Introduction
A number of mechanisms are thought to be involved in
the pathogenesis of chronic renal failure, including excessive
deposition of extracellular matrix, exposure to increased cytokine
levels, inhibition of apoptosis and tubular
epithelial-myofibroblast transdifferentiation (TEMT) (1,2). The
latter process is characterized by the activation of tubular
epithelial cells and their differentiation into myofibroblasts
(3–5), which is considered to be the primary
underlying mechanism of renal failure (6). α-smooth muscle actin (α-SMA) is
expressed in smooth muscle cells and myofibroblasts (7). Increased expression of α-SMA in renal
tubular epithelial cells has been suggested to be a potential
marker of transdifferentiation into tubular
epithelial-myofibroblasts. Hepatocyte growth factor (HGF) is a
pleiotropic cytokine with multiple biological functions, including
promoting karyomitosis, accelerating cell locomotion and
anti-apoptotic regulation (8,9). HGF
is considered to possess renoprotective effects by accelerating the
degradation of excessive extracellular matrix (10,11),
restricting TEMT and promoting hyperplasia of tubular epithelial
cells (12–14).
Transforming growth factor-β1 (TGF-β1) is an
important factor involved in TEMT (15). HGF is thought to antagonize the
effect of TGF-β1 by inhibiting its expression (16). In addition, angiotensin II (AngII)
is thought to promote TEMT (17,18).
Benazepril is an angiotensin converting enzyme inhibitor that has
been demonstrated to confer substantial benefits in patients with
advanced renal insufficiency, particularly in those with increased
urinary protein excretion (19,20).
In a previous study, combined treatment of HGF and benazepril
demonstrated highly effective renal protection when compared to
treatment with either drug alone (16). As the renoprotective effects of
benazepril are mediated through inhibition of AngII expression, the
authors of the present study investigated the association between
HGF and AngII. It was hypothesized that TGF-β1 repression and AngII
inhibition may reduce TEMT via HGF. As renal protection by
benazepril was previously observed to be mediated by repression of
AngII expression (20), the
authors investigated whether the reduction of TEMT by HGF may be
mediated by AngII inhibition.
The Janus kinase 2/signal transducer and activator
of transcription 3 (JAK2/STAT3) signaling pathway serves an
important role in the AngII-induced proliferation of smooth muscle
cells (7). A previous study
demonstrated that AngII activated the JAK2/STAT3 signaling pathway
and increased the expression of TGF-β1, as well as connective
tissue growth factors (21). These
results indicate that AngII may contribute to renal interstitial
fibrosis through the JAK2/STAT3 signaling pathway. Therefore, the
association between HGF, AngII and the JAK2/STAT3 signaling pathway
was investigated in the present study.
In the current study, the effect of HGF in reversing
TEMT was first investigated. The association between the effects of
HGF and AngII treatment, together with the potential signaling
pathways involved, was subsequently examined.
Materials and methods
Materials
Human kidney proximal tubular cells (HK-2) were
obtained from The Cell Bank of Type Culture Collection of Chinese
Academy of Sciences (Shanghai, China). α-SMA (cat. no. BM0002),
JAK2 (cat. no. BM1219), phosphorylated (p)-JAK2 (cat. no. BA3398),
STAT3 (cat. no. BA0621), p-STAT3 (cat. no. BA1709) and β-actin
(cat. no. BA2305) primary antibodies were obtained from Wuhan
Boster Biological Technology, Ltd. (Wuhan, China). HGF and AngII
were obtained from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA).
Cell culture
HK-2 cells were cultured as described previously
(22). The cells were maintained
in RPMI-1640 (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany),
and supplemented with 100 IU penicillin, 100 µg/ml streptomycin
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), in
a humidified incubator at 37°C in 5% CO2. Cells were
divided into 4 groups and treated with AngII (1×10−6 M),
HGF (8×10−3 M), AngII plus HGF or control conditions
(RPMI-1640 with 10% FBS) for 24 h.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was isolated from cells (1×106)
using Trizol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. First-strand cDNA was
reverse transcribed using PrimeScript™ RT reagent kit
(Perfect Real Time; Takara Bio, Inc., Otsu, Japan). The protocol
for conducting RT-PCR was identical to that described in a previous
study (23). PCR products were
separated by 1% agarose electrophoresis and DNA band intensities
were quantified using Quantity One software (version no. 4.62;
Bio-Rad Laboratories, Inc., Hercules, CA, USA). Target gene band
densities were normalized to β-actin. The primers and parameters
used for PCR are listed in Table
I.
 | Table I.Primer sequences and thermal cycling
conditions. |
Table I.
Primer sequences and thermal cycling
conditions.
| Gene | Primer | Sequence
(5′-3′) | Annealing
temperature (°C) | Number of
cycles | Product (bp) |
|---|
| α-SMA | Sense |
ACTGGGACGACTAGGAAAAA | 58 | 28 | 240 |
|
| Antisense |
CATCTCCAGAGTCCAGCACA |
| β-actin | Sense |
ATCATGTTTGAGACCTTCAACA | 58 | 28 | 552 |
|
| Antisense |
CATGGTGGTGCCGCCAGACAG |
Western blot analysis
Cells (1×106) were lysed in a sodium
dodecyl sulfate (SDS) sample buffer containing 2% SDS, 10 mmol/l
Tris-HCl (pH 6.8) and 10% (v/v) glycerol. The lysates were
centrifuged at 12,000 × g for 15 min at 4°C, and the supernatant
was stored at −70°C. Protein concentration was determined using a
bicinchoninic acid assay kit (Bio-Rad Laboratories, Inc.). Total
protein (50 µg) was loaded in each lane, before it was separated in
a 10% SDS-PAGE gel and transferred to a nitrocellulose membrane.
Following blocking in 4% non-fat dry milk in TBS, the membranes
were incubated with primary antibodies (α-SMA, JAK2, p-JAK2, STAT3
or p-STAT3) at a 1:1,000 dilution in TBS overnight at 4°C.
Following washing with TBS-0.5% Tween-20, the membranes were
incubated for 1 h at 37°C with a horseradish peroxidase-conjugated
anti-mouse IgG secondary antibody (cat. no. 7076; Cell Signaling
Technology, Inc., Danvers, MA, USA) at 1:2,000 dilution, and
immunoreactive proteins were detected using SuperSignal
chemiluminescence reagent (Pierce; Thermo Fisher Scientific, Inc.).
The blots were stripped and reprobed with β-actin antibody
(dilution, 1:5,000). The immunoblots were analyzed by densitometry,
and protein band densities were quantified using Quantity One
software (version no. 4.62; Bio-Rad Laboratories, Inc.).
Acridine orange/ethidium bromide
staining
HK-2 cells (5×104) were cultured in
24-well plates and divided into 4 groups that were treated with
AngII (1×10−6 M), HGF (8×10−3 M), AngII plus
HGF or control conditions. Following incubation for 24 h at 37°C in
5% CO2, 5 µl (10 µg/ml) acridine orange and 5 µl (10
µg/ml) ethidium bromide were applied to each well, before the cells
were incubated for 5 min at room temperature. The stained cells
were analyzed using a fluorescence microscope (Olympus Corporation,
Tokyo, Japan). The experiments were repeated three times.
Analysis of apoptosis by Annexin V
staining
In order to determine the level of apoptosis in HK-2
cells in each treatment group, Annexin V staining was performed
using the Annexin V-FITC Apoptosis Detection kit (cat. no. ab14085;
Abcam, Cambridge, UK) according to the manufacturer's instructions.
Briefly, HK-2 cells (5×105) were stained with Annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide (PI), and
incubated in the dark at room temperature for 30 min following
exposure to AngII, HGF, or AngII plus HGF for 24 h. Cells
(1×105) were subsequently analyzed using a flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA) following the
addition of binding buffer and the results were analyzed with
Navios tetra software (version no. 1.1; Beckman Coulter, Inc.).
Annexin V+/PI− cells were defined as cells in
early apoptosis and Annexin V+/PI+ cells were
defined as cells in late apoptosis or necrosis.
Statistical analysis
Data are expressed as mean ± standard error. One-way
analysis of variance followed by the Tukey test for multiple
comparisons was conducted to assess the differences among multiple
groups. SPSS 17.0 (SPSS, Inc., Chicago, IL, USA) was used for data
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
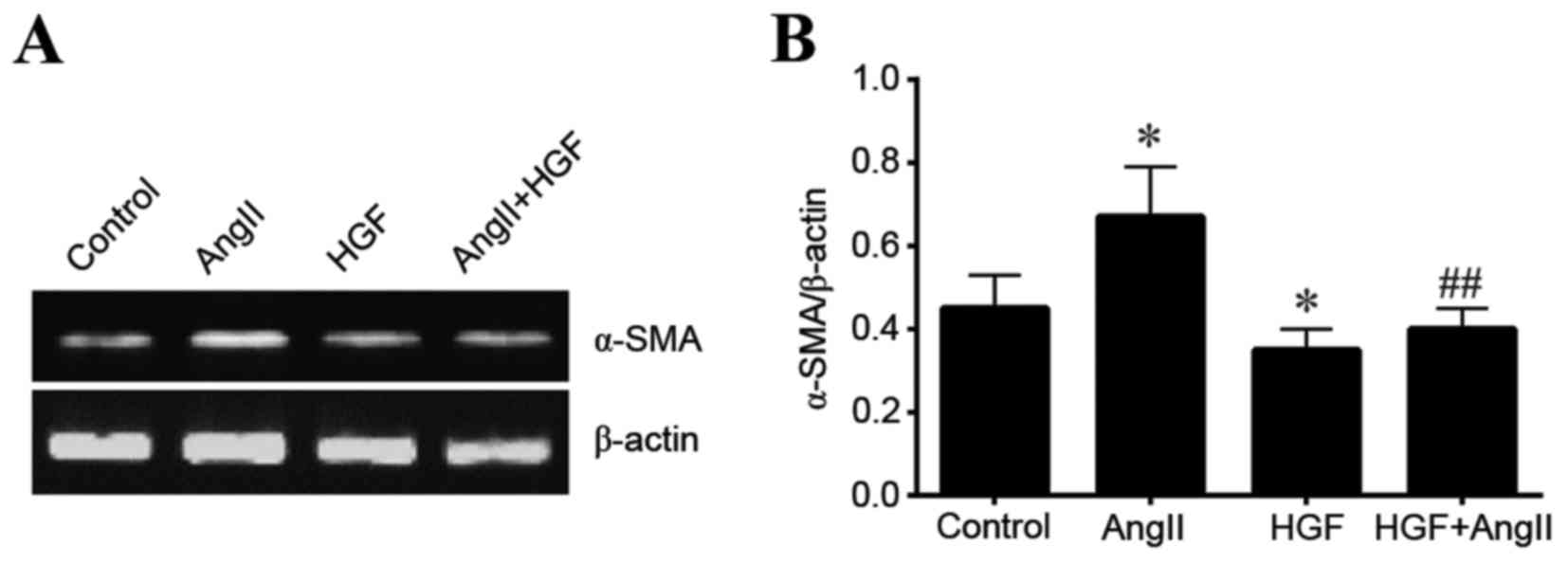
HGF decreases α-SMA expression
To investigate the role of HGF and AngII in TEMT,
the expression of α-SMA in HK-2 cells was examined at the RNA and
protein levels. HGF significantly decreased the expression of α-SMA
at the mRNA level when compared with the controls (P<0.05;
Fig. 1). By contrast, AngII
increased α-SMA expression when compared to control conditions
(P<0.05; Fig. 1). In addition,
HGF significantly attenuated AngII-induced expression of α-SMA mRNA
(P<0.01 vs. AngII-only treated cells; Fig. 1). A similar α-SMA expression
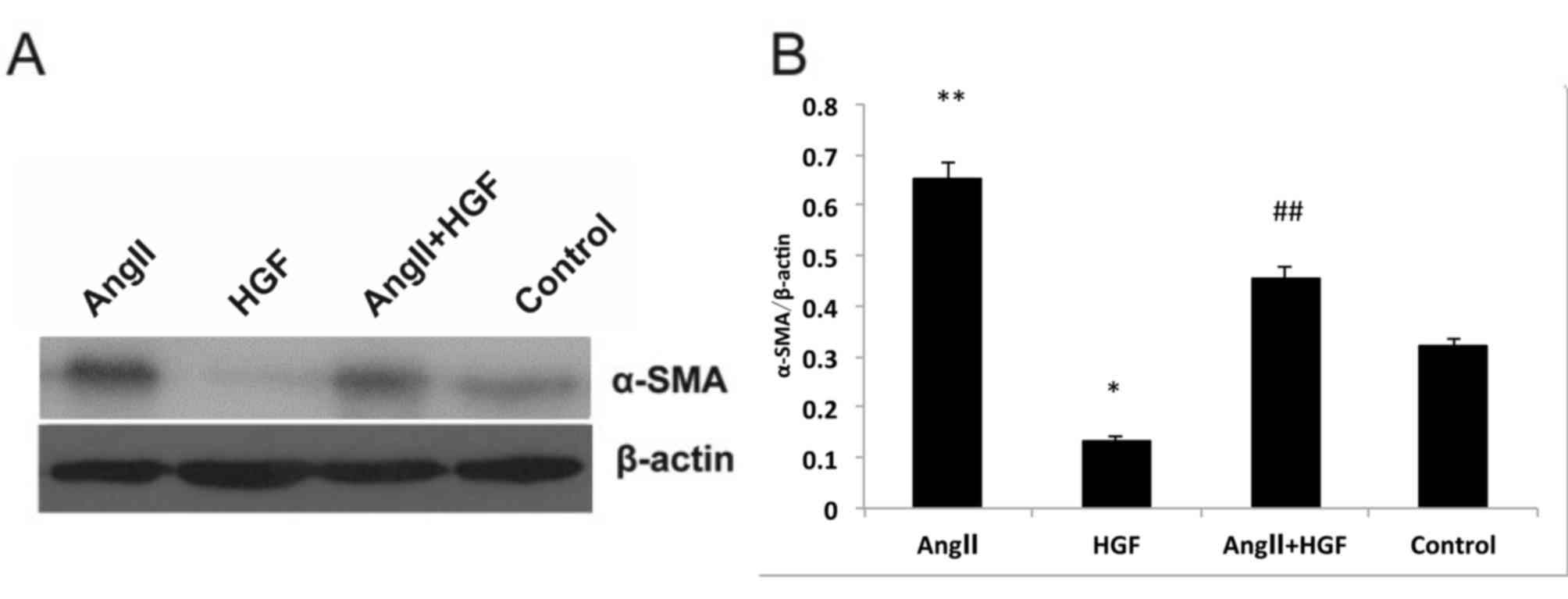
profile was observed at the protein level. Exposure to HGF
significantly decreased α-SMA protein expression when compared with
the controls (P<0.05; Fig. 2).
However, AngII treatment significantly increased α-SMA expression
relative to that of control cells (P<0.01; Fig. 2). In addition, exposure to HGF
significantly attenuated AngII-induced increase α-SMA expression
(P<0.01 vs. AngII-only treated cells; Fig. 2). As α-SMA expression is considered
to provide a measure of TEMT, it is possible that AngII may promote
the transdifferentiation process, whilst HGF may have the opposite
effect. It is therefore possible that HGF may regulate TEMT by
inhibiting AngII.
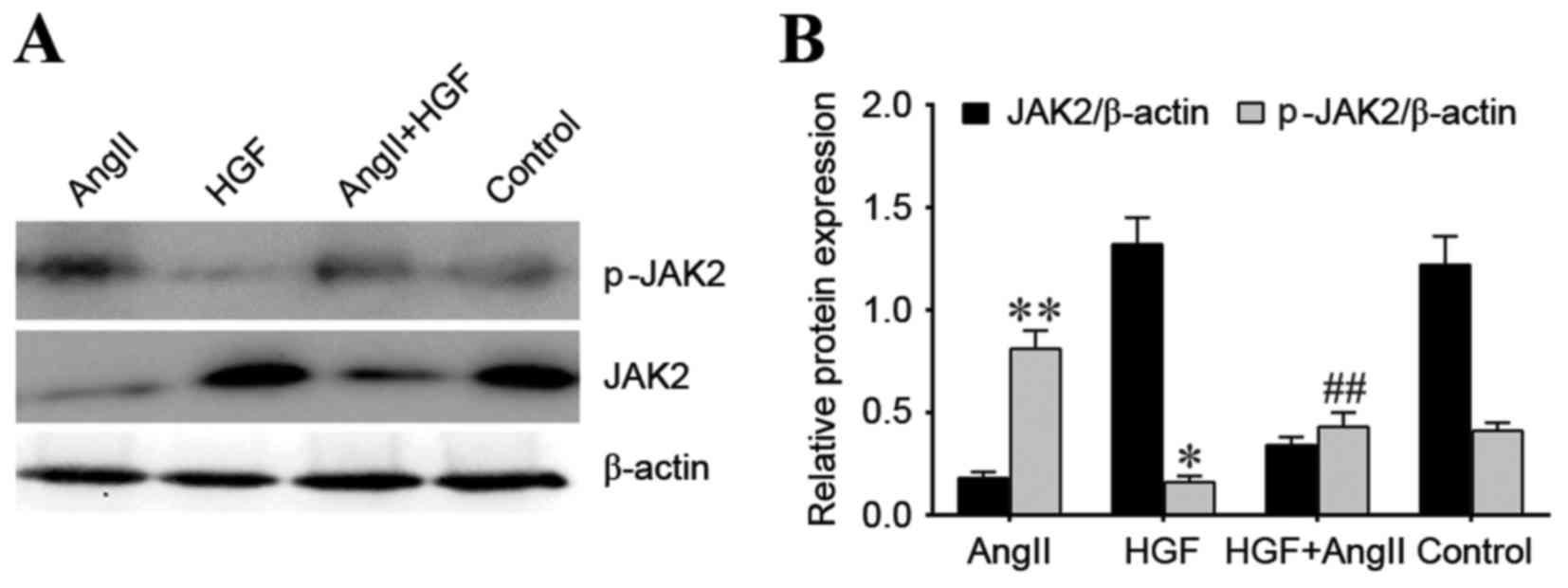
Regulation of JAK2 and p-JAK2
proteins
To investigate the association between HGF, AngII
and the JAK2/STAT3 signaling pathway, the protein expression levels
of JAK2 and STAT3, as well as the phosphorylated forms of these
proteins, were examined. p-JAK2 protein expression was
significantly decreased in the HGF treatment group when compared
with the control group (P<0.05), whereas p-JAK2 protein
expression was significantly increased in the AngII treatment group
when compared with the controls (P<0.01; Fig. 3). p-JAK2 protein expression was
significantly decreased in the AngII plus HGF treatment group when
compared to the AngII-only treatment group (P<0.01; Fig. 3). These effects were comparable to
those observed with α-SMA expression. However, a similar trend was
not observed for JAK2 protein expression. JAK2 protein expression
was higher in the HGF and control groups compared with the AngII
and AngII plus HGF groups (Fig.
3).
Regulation of STAT3 and p-STAT3
protein expression
HGF treatment significantly decreased p-STAT3
protein expression levels when compared with controls (P<0.05;
Fig. 4). However, AngII treatment
increased p-STAT3 expression when compared with controls
(P<0.01; Fig. 4). In addition,
expression of p-STAT3 protein following exposure to AngII and HGF
was significantly decreased when compared to AngII-only treated
cells (P<0.01; Fig. 4). This
was similar to the trend in expression of α-SMA mRNA and protein
among treatment groups. However, a similar trend to p-STAT3 was not
observed for STAT3 protein expression. STAT3 protein expression was
higher in the HGF and control groups compared with the AngII and
AngII plus HGF groups. These results suggest that HGF may inhibit
TEMT through the inhibition of AngII, and this effect may be
mediated by inhibition of the p-JAK2/p-STAT3 signaling pathway.
Acridine orange/ethidium bromide
staining
Following exposure to AngII, HGF or AngII plus HGF,
HK-2 cells were stained with acridine orange and ethidium bromide
to determine the level of apoptosis in HK-2 cells exposed to
different treatments. The results demonstrated that treatment of
cells with AngII was associated with induction of apoptosis when
compared with controls (Fig. 5).
By contrast, treatment with HGF attenuated AngII-induced apoptosis
(Fig. 5).
Annexin V analysis
The results of the Annexin V-FITC/PI
double-fluorescence staining assay revealed that AngII treatment
significantly induced apoptosis in HK-2 cells when compared with
controls (P<0.01; Fig. 6). This
was demonstrated by an increase in the percentage of Annexin
V+/PI− and Annexin
V+/PI+ subpopulations. In addition, the
percentage of apoptotic cells significantly decreased following
exposure to AngII plus HGF (P<0.01 vs. the AngII-only treated
group; Fig. 6). The present study
demonstrated that 15.2% of cells underwent apoptosis (Annexin
V+/PI−) following treatment with AngII plus
HGF (Fig. 6). By contrast, 28.4%
of cells underwent apoptosis following treatment with AngII alone
(Fig. 6). This indicated that HGF
may prevent apoptosis induced by AngII.
Discussion
It has been previously demonstrated that HGF
represses renal interstitial fibrosis (24–27).
In addition, previous studies have revealed that HGF exhibits
renoprotective effects in a number of animal models, such as acute
renal failure and diabetic nephropathy models (28–30).
Induction of TGF-β1 is one of the key mechanisms responsible for
increased fibrosis (31). Previous
studies have indicated that HGF may induce unfavorable conditions
for TEMT (10). Furthermore,
increased expression of α-SMA in the kidney has been reported to be
a marker of TEMT pathology (4).
AngII is an important component of the
renin-angiotensin system, and has been reported to serve an
important role in a number of renal diseases (20). AngII-induced renal injury is
mediated by its systemic effect on blood pressure regulation,
and/or by its regulatory effect on TGF-β1 (32,33).
HGF and AngII have opposing effects, and in vascular smooth muscle
cells it has been demonstrated that AngII may repress the
production of HGF in a dose-dependent manner (34). Lotensin is an angiotensin
converting enzyme inhibitor, that inhibits AngII production.
Previous studies have demonstrated the renoprotective effect of
Lotensin (19,20). It is commonly used in clinical
practice to decrease urinary protein excretion and to stabilize
renal function during the early stages of chronic renal failure. In
the present study, AngII increased α-SMA expression at the mRNA and
protein level in HK-2 cells, whereas HGF suppressed the
AngII-induced expression of α-SMA. These results indicated that,
although AngII promotes TEMT, HGF may function to alleviate this
process. Acridine orange/ethidium bromide staining was performed to
determine functional activity. The results demonstrated that AngII
induced apoptosis in HK-2 cells, whereas the addition of HGF was
able to attenuate this effect.
The JAK2/STAT3 signaling pathway participates in the
propagation of cell division, apoptosis and the regulation of
immune cells, and serves an important function in diabetic kidney
disease (35,36). The JAK2/STAT3 signaling pathway is
activated during smooth muscle cell proliferation induced by AngII.
It has been suggested that the coupling of AngII with the
angiotensin type 1 receptor on the surface of mesangial cells may
lead to JAK2 phosphorylation, thereby binding the downstream
factor, STAT3. Dimerization of STAT3 and transfer into the cell
nucleus may lead to altered gene expression (37,38).
It was also demonstrated that AngII may increase the expression of
TGF-β1 and connective tissue growth factor by the JAK2/STAT3
signaling pathway (21). As a
result of this research, the authors of the present study
investigated whether AngII may be involved in the process of renal
fibrosis by activating the JAK2/STAT3 signaling pathway (21). The results of the present study
revealed that HGF may reduce TEMT by inhibiting AngII via by the
p-JAK2/p-STAT3 signaling pathway. However, further investigation
involving loss- and gain-of-function experiments, using small
interfering-RNAs and/or expression vectors, are required to test
this hypothesis.
Acknowledgements
The present study was supported by the Jilin
Provincial Department of Health (grant no. 2009ZC041).
References
|
1
|
Yamaguchi Y, Suzuki T, Arita S, Iwashita
C, Sakamoto K, Hatakeyama E, Shimmura H, Tanabe K, Ichinose M,
Suzuki N and Yamada K: Possible involvement of urokinase-type
plasminogen activator release from human peripheral blood
lymphocytes in the pathophysiology of chronic allograft
nephropathy. Transplant Proc. 37:4276–4281. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang HY, Yang LZ, Cui MJ, Gu CM, Zhao Y,
Chen Y, Zhao D, Li TS and Chi B: Hepatocyte growth factor-induced
amelioration in chronic renal failure is associated with reduced
expression of α-smooth muscle actin. Ren Fail. 34:862–870. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grupp C, Troche I, Klass C, Köhler M and
Müller GA: A novel model to study renal myofibroblast formation in
vitro. Kidney Int. 59:543–553. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ruiz-Ortega M, Ruperez M, Lorenzo O,
Esteban V, Blanco J, Mezzano S and Egido J: Angiotensin II
regulates the synthesis of proinflammatory cytokines and chemokines
in the kidney. Kidney Int Suppl. 82:S12–S22. 2002. View Article : Google Scholar
|
|
5
|
Okada H, Inoue T, Suzuki H, Strutz F and
Neilson EG: Epithelial-mesenchymal transformation of renal tubular
epithelial cells in vitro and in vivo. Nephrol Dial Transplant.
15:(Suppl 6). S44–S46. 2000. View Article : Google Scholar
|
|
6
|
Badid C, Mounier N, Costa AM and
Desmoulière A: Role of myofibroblasts during normal tissue repair
and excessive scarring: Interest of their assesment in
nephropathies. Histol Histopathol. 15:269–280. 2000.PubMed/NCBI
|
|
7
|
Jiang T, Zhou QS, Pi L and Huang B: Role
of angiotensin II and JAK2 signal pathway in transdifferentiation
of renal tubular cells in mice after acute ischemic followed by
reperfusion. Zhonghua Bing Li Xue Za Zhi. 38:466–471. 2009.(In
Chinese). PubMed/NCBI
|
|
8
|
Funakoshi H and Nakamura T: Hepatocyte
growth factor: From diagnosis to clinical applications. Clin Chim
Acta. 327:1–23. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Forte G, Minieri M, Cossa P, Antenucci D,
Sala M, Gnocchi V, Fiaccavento R, Carotenuto F, De Vito P, Baldini
PM, et al: Hepatocyte growth factor effects on mesenchymal stem
cells: Proliferation, migration, and differentiation. Stem Cells.
24:23–33. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dai C and Liu Y: Hepatocyte growth factor
antagonizes the profibrotic action of TGF-beta1 in mesangial cells
by stabilizing Smad transcriptional corepressor TGIF. J Am Soc
Nephrol. 15:1402–1412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang J, Dai C and Liu Y: Hepatocyte growth
factor suppresses renal interstitial myofibroblast activation and
intercepts Smad signal transduction. Am J Pathol. 163:621–632.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Yang J, Dai C, Wu C and Liu Y: Role
for integrin-linked kinase in mediating tubular epithelial to
mesenchymal transition and renal interstitial fibrogenesis. J Clin
Invest. 112:503–516. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimamura M, Sato N, Yoshimura S, Kaneda Y
and Morishita R: HVJ-based non-viral gene transfer method:
Successful gene therapy using HGF and VEGF genes in experimental
ischemia. Front Biosci. 11:753–759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Li C, Summer SN, Falk S, Wang W,
Ljubanovic D and Schrier RW: Role of AQP1 in endotoxemia-induced
acute kidney injury. Am J Physiol Renal Physiol. 294:F1473–F1480.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Reeves WB and Andreoli TE: Transforming
growth factor beta contributes to progressive diabetic nephropathy.
Proc Natl Acad Sci USA. 97:7667–7669. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang HY, Wang YJ, Cui MJ, Gu CM, Yang LZ,
Zhao Y, Chen Y, Zhao D, Li TS and Chi BR: Hepatocyte growth
factor-induced amelioration in renal interstitial fibrosis is
associated with reduced expression of alpha-smooth muscle actin and
transforming growth factor-beta1. Indian J Biochem Biophys.
48:308–315. 2011.PubMed/NCBI
|
|
17
|
Yan Z, Yao F and Shi YH: Effects of
irbesartan on expression of glycogen synthase kinase-3β in tubular
epithelial-mesenchymal transition induced by high glucose. Chinese
Pharmacological Bulletin. 25:225–229. 2009.
|
|
18
|
Chen J, Chen JK and Harris RC: Angiotensin
II induces epithelial-to-mesenchymal transition in renal epithelial
cells through reactive oxygen species/Src/caveolin-mediated
activation of an epidermal growth factor receptor-extracellular
signal-regulated kinase signaling pathway. Mol Cell Biol.
32:981–991. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ihle BU, Whitworth JA, Shahinfar S, Cnaan
A, Kincaid-Smith PS and Becker GJ: Angiotensin-converting enzyme
inhibition in nondiabetic progressive renal insufficiency: A
controlled double-blind trial. Am J Kidney Dis. 27:489–495. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hou FF, Zhang X, Zhang GH, Xie D, Chen PY,
Zhang WR, Jiang JP, Liang M, Wang GB, Liu ZR and Geng RW: Efficacy
and safety of benazepril for advanced chronic renal insufficiency.
N Engl J Med. 354:131–140. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Fan Q and Wang L: Significance of
JAK2/STAT3 in angiotensin II up-regulation of TGF-β1, CTGF and FN
mRNA expression on mesangial cells under hyperglucose. J Nephrol
Dialy Transplant. 18:44–48. 2009.
|
|
22
|
Kim YS, Xu ZG, Reddy MA, Li SL, Lanting L,
Sharma K, Adler SG and Natarajan R: Novel interactions between
TGF-{beta}1 actions and the 12/15-lipoxygenase pathway in mesangial
cells. J Am Soc Nephrol. 16:352–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ionescu E, Sauter JF and Jeanrenaud B:
Abnormal oral glucose tolerance in genetically obese (fa/fa) rats.
Am J Physiol. 248:E500–E506. 1985.PubMed/NCBI
|
|
24
|
Liu Y, Rajur K, Tolbert E and Dworkin LD:
Endogenous hepatocyte growth factor ameliorates chronic renal
injury by activating matrix degradation pathways. Kidney Int.
58:2028–2043. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mizuno S, Kurosawa T, Matsumoto K,
Mizuno-Horikawa Y, Okamoto M and Nakamura T: Hepatocyte growth
factor prevents renal fibrosis and dysfunction in a mouse model of
chronic renal disease. J Clin Invest. 101:1827–1834. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Azuma H, Takahara S, Matsumoto K, Ichimaru
N, Wang JD, Moriyama T, Waaga AM, Kitamura M, Otsuki Y and Okuyama
A: Hepatocyte growth factor prevents the development of chronic
allograft nephropathy in rats. J Am Soc Nephrol. 12:1280–1292.
2001.PubMed/NCBI
|
|
27
|
Zhang TXL: Hepatocyte growth factor and
its effect on the kidney damage. Chemistry of Life. 25:399–401.
2005.
|
|
28
|
Yamasaki N, Nagano T, Mori-Kudo I,
Tsuchida A, Kawamura T, Seki H, Taiji M and Noguchi H: Hepatocyte
growth factor protects functional and histological disorders of
HgCl(2)-induced acute renal failure mice. Nephron. 90:195–205.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cruzado JM, Lloberas N, Torras J, Riera M,
Fillat C, Herrero-Fresneda I, Aran JM, Alperovich G, Vidal A and
Grinyó JM: Regression of advanced diabetic nephropathy by
hepatocyte growth factor gene therapy in rats. Diabetes.
53:1119–1127. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang J and Liu Y: Delayed administration
of hepatocyte growth factor reduces renal fibrosis in obstructive
nephropathy. Am J Physiol Renal Physiol. 284:F349–F357. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsuchida K, Zhu Y, Siva S, Dunn SR and
Sharma K: Role of Smad4 on TGF-beta-induced extracellular matrix
stimulation in mesangial cells. Kidney Int. 63:2000–2009. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rekola S, Bergstrand A and Bucht H:
Deterioration rate in hypertensive IgA nephropathy: Comparison of a
converting enzyme inhibitor and beta-blocking agents. Nephron.
59:57–60. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Taal MW and Brenner BM: Renoprotective
benefits of RAS inhibition: From ACEI to angiotensin II
antagonists. Kidney Int. 57:1803–1817. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakano N, Morishita R, Moriguchi A,
Nakamura Y, Hayashi SI, Aoki M, Kida I, Matsumoto K, Nakamura T,
Higaki J and Ogihara T: Negative regulation of local hepatocyte
growth factor expression by angiotensin II and transforming growth
factor-beta in blood vessels: Potential role of HGF in
cardiovascular disease. Hypertension. 32:444–451. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lu TC, Wang ZH, Feng X, Chuang PY, Fang W,
Shen Y, Levy DE, Xiong H, Chen N and He JC: Knockdown of Stat3
activity in vivo prevents diabetic glomerulopathy. Kidney Int.
76:63–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Pang M, Ma L, Gong R, Tolbert E, Mao H,
Ponnusamy M, Chin YE, Yan H, Dworkin LD and Zhuang S: A novel STAT3
inhibitor, S3I-201, attenuates renal interstitial fibroblast
activation and interstitial fibrosis in obstructive nephropathy.
Kidney Int. 78:257–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Levy O and Granot Y: Arginine-Vasopressin
activates the JAK-STAT pathway in vascular smooth muscle cells. J
Biol Chen. 281:15597–15604. 2006. View Article : Google Scholar
|
|
38
|
Banes AK, Shaw S, Jenkins J, Redd H, Amiri
F, Pollock DM and Marrero MB: Angiotensin II blockade prevents
hyperglycemia-induced activation of JAK and STAT proteins in
diabetic rat kidney glomeruli. Am J Physiol Renal Physiol.
286:F653–F657. 2004. View Article : Google Scholar : PubMed/NCBI
|