Introduction
Macrophages are tissue-resident innate immune cells
that are important in the host's defense against pathogens.
Furthermore, macrophages are involved in maintaining tissue
homeostasis and promoting wound healing of injured tissues
(1). However, recent evidence has
demonstrated that macrophages are also involved in many diseases,
including obesity, diabetes, and cancer (2). In cancer, macrophages that reside in
the tumor microenvironment are referred to as tumor-associated
macrophages, and are associated with poor therapeutic efficacy
because they enable drug resistance in cancer cells and promote
tumor growth, immunosuppression and metastasis (3–5).
Therefore, the development of strategies to selectively regulate
tumor-associated macrophages may result in more effective cancer
treatments.
Radiation therapy is extensively used for various
cancers. Ionizing radiation directly or indirectly causes DNA
damage, leading to cell cycle arrest and death. In general, rapidly
proliferating and undifferentiated cells are relatively sensitive
to ionizing radiation, whereas non-proliferating and highly
differentiated cells exhibit radioresistance. Consistently,
although monocytes are sensitive to DNA damage by oxidative stress,
including ionizing radiation, macrophages or dendritic cells, that
are differentiated from monocytes, are resistant to DNA damage
(6). Therefore, although radiation
therapy can target the macrophages that reside in the tumor
microenvironment as well as the cancer cells, macrophages are not
killed by ionizing radiation. Furthermore, since activation of DNA
damage response pathways, including tumor protein p53 (p53)
(7), is very weak in macrophages
compared with monocytes (6), cell
death mechanisms that are independent of DNA damage and the p53
pathway are preferable to induce macrophage apoptosis as a
therapeutic intervention.
Endoplasmic reticulum (ER) stress is induced by the
accumulation of unfolded and/or misfolded proteins in the ER lumen
(8,9). To deal with these accumulated
unfolded and/or misfolded proteins, cells activate the unfolded
protein response (UPR) signaling pathway. UPR triggers the
following three main cellular responses: i) inhibition of protein
translation by phosphorylating eukaryotic initiation factor 2-α
(eIF2α), ii) expression of ER chaperone proteins such as binding
immunoglobulin protein (BiP), and iii) degradation of unfolded
and/or misfolded proteins in the ER (8,9).
When cells are exposed to excessive and/or prolonged ER stress,
apoptotic pathways are initiated in both p53-dependent and
independent manners (10).
Therefore, ER stress inducers may cause apoptosis in macrophages.
Furthermore, because previous studies have demonstrated that ER
stress sensitizes cancer cells to radiation (11,12),
combination treatment with an ER stress inducer and ionizing
radiation may effectively induce apoptosis in macrophages. However,
the synergistic effects of ER stress and ionizing radiation on
apoptosis induction in macrophages remain unknown. Therefore, in
the present study, the effects of chemical ER stress inducers
thapsigargin and tunicamycin were examined on the UPR pathway and
apoptosis in THP-1-derived macrophage-like cells. Furthermore, the
effect of combination treatment with ER stress inducers and
ionizing radiation was examined on macrophage apoptosis
induction.
Materials and methods
Reagents
Phorbol 12-myristate 13-acetate (PMA), dimethyl
sulfoxide (DMSO), propidium iodide (PI), and thapsigargin were
purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
Tunicamycin was purchased from Enzo Life Sciences (Farmingdale, NY,
USA). Carbobenzoxy-
valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK) was
purchased from Peptide Institute, Inc. (Osaka, Japan). Primary
antibodies targeting phosphorylated (p-) eIF2α (Ser51; cat. no.
3597), BiP (cat. no. 3177), caspase-3 (cat. no. 9662), poly
(ADP-ribose) polymerase (PARP; cat. no. 9532), and β-actin (cat.
no. 4967), and secondary anti-rabbit immunoglobulin (Ig) G
horseradish peroxidase (HRP)-linked antibody (cat. no. 7074) and
anti-mouse IgG HRP-linked antibody (cat. no. 7076) were purchased
from Cell Signaling Technology Japan, K.K. (Tokyo, Japan).
Cell culture and treatments
Since the THP-1 human acute monocytic leukemia cell
line is negative for p53 expression due to a 26 bp deletion
beginning at codon 174 of the p53 coding sequence (13), THP-1-derived macrophage-like cells
are suitable for exploring apoptosis in macrophages independent of
the p53 pathway. THP-1 human acute monocytic leukemia cells were
obtained from RIKEN BioResource Center (Tsukuba, Japan). The cells
were cultured in Gibco RPMI1640 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 1% penicillin and streptomycin
and 10% heat-inactivated fetal bovine serum (Japan Bio Serum Co.,
Ltd., Nagoya, Japan) at 37°C in a humidified atmosphere containing
5% CO2. THP-1-derived macrophages (hereafter termed
macrophages) were prepared as previously described (14). THP-1 cells (2×105
cells/ml) were plated in 35-mm dishes with 2 ml of medium
containing 100 ng/ml PMA and cultured for 48 h, then the medium was
replaced with fresh PMA-free medium, and the macrophage-like cells
were used in experiments.
For the treatments, THP-1 cells (2×105
cells/dish) or macrophages were cultured in the presence of either
50 or 500 nM thapsigargin, or 5 or 20 µg/ml tunicamycin for the
indicated time periods. Following treatment, cells were harvested
by trypsinization followed by centrifugation (300 × g at 4°C
for 5 min) for further analysis. Z-VAD-FMK (50 µM), a general
caspase inhibitor, was added 1 h prior to thapsigargin treatment.
Irradiation (10-Gy) was performed at 1 h following thapsigargin
treatment. Since thapsigargin, tunicamycin, and Z-VAD-FMK were
dissolved in DMSO, DMSO was used as vehicle control.
In vitro X-ray irradiation
X-ray irradiation (150 kVp, 20 mA, 0.5 mm Al, and
0.3-mm Cu filters) was performed using an MBR-1520R-3 X-ray
generator (Hitachi, Ltd., Tokyo, Japan) at a distance of 45 cm from
the focus, with a dose rate of 1.02–1.05 Gy/min.
Cell cycle analysis
Harvested cells were fixed with 70% ethanol
overnight at −20°C. Fixed cells were washed with Ca2+-
and Mg2+-free phosphate-buffered saline [PBS(−)] and
then treated with RNase A (200 µg/ml) at 37°C for 30 min to
hydrolyze RNA. Following treatment, cells were washed with PBS(−)
and stained with propidium iodide (PI; 30 µg/ml) for 30 min in the
dark. Finally, the cells were filtered with a 40 µm cell strainer
(BD Biosciences, Franklin Lakes, NJ, USA) and cell cycle
distribution was analyzed by flow cytometry (Cytomics FC500 with
CXP software ver. 2; Beckman Coulter, Inc., Brea, CA, USA).
SDS-PAGE and western blotting
Harvested cells were lysed in 1X Laemmli Sample
Buffer (Bio-Rad Laboratories, Inc., Hercules, CA, USA) containing
2.5% 2-mercaptoethanol, by sonication and boiling for 10 min.
Protein concentration was determined using the XL-Bradford assay
kit (APRO Science, Tokushima, Japan) and a SmartSpec Plus
spectrophotometer (Bio-Rad Laboratories, Inc.). The proteins (~5
µg/lane) were separated using 4–20% Mini-PROTEAN TGX precast gels
(Bio-Rad Laboratories, Inc.) and were transferred onto
polyvinylidene difluoride (PVDF) membranes from Trans-Blot Turbo
Mini PVDF Transfer Packs (Bio-Rad Laboratories, Inc.) using the
Trans-Blot Turbo transfer system (Bio-Rad Laboratories, Inc.).
Membranes were blocked in TBST buffer (10 mM HCl, pH 7.5, 100 mM
NaCl, and 0.1% Tween-20) that contained 5% non-fat milk or PVDF
Blocking Reagent for Can Get Signal (Toyobo Co., Ltd., Osaka,
Japan) at room temperature for 1 h. The membranes were probed with
each primary antibody in the TBST buffer containing 5% non-fat skim
milk or Can Get Signal immunoreaction enhancer solution 1 (Toyobo
Co., Ltd.) overnight at 4°C. The following primary antibodies were
used: Anti-p-eIF2α antibody (1:4,000), anti-BIP antibody (1:4,000),
anti-caspase-3 antibody (1:3,000), anti-PARP antibody (1:3,000), or
anti-actin antibody (1:4,000). The membranes were then incubated
with HRP-conjugated secondary antibodies in TBST buffer containing
5% non-fat milk or Can Get Signal immunoreaction enhancer solution
2 (Toyobo Co., Ltd.) for 1 h. The following secondary antibodies
were used: HRP-linked anti-rabbit IgG antibody (1:10,000) or
HRP-linked anti-mouse IgG antibody (1:10,000). Antigens were
visualized using the Clarity enhanced chemiluminescence western
blotting substrate (Bio-Rad Laboratories, Inc.). Blot stripping was
performed using Stripping Solution (Wako Pure Chemical Industries,
Ltd., Osaka, Japan).
Statistical analysis
Data are presented as the mean ± standard error.
Comparisons between the control and experimental groups were
performed using a two-sided Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Excel 2010 (Microsoft Corporation, Redmond, WA, USA) with the
add-in Statcel 3 software (The Publisher OMS Ltd., Tokyo, Japan)
was used to perform statistical analyses.
Results
ER stress-related protein expression
in macrophages treated with ER stress inducers
The effects of ER stress inducers thapsigargin and
tunicamycin were investigated in THP-1-derived macrophages. As
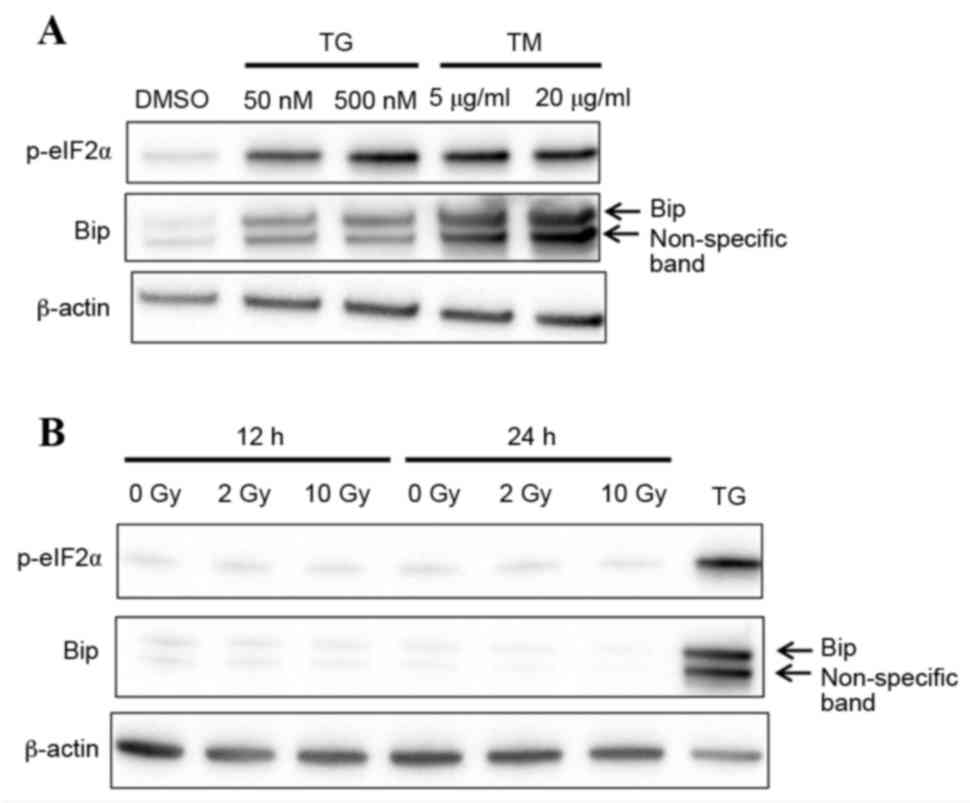
demonstrated in Fig. 1A,
expression levels of the ER stress protein markers p-eIF2α and BiP
were visibly increased in macrophages following treatment with
thapsigargin and tunicamycin for 24 h, compared with macrophages
treated with DMSO vehicle control. This finding suggests that
macrophages responded to ER stress. Ionizing radiation has been
reported to induce ER stress in cancer cells (15,16),
therefore the expression levels of p-eIF2α and BiP were examined in
macrophages following X-ray irradiation. No change was observed in
p-eIF2α and BiP protein expression in irradiated compared with
untreated cells, suggesting that X-ray irradiation does not induce
ER stress in macrophages (Fig.
1B).
ER stress inducers effect on
apoptosis
The effect of X-ray irradiation and ER stress
induction on apoptosis was then examined in THP-1 cells and
THP-1-derived macrophages, by cell cycle phase distribution
analysis using flow cytometry. As demonstrated in Fig. 2A, X-ray irradiation did not
increase the % of macrophages in the sub-G1 fraction, which is a
hallmark of apoptosis, compared with untreated macrophages.
However, when the immature monocytic THP-1 cells were irradiated, a
significant increase in the % of cells in the sub-G1 fraction was
observed compared to untreated THP-1 cells (Fig. 2A). These data suggest that the
differentiation status of macrophages renders them resistant to
ionizing radiation. By contrast, treatment with ER stress inducers
thapsigargin and tunicamycin significantly induced the % of cells
in the sub-G1 fraction in both macrophages and THP-1 cells,
compared to their respective untreated cell control group (Fig. 2B and C). Finally, thapsigargin
exhibited a more potent effect in inducing apoptosis in macrophages
than tunicamycin, as exhibited by markedly higher numbers of
macrophages in the sub-G1 fraction following 72 h of thapsigargin
treatment compared with 72 h of tunicamycin treatment (Fig. 2B and C).
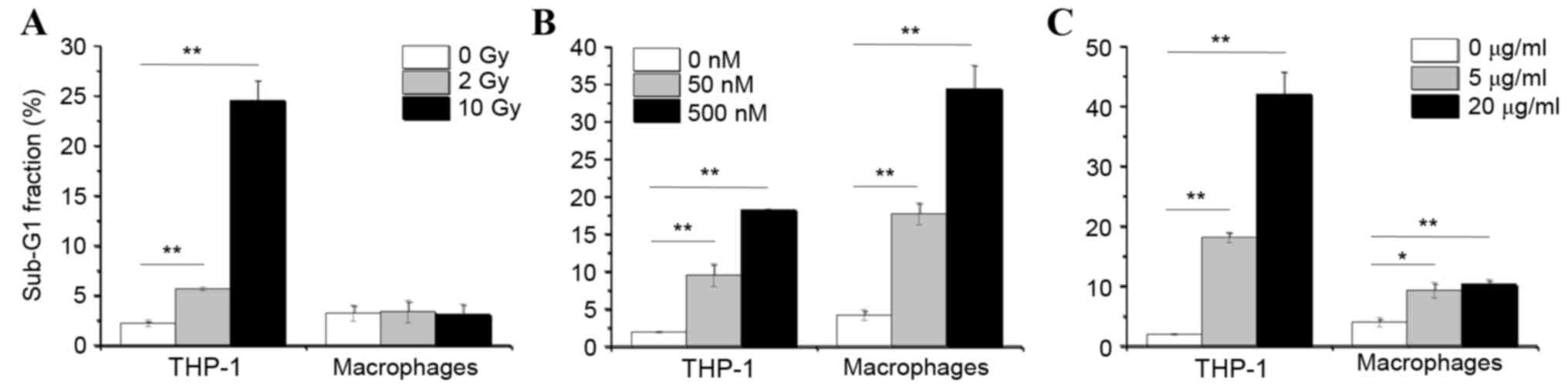 | Figure 2.Effects of X-ray irradiation and ER
stress inducers on apoptosis. THP-1 cells and THP-1-derived
macrophages were cultured for 48 and 72 h respectively, following
(A) irradiation with 0, 2 or 10 Gy, (B) treatment with 0, 50 or 500
nM TG or (C) treatment with 0, 5 or 20 µg/ml TM. Following
treatments, cells were analyzed for cell cycle distribution by flow
cytometry. Data are presented as the mean % of cells in the Sub-G1
fraction ± standard error of 3 independent experiments. *P<0.05
and **P<0.01 with comparisons indicated by lines. ER,
endoplasmic reticulum; TG, thapsigargin; TM, tunicamycin; Gy, gray
(unit). |
Involvement of caspases in
thapsigargin-induced apoptosis
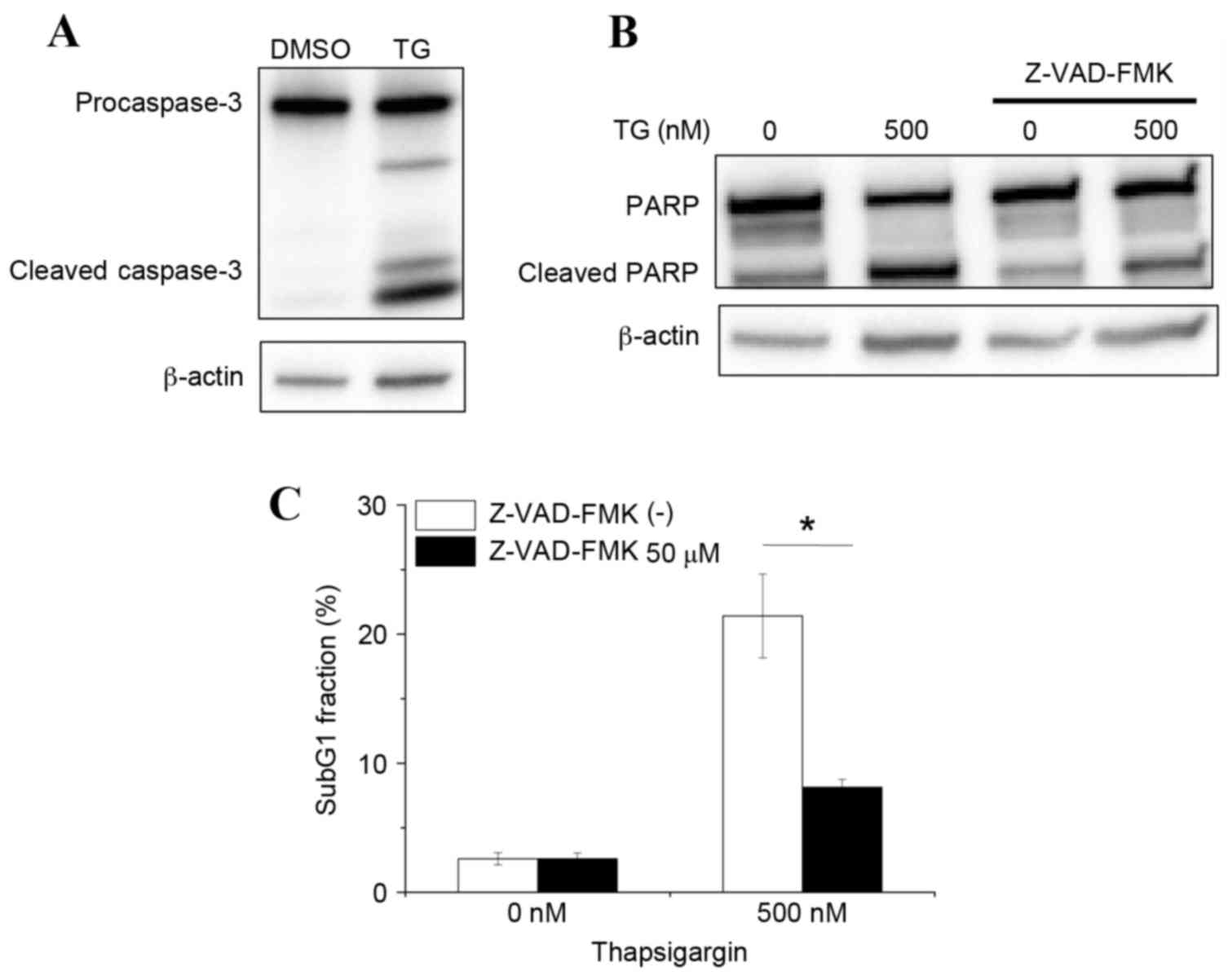
Caspase-3 is a key effector protease that is
responsible for DNA fragmentation during apoptosis. Therefore, the
hypothesis that thapsigargin-induced apoptosis may be mediated by
caspase-3 activation was further examined. As demonstrated in
Fig. 3A, treatment of macrophages
with 500 nM thapsigargin induced caspase-3 cleavage, compared with
macrophages treated with DMSO vehicle control. Furthermore,
increased cleavage of PARP, which is a target of cleaved caspase-3,
was observed in thapsigargin-treated macrophages compared with
DMSO-treated macrophages (Fig.
3B). Finally, thapsigargin-induced apoptosis in macrophages was
demonstrated to be caspase-dependent, as treatment with the
pan-caspase inhibitor Z-VAD-FMK effectively prevented PARP cleavage
and reduced the % of cells in the sub-G1 fraction in
thapsigargin-treated macrophages (Fig.
3B and C). Taken together, these results suggest that
thapsigargin induces caspase-mediated apoptosis.
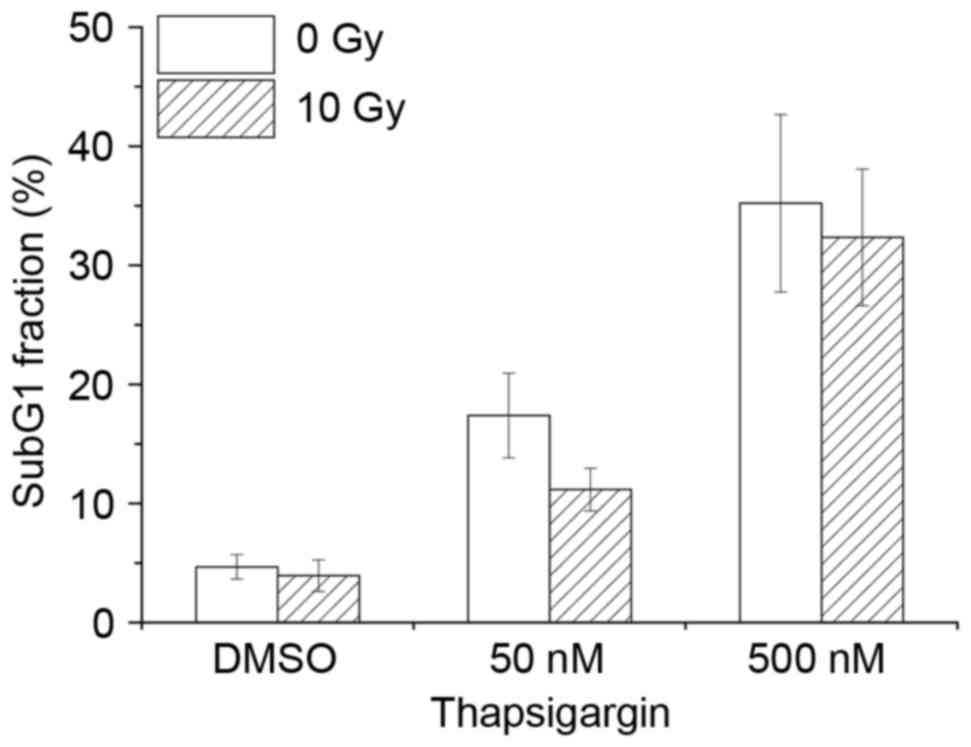
Effect of combination treatment in
macrophages with thapsigargin and X-ray irradiation
Previous studies have demonstrated that ER stress
causes radiosensitization in cancer cells (11,12).
Therefore, the hypothesis that combination treatment with
thapsigargin and X-ray irradiation may enhance apoptosis in
THP-1-derived macrophages was examined. Treatment with thapsigargin
for 48 h induced apoptosis in macrophages, however, combination
treatment with 10-Gy irradiation did not affect the % of apoptotic
cells compared with thapsigargin treatment alone (Fig. 4). Similarly, combination treatment
of macrophages with tunicamycin and 10-Gy irradiation had no effect
on apoptosis rate compared with treatment with tunicamycin alone
(data not shown). These data suggest that there is no synergistic
effect of ER inducers and X-ray irradiation on apoptosis of THP-1
derived macrophages.
Discussion
Although macrophages are important in the host's
immune defense against pathogens, they are also associated with
many diseases, including cancer (2). Therefore, it is crucial for effective
cancer treatment to regulate macrophage apoptosis. In the present
study, the potential of ER stress as a regulator of apoptosis in
radioresistant macrophages was explored. The present findings
demonstrated that radioresistant macrophages responded to ER stress
inducers, leading to enhanced apoptosis through caspase activation
in a p53-independent manner. Consistent with the present results,
mouse macrophage-like RAW 264.7 cells that are treated with
lipopolysaccharide and interferon-γ, undergo apoptosis through ER
stress-mediated signaling pathways without the accumulation of p53
protein (17). Furthermore, free
cholesterol induces ER stress-mediated apoptosis in mouse
macrophages (18). Therefore, it
is considered that ER stress is a promising target pathway to
regulate macrophage apoptosis.
In the present study, two types of ER
stress-inducing agents were used: Thapsigargin and tunicamycin.
Thapsigargin induces ER stress by irreversibly inhibiting the
sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), while
tunicamycin inhibits N-linked glycosylation (19–21).
The present study demonstrated that thapsigargin was more effective
in inducing apoptosis in radioresistant macrophages than
tunicamycin. SERCA pumps are important in maintaining low free
cytosolic Ca2+ levels, and their inhibition activates
the Ca2+-mediated apoptotic pathways (22). Buckley and Whorton (23) have reported that tunicamycin, as
well as thapsigargin, induces Ca2+ release from the
intracellular Ca2+ stores in bovine aortic endothelial
cells. They demonstrated that partial refilling of the
intracellular Ca2+ stores occurred in the cells treated
with tunicamycin in the presence of extracellular Ca2+,
whereas this effect was not observed in the cells treated with
thapsigargin. This result indicates that endoplasmic reticulum
Ca2+-ATPase is active following tunicamycin treatment.
Therefore, it is thought that the inhibition of SERCA pumps may be
an attractive strategy for regulating macrophage apoptosis.
Ionizing radiation induces ER stress in both normal
and cancer cells (15,16,24).
For example, ionizing radiation increases BiP mRNA expression
levels in the IEC-6 rat small intestinal epithelium cell line
(16) and eIF2α phosphorylation in
the MDA-MB-231 human breast cancer cell line (15,24).
In the present study, expression levels of p-eIF2α and BiP in
macrophages were not affected by ionizing radiation, suggesting
that radiation-induced ER stress depends on cell type. IEC-6 and
MDA-MB-231 cells demonstrate apoptotic features following ionizing
radiation (16,24), whereas the present study
demonstrated that irradiated macrophages did not undergo apoptosis.
Therefore, there may be an association between the sensitivity of
cells to radiation-induced ER stress and radiation-induced
apoptosis.
Tunicamycin or other ER stress inducers enhance the
radiosensitivity of cancer cells (11,12,24,25).
Furthermore, Lee et al (16) have reported that tunicamycin and
thapsigargin enhance ionizing radiation-induced caspase-3
activation in IEC-6 cells. Therefore, the hypothesis that
combination treatment of ER stress inducers and ionizing radiation
may kill macrophages in the tumor microenvironment was examined. In
the present study, no synergistic effect of ER stress inducers and
irradiation on apoptosis induction in macrophages was observed.
Yamamori et al (12)
demonstrated that ER stress sensitizes cells to radiation by
impairing DNA double-strand break repair through degradation of the
DNA repair protein RAD51 recombinase. Because p53 is important in
apoptosis induction following DNA damage, synergistic effects of ER
stress and X-ray irradiation might not be feasible in macrophages
derived from p53-negative THP-1 cells. However, considering that
ionizing radiation did not attenuate ER stress-induced apoptosis in
macrophages (Fig. 4), combination
treatment of ER stress inducers with radiotherapy may still be an
effective cancer therapy due to its synergistic effect on cancer
cell radiosensitization.
In conclusion, the present study demonstrated that
ER stress induced apoptosis in radioresistant macrophages. In the
present study, macrophages were produced by differentiation of
monocytic THP-1 cells by PMA stimulation, allowing investigations
of macrophage apoptosis mechanisms in a p53-independent background.
However, there are many subsets of macrophages in vivo and
their phenotype and function varies widely depending on the subset
(1). Therefore, future studies
will be needed to address in more detail whether ER stress induces
apoptosis in disease-related macrophages, and in particular
tumor-associated macrophages.
Acknowledgements
The present study was supported by a Hirosaki
University Grant for Exploratory Research by Young Scientists and
Newly Appointed Scientists. This work was also partially supported
by JSPS KAKENHI, Grant-in-Aid for Scientific Research (grant no.
15K09985), a Grant for Hirosaki University Institutional Research,
and the Takeda Science Foundation [HY]. The authors would like to
thank Enago (www.enago.jp) for the English language
review.
Glossary
Abbreviations
Abbreviations:
|
BiP
|
binding immunoglobulin protein
|
|
DMSO
|
dimethyl sulfoxide
|
|
eIF2
|
eukaryotic initiation factor 2
|
|
ER
|
endoplasmic reticulum
|
|
FBS
|
fetal bovine serum
|
|
HRP
|
horseradish peroxidase
|
|
PBS(−)
|
Ca2+- and Mg2+-
free phosphate-buffered saline
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
PI
|
propidium iodide
|
|
SERCA
|
sarco/endoplasmic reticulum
Ca2+-ATPase
|
|
UPR
|
unfolded protein response
|
|
Z-VAD-FMK
|
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
|
References
|
1
|
Murray PJ and Wynn TA: Protective and
pathogenic functions of macrophage subsets. Nat Rev Immunol.
11:723–737. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schultze JL, Schmieder A and Goerdt S:
Macrophage activation in human diseases. Semin Immunol. 27:249–256.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qian BZ and Pollard JW: Macrophage
diversity enhances tumor progression and metastasis. Cell.
141:39–51. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jinushi M, Chiba S, Yoshiyama H, Masutomi
K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A and Tahara H:
Tumor-associated macrophages regulate tumorigenicity and anticancer
drug responses of cancer stem/initiating cells. Proc Natl Acad Sci
USA. 108:12425–12430. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ostuni R, Kratochvill F, Murray PJ and
Natoli G: Macrophages and cancer: From mechanisms to therapeutic
implications. Trends Immunol. 36:229–239. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bauer M, Goldstein M, Christmann M, Becker
H, Heylmann D and Kaina B: Human monocytes are severely impaired in
base and DNA double-strand break repair that renders them
vulnerable to oxidative stress. Proc Natl Acad Sci USA.
108:21105–21110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Norbury CJ and Zhivotovsky B: DNA
damage-induced apoptosis. Oncogene. 23:2797–2808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu J and Kaufman RJ: From acute ER stress
to physiological roles of the unfolded protein response. Cell Death
Differ. 13:374–384. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Naidoo N: Cellular stress/the unfolded
protein response: Relevance to sleep and sleep disorders. Sleep Med
Rev. 13:195–204. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Lee B and Lee AS: Endoplasmic
reticulum stress-induced apoptosis: Multiple pathways and
activation of p53-up-regulated modulator of apoptosis (PUMA) and
NOXA by p53. J Biol Chem. 281:7260–7270. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Contessa JN, Bhojani MS, Freeze HH,
Rehemtulla A and Lawrence TS: Inhibition of N-linked glycosylation
disrupts receptor tyrosine kinase signaling in tumor cells. Cancer
Res. 68:3803–3809. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yamamori T, Meike S, Nagane M, Yasui H and
Inanami O: ER stress suppresses DNA double-strand break repair and
sensitizes tumor cells to ionizing radiation by stimulating
proteasomal degradation of Rad51. FEBS Lett. 587:3348–3353. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugimoto K, Toyoshima H, Sakai R, Miyagawa
K, Hagiwara K, Ishikawa F, Takaku F, Yazaki Y and Hirai H: Frequent
mutations in the p53 gene in human myeloid leukemia cell lines.
Blood. 79:2378–2383. 1992.PubMed/NCBI
|
|
14
|
Yoshino H, Saitoh T, Kozakai M and
Kashiwakura I: Effects of ionizing radiation on retinoic
acid-inducible gene-I-like receptors. Biomed Rep. 3:59–62.
2015.PubMed/NCBI
|
|
15
|
Nagelkerke A, Bussink J, van der Kogel AJ,
Sweep FC and Span PN: The PERK/ATF4/LAMP3-arm of the unfolded
protein response affects radioresistance by interfering with the
DNA damage response. Radiother Oncol. 108:415–421. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee ES, Lee HJ, Lee YJ, Jeong JH, Kang S
and Lim YB: Chemical chaperones reduce ionizing radiation-induced
endoplasmic reticulum stress and cell death in IEC-6 cells. Biochem
Biophys Res Commun. 450:1005–1009. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gotoh T, Oyadomari S, Mori K and Mori M:
Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated
by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J
Biol Chem. 277:12343–12350. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Devries-Seimon T, Li Y, Yao PM, Stone E,
Wang Y, Davis RJ, Flavell R and Tabas I: Cholesterol-induced
macrophage apoptosis requires ER stress pathways and engagement of
the type A scavenger receptor. J Cell Biol. 171:61–73. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sagara Y and Inesi G: Inhibition of the
sarcoplasmic reticulum Ca2+ transport ATPase by thapsigargin at
subnanomolar concentrations. J Biol Chem. 266:13503–13506.
1991.PubMed/NCBI
|
|
20
|
Lytton J, Westlin M and Hanley MR:
Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum
Ca-ATPase family of calcium pumps. J Biol Chem. 266:17067–17071.
1991.PubMed/NCBI
|
|
21
|
Duksin D and Mahoney WC: Relationship of
the structure and biological activity of the natural homologues of
tunicamycin. J Biol Chem. 257:3105–3109. 1982.PubMed/NCBI
|
|
22
|
Dubois C, Prevarskaya N and Vanden Abeele
F: The calcium-signaling toolkit: Updates needed. Biochim Biophys
Acta. 1863:1337–1343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Buckley BJ and Whorton AR: Tunicamycin
increases intracellular calcium levels in bovine aortic endothelial
cells. Am J Physiol. 273:C1298–C1305. 1997.PubMed/NCBI
|
|
24
|
Suzuki K, Gerelchuluun A, Hong Z, Sun L,
Zenkoh J, Moritake T and Tsuboi K: Celecoxib enhances
radiosensitivity of hypoxic glioblastoma cells through endoplasmic
reticulum stress. Neuro Oncol. 15:1186–1199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yasui H, Takeuchi R, Nagane M, Meike S,
Nakamura Y, Yamamori T, Ikenaka Y, Kon Y, Murotani H, Oishi M, et
al: Radiosensitization of tumor cells through endoplasmic reticulum
stress induced by PEGylated nanogel containing gold nanoparticles.
Cancer Lett. 347:151–158. 2014. View Article : Google Scholar : PubMed/NCBI
|