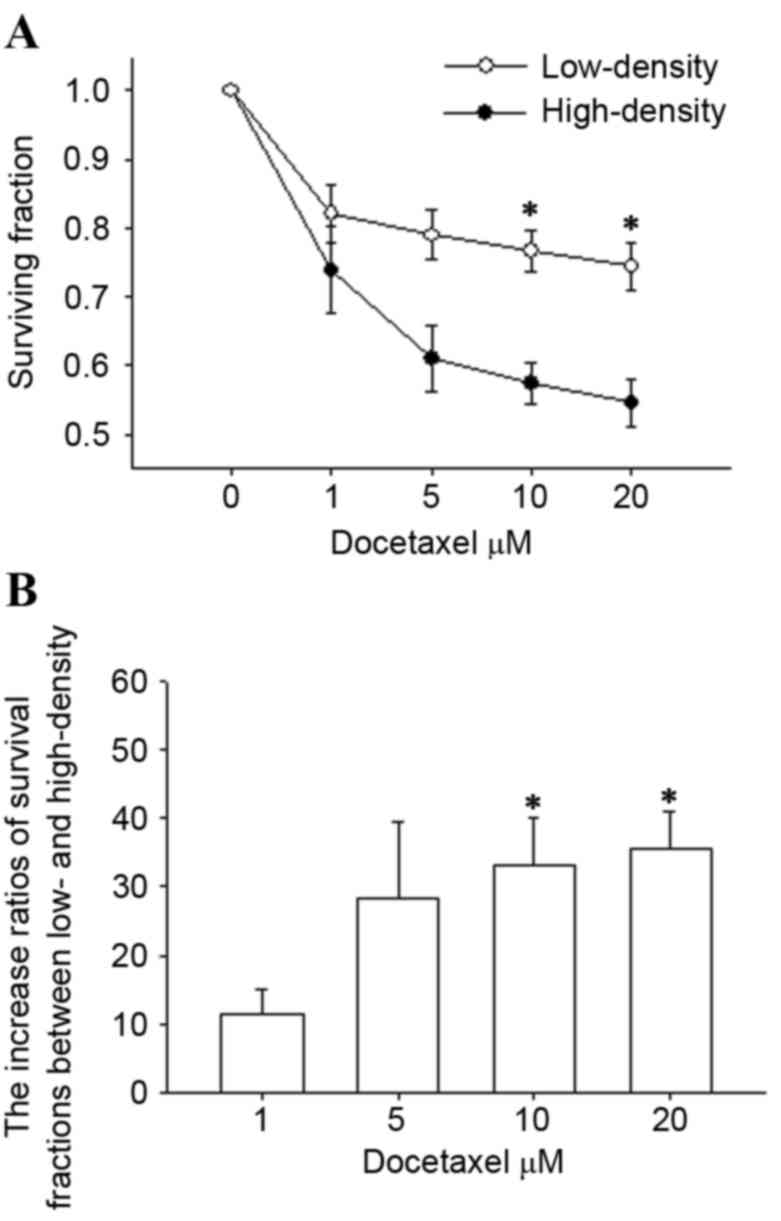
evaluated by CCK-8 assay. As demonstrated in Fig. 1A, docetaxel reduced cell survival
in a concentration-dependent manner in the cases of low- and
high-density. However, the survival fractions in low-density (GJ
absence) were higher than that of cells in high-density (GJ
presence) at concentrations of docetaxel up to 20 µM. In addition,
a significant difference of cell viability was observed at 10 and
20 µM (P<0.05). The increased ratios of the survival fractions
between low- and high-density conditions were more than 30.0% at
docetaxel concentrations of 10 and 20 µM (P<0.05; Fig. 1B). These results indicate that
docetaxel hepatotoxicity is dependent on cell density and the toxic
effect is decreased in blocking intercellular communication.
Inhibition of GJIC reduces the
docetaxel-induced hepatotoxicity
Since the reduced toxicity of docetaxel was
attributed to the low-density culture, the next step involved
further investigating the role of GJIC on docetaxel hepatotoxicity.
Two methods were applied to regulate the GJs composed of Cx32 (Cx32
GJ) in BRL-3A cells: i) knockdown Cx32 expression by siRNA and ii)
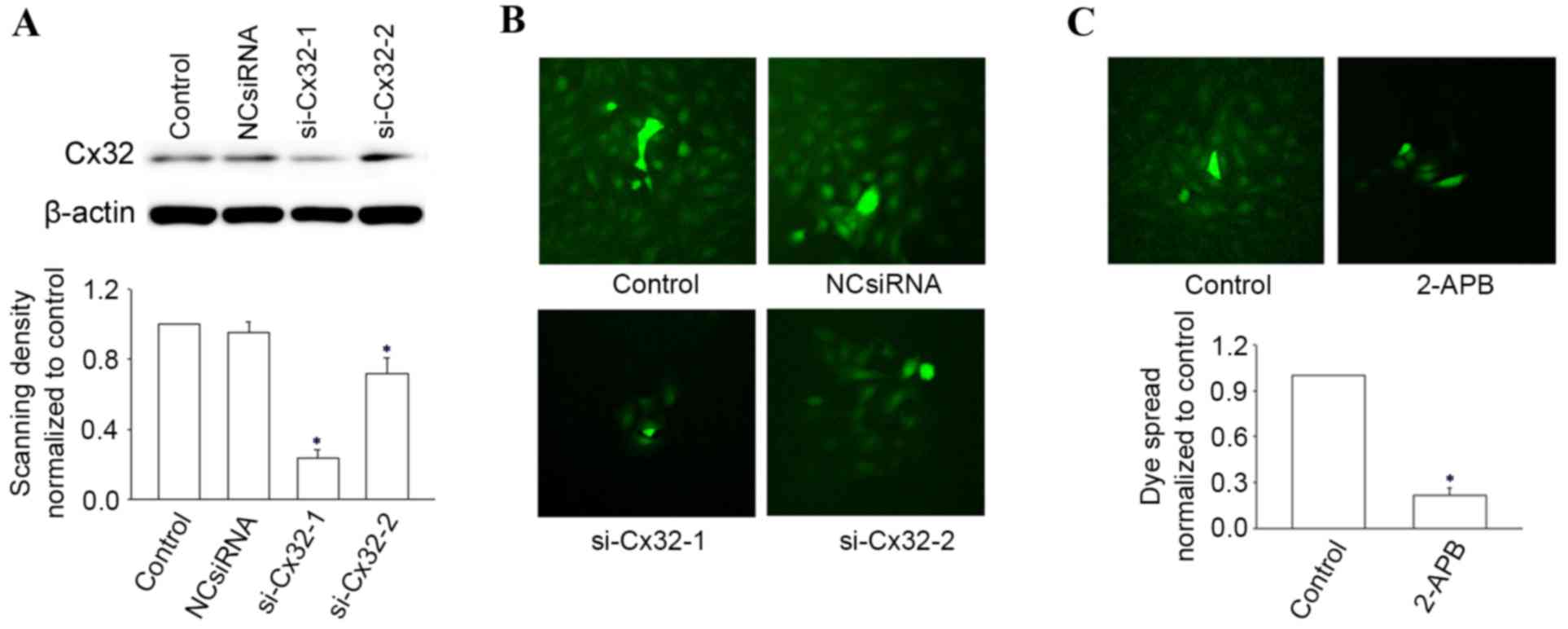
using the chemical inhibitor 2-APB (26). The expression of Cx32 was confirmed
by western blot analysis and was markedly downregulated by
transfection with si-Cx32-1 relative to the vehicle control and
NCsiRNA (Fig. 2A). The GJIC
inhibition of si-Cx32-1 (Fig. 2B)
and 2-APB (Fig. 2C), were assessed
by ‘parachute’ dye-coupling assay.
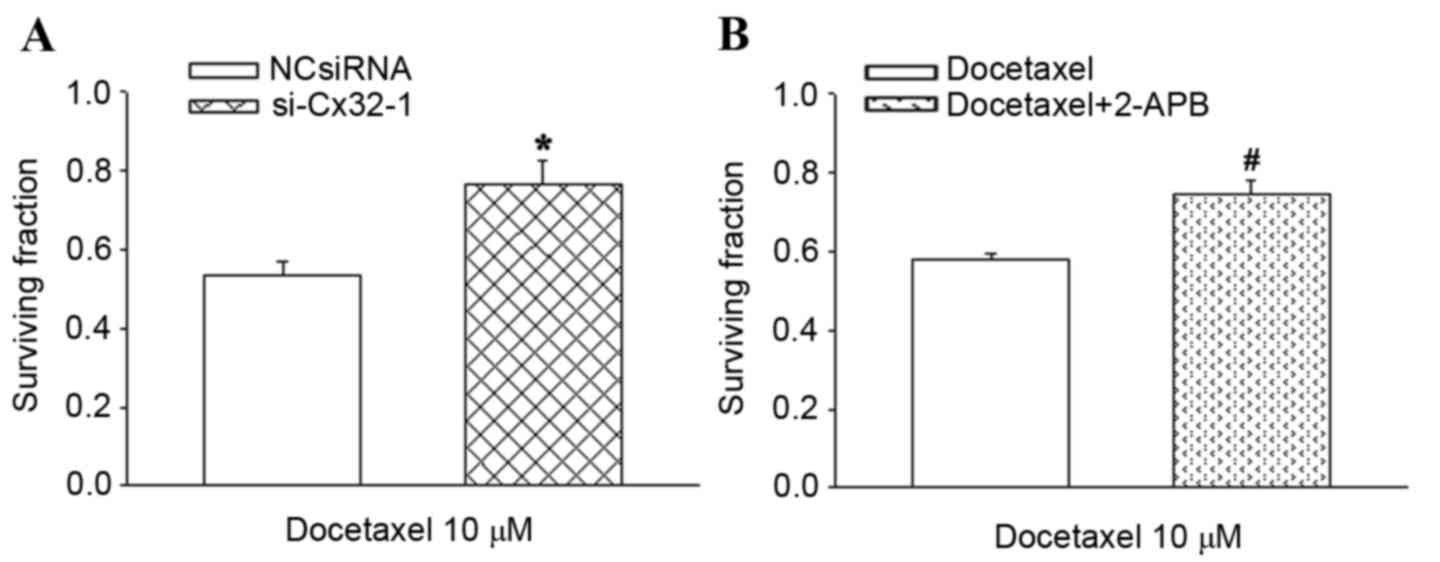
In the high-density group, the survival of
Cx32-knockdown cells was significantly increased compared with the
negative control treated cells in the presence of 10 µM docetaxel,
by a factor of 1.4 (P<0.05; Fig.
3A). Treatment of BRL-3A cells with 50 µM 2-APB under high
density conditions increased the cell viability from 57.3 to 75.5%
during treatment with 10 µM docetaxel (P<0.05; Fig. 3B). These results demonstrated that
inhibition of GJ function by either Cx32-knockdown or chemical
inhibitor significantly reduces the hepatotoxicity of
docetaxel.
Influence of GJ on docetaxel-induced
apoptosis
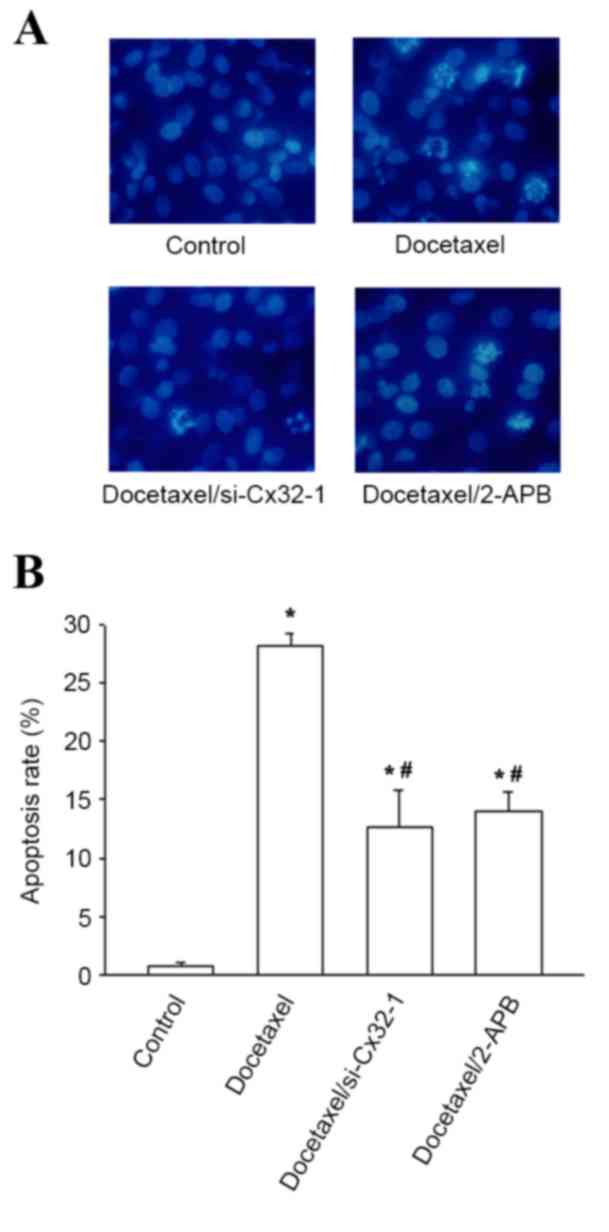
To illustrate whether apoptosis is involved in the
protective effect against docetaxel cytotoxicity by blocking GJs,
Hoechst 33258 staining was used to evaluate the apoptosis rates of
BRL-3A cells with or without GJIC. As demonstrated in Fig. 4A, the cell nuclei showed uniformly
blue and smooth edges when cells were incubated with vehicle
control. However, following treatment with docetaxel, some cells
exhibited typical apoptosis characteristics, such as nuclei
shrinkage and fragmentation leading to strong blue fluorescence.
The apoptosis rates in BRL-3A cells pretreated with siRNA or 2-APB
prior to docetaxel treatment was significantly decreased, by 55.0
and 50.4%, respectively, compared with docetaxel only (P<0.05;
Fig. 4B). These observations
demonstrated that reduced apoptosis is largely responsible for the
protective effect of GJ inhibition on docetaxel hepatotoxicity.
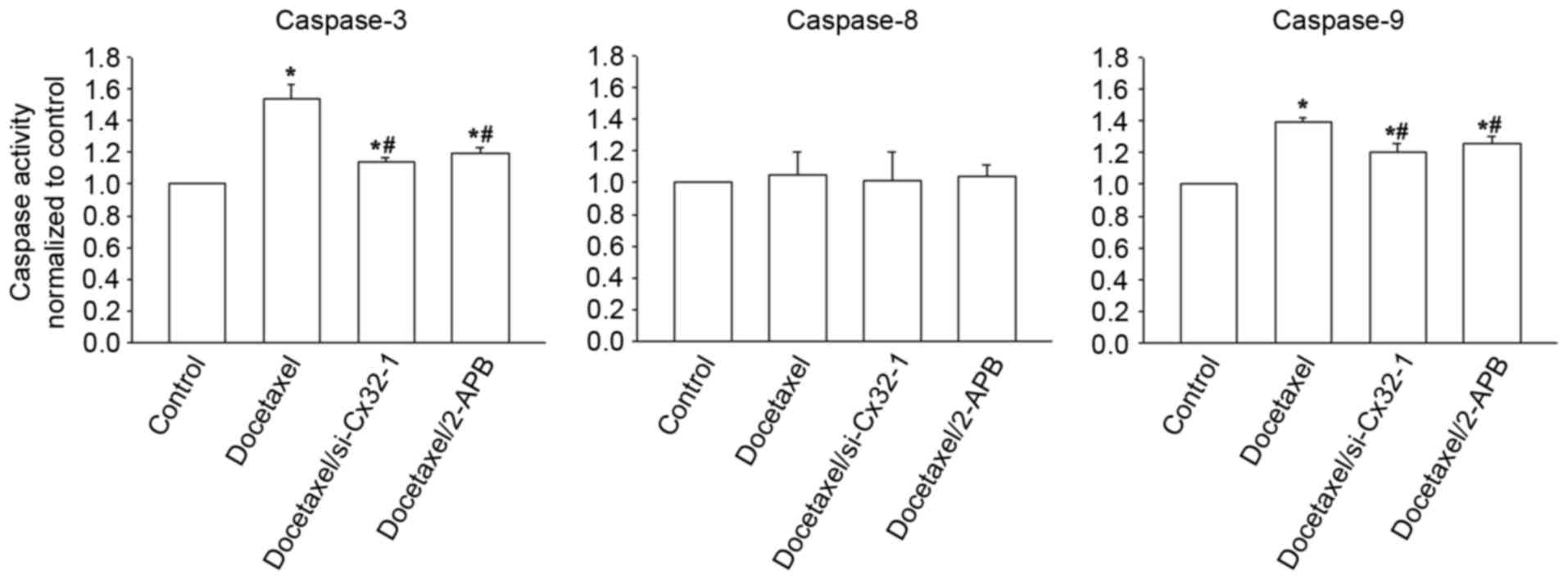
Effects of GJ on caspase-3, −8, −9
activities
The caspase cascade system serves a vital role in
the process of apoptosis. To investigate the possible apoptotic
pathways, the activities of caspase-3, −8 and −9 in BRL-3A cells
exposed to 10 µM docetaxel were examined in the presence or absence
of GJIC. As presented in Fig. 5,
caspase-3 and caspase-9, but not caspase-8, were activated
following treatment of cells with 10 µM docetaxel for 24 h
(P<0.05, control vs. docetaxel). However, knockdown of Cx32
expression and 2-APB treatment both significantly reduced the
increased caspase-3 and caspase-9 activities, compared with
docetaxel treatment only (P<0.05; Fig. 5), while having no effect on
caspase-8 activity (P>0.05; Fig.
5). These observations indicate that docetaxel-induced
cytotoxicity may be associated with caspase-3 and caspase-9
activations, and is attenuated through this apoptosis pathway by
blocking GJIC.
Discussion
The present study illustrates that the toxicity of
docetaxel is mediated by GJIC in BRL-3A cells. GJIC was
downregulated using 3 methods: Low-density culture, knockdown of
Cx32 expression through siRNA transfection, and application of the
GJ chemical inhibitor, 2-APB. All 3 methods led to reduced
docetaxel cytotoxicity, which reduced the toxic effect to BRL-3A
cells. To the best of our knowledge, the present findings revealed
for the first time that inhibition GJIC exerts a protective effect
on liver injury caused by docetaxel.
Apoptosis is an orderly cell death program and is
critical for the maintenance of cell homeostasis, which is one of
the main mechanisms in antineoplastic agents' cytotoxicity
(27–29). The caspase cascade system exerts an
important role in initiating and amplifying apoptotic signals.
There are 2 major pathways resulting in caspase activation: One is
the mitochondrial pathway, mainly mediated by the caspase-9; the
other is death receptor pathway, mainly mediated by the caspase-8
(30). The two pathways both go on
to activate caspase-3, thereby causing the morphological and
biochemical changes (31). In the
current study, docetaxel-induced apoptosis was demonstrated to be
related to its hepatotoxicity. Furthermore, docetaxel increases
caspase-9, rather than caspase-8, to activate downstream caspase-3,
indicating that mitochondrial pathway is largely responsible for
the docetaxel hepatotoxicity. While this hepatotoxicity was
attenuated when blocking GJs activities mainly via decreasing
apoptosis, for caspase-3 (the important executive factor of
apoptosis) and the upstream factor of caspase-9, not caspase-8,
were influenced. Results of the current study demonstrated that
GJIC regulated the biochemical factors induced by docetaxel through
the mitochondrial pathway but not the death receptor pathway.
GJ channels composed of different Cx exhibit
distinct permeability for signal molecules. For instance, adenosine
permeates Cx32 channels approximately 12 times more effectively
than Cx43 channel; the permeability of inositol 1,4,5-trisphosphate
(IP3) through Cx32 channels is higher than that of Cx26
channels (32). The present
results indicated that docetaxel hepatotoxicity was reduced when
Cx32 GJ function was suppressed, suggesting some ‘injury signals’
induced by docetaxel were prevented transmission through Cx32 GJs.
Free radicals and parent drugs are likely candidates. Previous
studies have reported that oxidative stress is a widely accepted
consequence of hepatotoxin exposure and has a close relationship
with mitochondrial function (33,34).
Free radicals as the oxidative stress signals can propagate through
Cx32 GJs and thus amplify this injury (20). Docetaxel may produce a direct toxic
effect, causing mitochondrial damage, which may pass through Cx32
GJs by passive transference due to its molecular weight (807.9
kDa), which is less than the limit of GJ permeable molecules.
Nevertheless, the properties of ‘injury signals’ and their
underlying mechanisms have yet to be explored further.
A previous study demonstrated that the cytotoxicity
of docetaxel was enhanced at presence of GJIC in Cx32-transfected
HeLa cells (23). Therefore, the
therapeutic efficacy and hepatotoxicity of docetaxel are likely to
be affected by GJ function. For lack of GJIC in numerous cancers
(35,36), inhibition of GJs in liver cells may
be a promising strategy for the treatment of docetaxel-induced
hepatotoxicity. However, in some forms of carcinoma with GJIC
retention (37,38), the impact on docetaxel efficacy
needs to be considered when GJs are used as the target for the
treatment of hepatic injury.
In summary, the results of the present study
demonstrate that downregulation of GJs derived from Cx32 could
elicit a protective role against docetaxel-induced hepatotoxicity,
which is mediated by GJIC. In addition, this hepatoprotection
appears to be due to reduced caspase-3, −9 activation, thereby
decreasing the apoptosis and cell toxicity of docetaxel. Further
studies are required to examine the effects of GJ on the
docetaxel-induced cytotoxicity in other hepatocyte strains and
in vivo.
Acknowledgements
The present study was supported by the grants of the
National Natural Science Foundation of China (grant no. 81400619),
the Guangdong Province Public Interest Research and Capacity
Building Special Fund (grant no. 2014A020212508) and the Science
Foundation for the Doctoral Program of Guangdong Medical University
(grant no. B2013011).
References
|
1
|
Corsini A and Bortolini M: Drug-induced
liver injury: The role of drug metabolism and transport. J Clin
Pharmacol. 53:463–474. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amacher DE: The primary role of hepatic
metabolism in idiosyncratic drug-induced liver injury. Expert Opin
Drug Metab Toxicol. 8:335–347. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Claesson A and Spjuth O: On mechanisms of
reactive metabolite formation from drugs. Mini Rev Med Chem.
13:720–729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Daly AK: Drug-induced liver injury: Past,
present and future. Pharmacogenomics. 11:607–611. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hussaini SH and Farrington EA:
Idiosyncratic drug-induced liver injury: An overview. Expert Opin
Drug Saf. 6:673–684. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaplowitz N: Idiosyncratic drug
hepatotoxicity. Nat Rev Drug Discov. 4:489–499. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Senior JR: Evolution of the food and drug
administration approach to liver safety assessment for new drugs:
Current status and challenges. Drug Saf. 37:(Suppl 1). S9–S17.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Regev A: Drug-induced liver injury and
drug development: Industry perspective. Semin Liver Dis.
34:227–239. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meier Y, Cavallaro M, Roos M, Pauli-Magnus
C, Folkers G, Meier PJ and Fattinger K: Incidence of drug-induced
liver injury in medical inpatients. Eur J Clin Pharmacol.
61:135–143. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Thatishetty AV, Agresti N and O'Brien CB:
Chemotherapy-induced hepatotoxicity. Clin Liver Dis. 17:671–686,
ix-x. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bahirwani R and Reddy KR: Drug-induced
liver injury due to cancer chemotherapeutic agents. Semin Liver
Dis. 34:162–171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim HS, Ryu MH, Zang DY, Ryoo BY, Yang DH,
Cho JW, Lim MS, Kim MJ, Han B, Choi DR, et al: Phase II study of
docetaxel, oxaliplatin, and S-1 therapy in patients with metastatic
gastric cancer. Gastric Cancer. 19:579–585. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gelmon K: The taxoids: Paclitaxel and
docetaxel. Lancet. 344:1267–1272. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pazdur R, Kudelka AP, Kavanagh JJ, Cohen
PR and Raber MN: The taxoids: Paclitaxel (Taxol) and docetaxel
(Taxotere). Cancer Treat Rev. 19:351–386. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dancey J, Shepherd FA, Gralla RJ and Kim
YS: Quality of life assessment of second-line docetaxel versus best
supportive care in patients with non-small-cell lung cancer
previously treated with platinum-based chemotherapy: Results of a
prospective, randomized phase III trial. Lung Cancer. 43:183–194.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Z, Liang X, Yu J, Zheng X, Zhu Y, Yan
Y, Dong N, Di L, Song G, Zhou X, et al: Non-genetic risk factors
and predicting efficacy for docetaxel-drug-induced liver injury
among metastatic breast cancer patients. J Gastroenterol Hepatol.
27:1348–1352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang X, Zhang J, Zhu Y, Lu Y, Zhou X,
Wang Z, Yu J, Yan Y, Di L, Che L, et al: Specific genetic
polymorphisms of IL10-592 AA and IL10-819 TT genotypes lead to the
key role for inducing docetaxel-induced liver injury in breast
cancer patients. Clin Transl Oncol. 15:331–334. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maeda S and Tsukihara T: Structure of the
gap junction channel and its implications for its biological
functions. Cell Mol Life Sci. 68:1115–1129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harris AL: Connexin channel permeability
to cytoplasmic molecules. Prog Biophys Mol Biol. 94:120–143. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Patel SJ, Milwid JM, King KR, Bohr S,
Iracheta-Vellve A, Li M, Vitalo A, Parekkadan B, Jindal R and
Yarmush ML: Gap junction inhibition prevents drug-induced liver
toxicity and fulminant hepatic failure. Nat Biotechnol. 30:179–183.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Naiki-Ito A, Asamoto M, Naiki T, Ogawa K,
Takahashi S, Sato S and Shirai T: Gap junction dysfunction reduces
acetaminophen hepatotoxicity with impact on apoptotic signaling and
connexin 43 protein induction in rat. Toxicol Pathol. 38:280–286.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Asamoto M, Hokaiwado N, Murasaki T and
Shirai T: Connexin 32 dominant-negative mutant transgenic rats are
resistant to hepatic damage by chemicals. Hepatology. 40:205–210.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tang N, Wang Q, Wu D, Zhang S, Zhang Y and
Tao L: Differential effects of paclitaxel and docetaxel on gap
junctions affects their cytotoxicities in transfected HeLa cells.
Mol Med Rep. 8:638–644. 2013.PubMed/NCBI
|
|
24
|
Goldberg GS, Bechberger JF and Naus CC: A
pre-loading method of evaluating gap junctional communication by
fluorescent dye transfer. Biotechniques. 18:490–497.
1995.PubMed/NCBI
|
|
25
|
Hong X, Wang Q, Yang Y, Zheng S, Tong X,
Zhang S, Tao L and Harris AL: Gap junctions propagate opposite
effects in normal and tumor testicular cells in response to
cisplatin. Cancer Lett. 317:165–171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tao L and Harris AL: 2-aminoethoxydiphenyl
borate directly inhibits channels composed of connexin26 and/or
connexin32. Mol Pharmacol. 71:570–579. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Todd RC and Lippard SJ: Inhibition of
transcription by platinum antitumor compounds. Metallomics.
1:280–291. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bhalla KN: Microtubule-targeted anticancer
agents and apoptosis. Oncogene. 22:9075–9086. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fulda S: Modulation of apoptosis by
natural products for cancer therapy. Planta Med. 76:1075–1079.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park HH, Lo YC, Lin SC, Wang L, Yang JK
and Wu H: The death domain superfamily in intracellular signaling
of apoptosis and inflammation. Annu Rev Immunol. 25:561–586. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tong X, Dong S, Yu M, Wang Q and Tao L:
Role of heteromeric gap junctions in the cytotoxicity of cisplatin.
Toxicology. 310:53–60. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Niessen H, Harz H, Bedner P, Krämer K and
Willecke K: Selective permeability of different connexin channels
to the second messenger inositol 1,4,5-trisphosphate. J Cell Sci.
113:1365–1372. 2000.PubMed/NCBI
|
|
33
|
Circu ML and Aw TY: Glutathione and
apoptosis. Free Radic Res. 42:689–706. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Friesen C, Kiess Y and Debatin KM: A
critical role of glutathione in determining apoptosis sensitivity
and resistance in leukemia cells. Cell Death Differ. 11:(Suppl 1).
S73–S85. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mesnil M, Crespin S, Avanzo JL and
Zaidan-Dagli ML: Defective gap junctional intercellular
communication in the carcinogenic process. Biochim Biophys Acta.
1719:125–145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Loewenstein WR and Kanno Y: Intercellular
communication and the control of tissue growth: Lack of
communication between cancer cells. Nature. 209:1248–1249. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hanna EA, Umhauer S, Roshong SL, Piechocki
MP, Fernstrom MJ, Fanning JD and Ruch RJ: Gap junctional
intercellular communication and connexin43 expression in human
ovarian surface epithelial cells and ovarian carcinomas in vivo and
in vitro. Carcinogenesis. 20:1369–1373. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang W, DeMattia JA, Song H and Couldwell
WT: Communication between malignant glioma cells and vascular
endothelial cells through gap junctions. J Neurosurg. 98:846–853.
2003. View Article : Google Scholar : PubMed/NCBI
|