Introduction
Chronic infection by Helicobacter pylori is
achieved through the colonization of an almost exclusive niche and
through evading detection by the host's cellular immune defense
mechanisms (1). H. pylori
is the only microorganism known to colonize the human stomach and
to inhabit gastric mucosal cells. To achieve this colonization,
H. pylori must escape detection by both innate and adaptive
immune responses (2). Macrophages,
phagocytic cells that are part of the innate immune system, are
located in various tissues. A great number of cytokines assist
macrophages in their role as custodians of the innate immune system
as they mediate the transition from innate to adaptive immunity
(3). Therefore, macrophages may
serve an important role in H. pylori infection.
Toll-like receptor (TLR) 4, a pattern recognition
receptor, recognizes pathogen-associated molecular patterns. It
activates macrophages to secrete cytokines, which initiate and
regulate the immune response (4).
T-cell immunoglobulin and mucin-domain-containing molecule 3
(Tim-3) is an important member of the TIM family, a recently
discovered family of trans-membrane proteins (5). Previous studies have demonstrated
that Tim-3 serves a role in the differentiation of T helper (Th) 1
cells from Th2 cells; upon activation, Tim-3 downregulates Th1 cell
function (5–8). Several studies have also demonstrated
that TLR4 and Tim-3 are expressed on macrophages and that they
participate in the regulation of cytokine secretion from
macrophages (9–11), suggesting that Tim-3 impacts
macrophage function by interacting with the TLR4 signaling pathway.
However, it is unclear to date how H. pylori infection
impacts the interaction of Tim-3 and TLR4 in macrophages.
Based on these previous studies, an investigation of
the interaction between Tim-3 and TLR4 signaling pathways in H.
pylori-associated inflammation was undertaken. The present
study aimed to provide theoretical and experimental evidence to
support Tim-3 as a potential therapeutic target for the control and
prevention of H. pylori infection-related diseases.
Materials and methods
Cell lines and bacteria
The murine macrophage RAW264.7 cell line was
obtained from the Gastroenterology Institute of Jiangxi Province
(Nanchang, China) and was cultured in DMEM with 10% FBS, 100 U/ml
penicillin (Gibco, Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and 100 µg/ml streptomycin (Gibco, Thermo Fisher Scientific,
Inc.) under 5% CO2 at 37°C in a humidified atmosphere.
The H. pylori standard strain SS1 (CagA+,
VacA+) was obtained from the Chinese Center for Disease
Control (Beijing, China) and was grown on a Campylobacter agar base
from the Chinese Research Center for Diarrhoeal Disease Control
(Shanghai, China), containing 10% sheep blood, under microaerobic
conditions (5% O2, 10% CO2 and 85%
N2) at 37°C for 2–3 days.
Cell transfection
Murine Tim-3 genes was amplified by polymerase chain
reaction (PCR) using the following primer: Forward,
5′-CAACTCGAGATGTTTTCAGGTCTTACCCT-3′ and reverse,
5′-AACGGATCCTCAGGATGGCTGCTGGCTGT-3′. The PCR products were purified
to generate the plasmid pLVX-IRES-ZsGreen1 (Clontech Laboratories,
Inc., Mountainview, CA, USA). The ligated products were transformed
into TOP10 chemically competent Escherichia coli
(Takara Bio, Inc., Otsu, Japan) and incubated on
Luria-Bertani plates containing 100 µg/ml ampicillin at 37°C
overnight. Subsequently, eight putative ampicillin-resistant
positive clones were selectedå to amplify, extract and purify for
PCR amplification and electrophoresis detection. The
pLVX-IRES-ZsGreen1-Tim-3 plasmid was constructed and the sequencing
was completed by Shanghai ShengGong Biotechnology Co., Ltd.
(Shanghai, China). Then the RAW264.7 cells were plated at a density
of 2×105 cells/ml in a 6-well plate, grown overnight,
and transferred to serum-free medium prior to Tim-3 transfection.
The RAW264.7 cells were transfected with 2.5 µg
pLVX-IRES-ZsGreen1-Tim-3 plasmid or empty vector using
Lipofectamine® LTX and Plus Reagent (15,338,100;
Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Cells were observed for green fluorescent
protein using fluorescence microscopy after 42 h transfection. The
control cells were grown in serum-free medium alone. Tim-3
overexpression was confirmed by dual-endonuclease digestion and
sequencing.
Reverse transcription (RT)-PCR
Total RNA was extracted from RAW264.7 cells in
6-well plate using an E.Z.N.A. Total RNA kit II (Omega BioTek,
Inc., Norcross, GA, USA) according to the manufacturer's protocols.
RNA quality was evaluated by 1% agarose gel electrophoresis. Using
a Quantiscript RT Kit (Tiangen Biotech Co., Beijing, China),
single-strand cDNA was synthesized and then used as a template for
PCR amplification of Tim-3, TLR4, myeloid differentiation factor 88
(MyD88) and β-actin (used for normalization). The 2−ΔΔCq
method (12) was used to determine
the relative quantities of products. Primer sequences were as
follows: Tim-3, forward 5′-ACTCTACCTACATCTGGGACACT-3′ and reverse
5′-TAGGTCCCATGGTCATCCAG-3′; TLR4, forward
5′-AGAAACGGCAACTTGGACCT-3′ and reverse 5′-GGCCTTAGCCTCTTCTCCT-3′;
MyD88, forward 5′-CTGGCCTTGTTAGACCGTGA-3′ and reverse
5′-TCGAAAAGTTCCGGCGTTTG-3′; and β-actin, forward
5′-GAGACCTTCAACACCCCAGC-3′ and reverse 5′-ATGTCACGCACGATTTCCC-3′.
Each 25 µl PCR consisted of 10 pmol of each primer, 10 ul 2xTaq
Master Mix, 3 ul template, and 10 µl ddH2O. β-actin and
MyD88 were amplified at 94°C for 90 sec (1 cycle); 94°C for 30 sec,
61°C for 30 sec and 72°C for 1 min (30 cycles); and 72°C for 5 min
(1 cycle). Tim3 and TLR4 were amplified at 94°C for 90 sec (1
cycle); 94°C for 30 sec, 60°C for 30 sec and 72°C for 1 min (30
cycles); 72°C for 5 min (1 cycle). The PCR products were visualized
following electrophoresis on 2% agarose gels. Densitometric
analysis of the bands was carried out using a ChemiDoc MP System
with Image Lab™ software version 5.1 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Results are expressed as the ratio of the
intensity of the band for Tim-3, TLR4, and MyD88 to the intensity
of the band for β-actin.
Western blot analysis
Total protein was extracted from cells using cell
lysis buffer (cat no. P0013; Beyotime Biotechnology, Shanghai,
China). Protein concentrations were measured using a bicinchoninic
acid (BCA) protein assay (Generay Biotech Co., Ltd., Shanghai,
China), according to the manufacturer's protocols. Equal amounts of
proteins (10 µg) of cell lysates were separated on a 10% SDS-PAGE
and electrotransferred onto a nitrocellulose membrane (EMD
Millipore, Billerica, MA, USA). The membranes were blocked in Tris
buffered saline Tween containing 5% fat-free dry milk and incubated
overnight at 4°C with primary antibodies, including rabbit
anti-Tim-3 antibody (M-171; sc-292390; 1:200; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), mouse anti-TLR4 antibody
(ab22048; 1:1,000; Abcam, Cambridge, UK), rabbit anti-MyD88
antibody (ab2068; 1:1,000; Abcam), rabbit anti-phosphorylated (p-)
nuclear factor κB (NF-κB) p65 antibody (3033; 1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA) and mouse
anti-β-actin antibody (ab1801; 1:1,000; Abcam). Goat anti-rabbit
horseradish peroxidase (HRP)-conjugated immunoglobulin G (IgG;
ZDR-5306; 1:2,000; ZSGB-BIO, Beijing, China) or rabbit anti-mouse
HRP-IgG (ZDR-5109; 1:2,000; ZSJB-BIO) were used as secondary
antibodies. Proteins were detected using a ChemiDoc MP System with
Image Lab™ Software version 5.1 (Bio-Rad Laboratories, Inc.). Data
were normalized to β-actin levels.
ELISA
The concentrations of interleukin 6 (IL-6), tumor
necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and interleukin 10
(IL-10) in the cell culture supernatants of RAW264.7 cells were
determined by ELISA, using a commercial human multiplex kit (IL-6,
TNF-α, IFN-γ and IL-10; 88-5083; Aushon Biosystems, Inc., Wuxi,
China) according to the manufacturer's protocol.
Statistical analysis
Data are expressed as the means ± standard deviation
of three independent experiments. Data were analyzed by paired or
unpaired t-tests when comparing differences between paired samples
or two independent groups respectively. One-way analysis of
variance with the LSD post hoc test was used for multiple
comparisons. Data analysis was performed using SPSS 17.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Effect of H
pylori infection on Tim-3, TLR4 and MyD88 mRNA
expression in macrophages. H. pylori infection of macrophages was
performed by co-culturing H. pylori with RAW264.7 cells for
12 h at various bacteria/cell ratios, i.e. multiplicities of
infection (MOI). H. pylori infection significantly elevated
Tim-3, TLR4 and MyD88 mRNA expression levels in RAW264.7
macrophages compared with control uninfected macrophages (P<0.01
compared with control; Fig. 1).
Tim-3, TLR4 and MyD88 mRNA levels increased in a MOI-dependent
manner (MOI range tested 25–200; Fig.
1). Tim-3 mRNA expression was significantly higher in the
infected cells compared with the control cells in all MOI tested
(P<0.01; Fig. 1), and no
significant difference was observed between different MOI effects
(P>0.05; Fig. 1). TLR4 mRNA
expression was significantly higher at MOI ratios 50–200 compared
with control uninfected cells (P<0.01; Fig. 1). Lastly, MyD88 mRNA expression was
significantly higher at MOI ratios 100 and 200 compared with
control uninfected cells (P<0.01; Fig. 1).
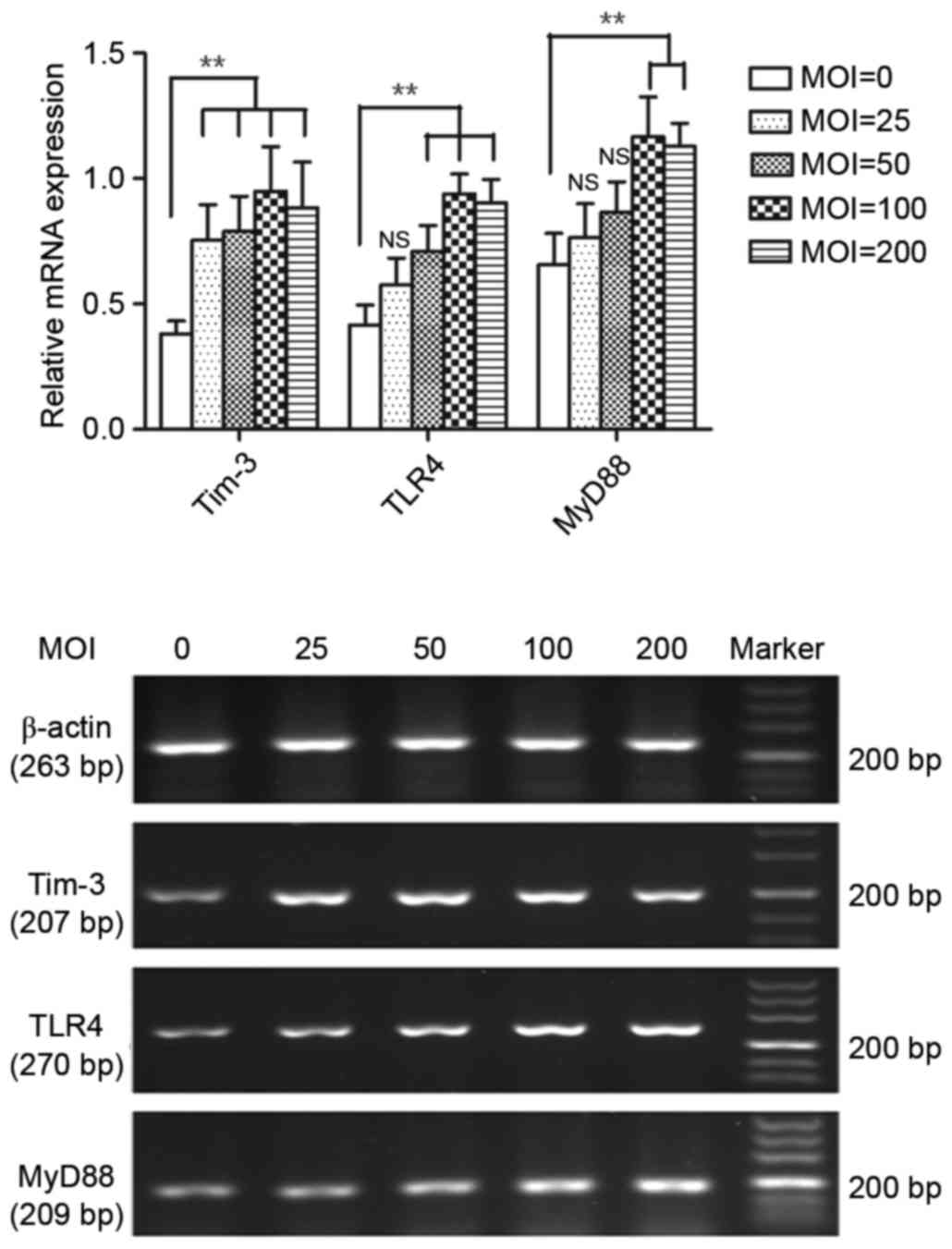 | Figure 1.Effect of Helicobacter pylori
infection on Tim-3, TLR4 and MyD88 mRNA expression. RAW264.7
macrophages were cocultured with H. pylori at 0, 25, 50,
100, or 200 MOI for 12 h, following which mRNA expression for
Tim-3, TLR4 and MyD88 was analyzed in the RAW264.7 cells by reverse
transcription-quantitative polymerase chain reaction. n=4
independent experiments performed in duplicate. *P<0.05 and
**P<0.01 vs. 0 MOI. Tim-3, T-cell immunoglobulin and
mucin-domain-containing molecule-3; TLR4, Toll-like receptor 4;
MyD88, myeloid differentiation factor 88; MOI, multiplicity of
infection; NS, not significant. |
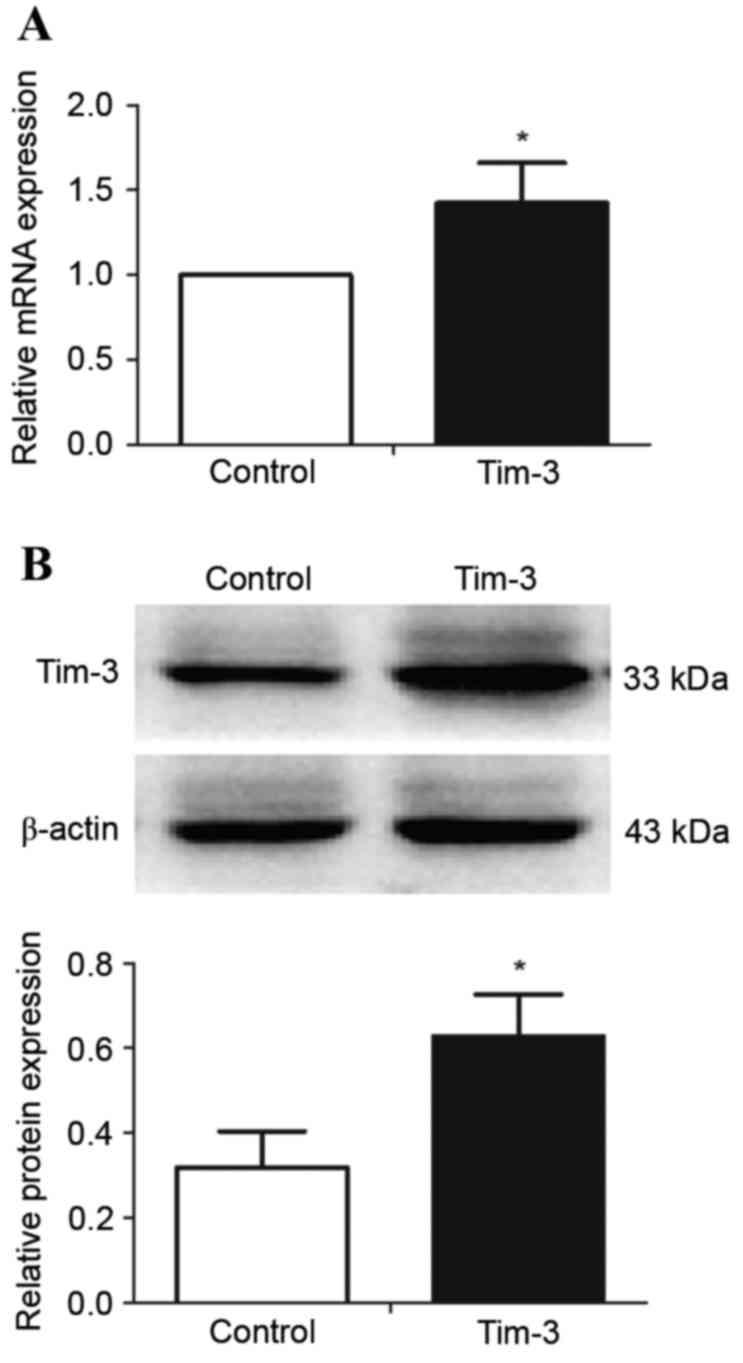
Construction of Tim-3-overexpressing
macrophages
RAW264.7 cells were either left untransfected
(control) or were transfected with the pLVX-IRES-ZsGreen1-Tim-3
expression plasmid, in order to establish Tim-3 overexpression in
macrophages. Tim-3 mRNA (Fig. 2A)
and protein (Fig. 2B) expression
levels were significantly higher in transfected RAW264.7 cells than
in the control cells (P<0.05; Fig.
2), suggesting that RAW264.7 cells were successfully
transfected.
Effect of H
pylori infection on Tim-3 expression and TLR4
pathway proteins in Tim-3-overexpressing macrophages. RAW264.7
cells were either left untransfected (control), or were transfected
with empty plasmid (mock) or Tim-3 overexpression plasmid (Tim-3),
then cultured for an additional 12 h either alone or with H.
pylori at an MOI of 100. In the control macrophages, Tim-3,
TLR4, MyD88 mRNA and protein levels, and p-NF-κBp65 protein levels,
were significantly higher in the H. pylori-infected cells
compared with the H. pylori-negative cells (P<0.05;
Fig. 3). In the Tim-3
overexpressing macrophages, TLR4 and MyD88 mRNA expression was
increased following infection (P<0.01; Fig. 3A), but no significant change was
observed in TLR4, MyD88 and p-NF-κBp65 protein levels between the
H. pylori-infected and uninfected cells (P>0.05; Fig. 3B). H. pylori infection
increased Tim-3 mRNA and protein expression in the
Tim-3-overexpressing cells compared with the untransfected cells
(P<0.05, Fig. 3). In addition,
infected by H. pylori, TLR4 and MyD88 mRNA expression levels
were significantly lower in the Tim-3-overexpressing cells compared
with the untransfected cells (P<0.01; Fig. 3A), but no significant change was
observed in TLR4, MyD88 and p-NF-κB p65 protein levels (P>0.05;
Fig. 3B).
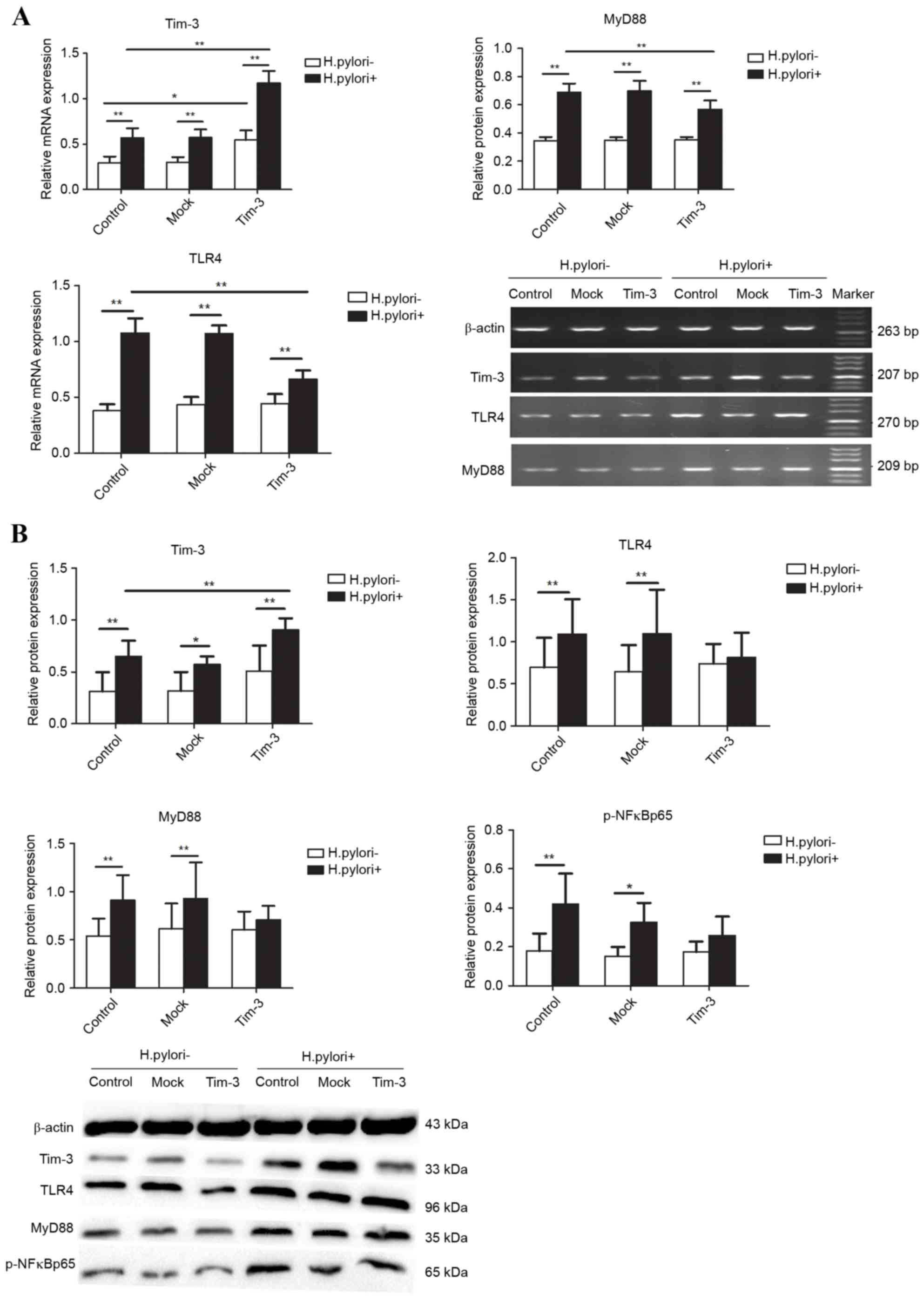 | Figure 3.Effect of Helicobacter pylori
infection on expression of Tim-3 and TLR4 signaling pathway
proteins in macrophages. RAW264.7 cells were either left
untransfected (control), or were transfected with empty plasmid
(Mock) or Tim-3 overexpression plasmid (Tim-3), and then they were
cultured for 12 h either alone (H. pylori-) or with H.
pylori at MOI 100 (H. pylori+). (A) mRNA expression was
measured for Tim-3, TLR4 and MyD88 by reverse
transcription-quantitative polymerase chain reaction and gel
electrophoresis. (B) Protein expression was measured for Tim-3,
TLR4, MyD88 and p-NFκBp65 mRNA by western blot analysis.
Densitometric quantification and representative images of the blots
are shown. Quantification for mRNA and protein was relative to
β-actin. n=3 independent experiments performed in duplicate.
*P<0.05 and **P<0.01, with comparisons indicated by lines.
Tim-3, T-cell immunoglobulin and mucin-domain-containing
molecule-3; TLR4, Toll-like receptor 4; MOI, multiplicity of
infection; MyD88, myeloid differentiation factor 88; p-NFκBp65,
phosphorylated nuclear factor κB p65 subunit. |
Effect of H
pylori infection on inflammatory cytokine secretion
in Tim-3-overexpressing macrophages. RAW264.7 cells were either
left untransfected (control), or were transfected with empty
plasmid (mock) or Tim-3 overexpression plasmid (Tim-3), then either
cultured alone or infected with H. pylori at MOI 100 for 12 h. The
culture supernatants were subsequently assayed by ELISA for levels
of the inflammatory cytokines TNF-α, IL-6, IFN-γ and IL-10. H.
pylori infection increased TNF-α, IL-6 and IFN-γ secretion in
the control and mock-transfected cells compared with the H.
pylori-negative cells (P<0.01; Fig. 4A-C), but no significant differences
were observed in the levels of these cytokines with or without
infection in the Tim-3-transfected cells (Fig. 4A-C). By contrast, H. pylori
infection significantly decreased IL-10 secretion in all cells
compared with the H. pylori-negative cells, independent of
whether they were control, or mock or Tim-3-transfected (P<0.01;
Fig. 4D). When comparing the H.
pylori-infected cells, TNF-α, IL-6, IFN-γ and IL-10
demonstrated significantly lower secretion in the Tim-3-transfected
cells compared with the control or mock-transfected cells
(P<0.01; Fig. 4).
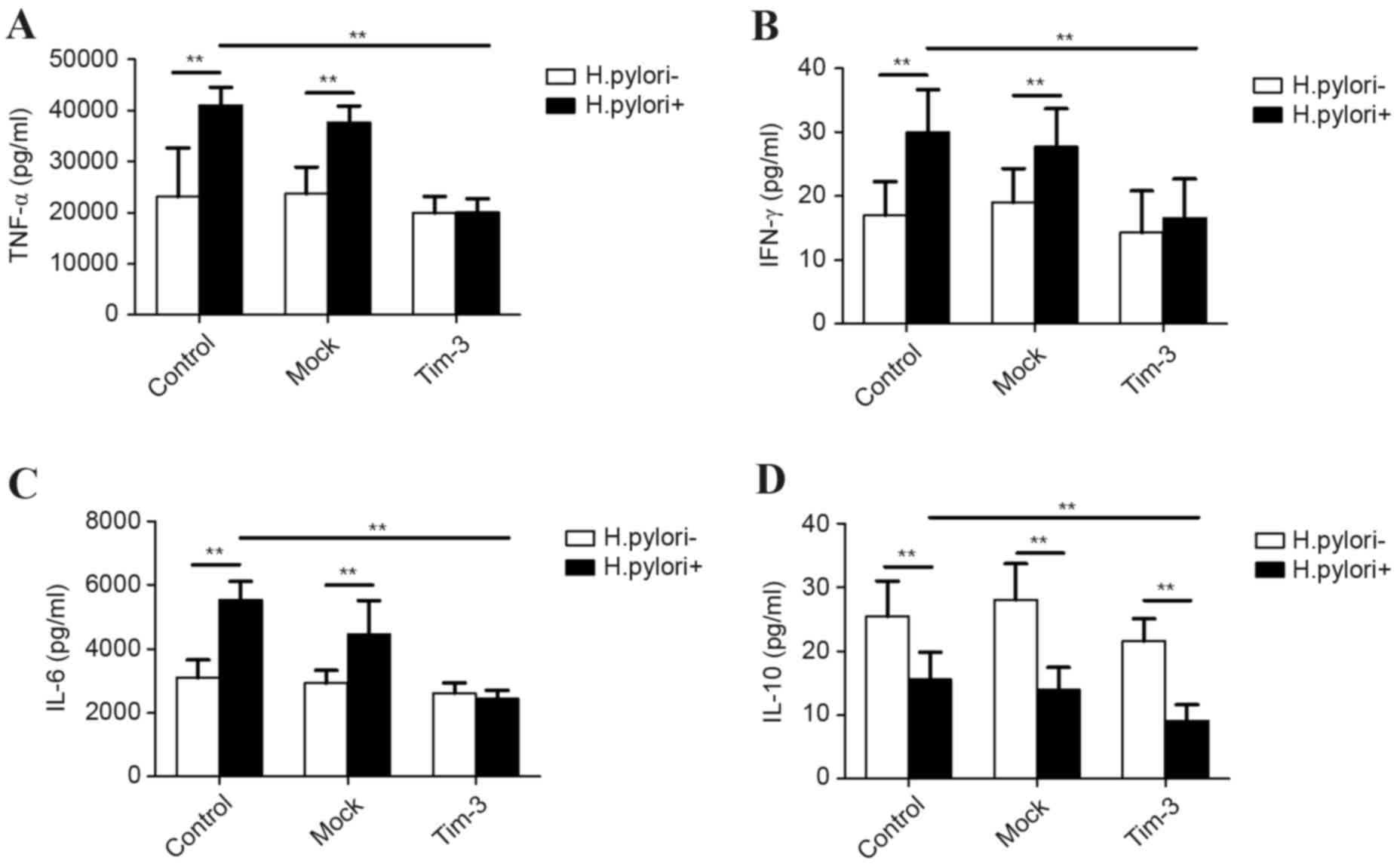 | Figure 4.Effect of Helicobacter pylori
infection on cytokine secretion in Tim-3-overexpressing
macrophages. RAW264.7 cells were either left untransfected
(control), or were transfected with empty plasmid (Mock) or Tim-3
overexpression plasmid (Tim-3), then cultured for 12 h either alone
(H. pylori-) or with H. pylori at MOI 100 (H.
pylori+). Levels of the inflammatory cytokines (A) TNF-α, (B)
IFN-γ, (C) IL-6 and (D) IL-10 were detected in the culture
supernatants by ELISA. n=3 independent experiments performed in
duplicate. *P<0.05 and **P<0.01, with comparisons indicated
by lines. Tim-3, T-cell immunoglobulin and mucin-domain-containing
molecule-3; MOI, multiplicity of infection; TNF-α, tumor necrosis
factor-α; IFN-γ, interferon-γ; IL-6, interleukin 6; IL-10,
interleukin 10. |
Discussion
H. pylori infection leads to the accumulation of a
large number of inflammatory cells in the gastric mucosa, including
macrophages (13,14). Previous studies have demonstrated
that TLR4 is a type of receptor that recognizes conserved microbial
products in macrophages (15,16).
Therefore, TLR4 is a signaling pathway that has important effects
on the inflammatory and immune responses to H. pylori
infections (17,18). It has been reported that H.
pylori lipopolysaccharide (LPS) activates NF-κB through the
TLR4 signaling pathway, which leads to the release of
pro-inflammatory cytokines, including IL-6, interleukin 1β (IL-1β)
and TNF-α, and to the promotion of inflammatory reactions (19). However, its regulation and the
exact mechanism by which it acts remain unclear. To determine the
effect of H. pylori infection on the TLR4 signaling pathway,
the expression of various proteins downstream of TLR4 was examined
in macrophages that were infected with different H. pylori
concentrations. In addition, the release of inflammatory cytokines
from the macrophages was also evaluated. The results revealed that
H. pylori infection at MOIs 25–100 significantly increased
TLR4 and MyD88 mRNA expression levels. When cells were infected
with H. pylori at MOI 100 for 12 h, the protein expression
levels of TLR4, MyD88 and p-NF-κBp65 were significantly
upregulated. H. pylori infection also significantly
increased the secretion of pro-inflammatory cytokines, including
IL-6, IFN-γ and TNF-α, but reduced the secretion of the
anti-inflammatory cytokine IL-10. The present results suggested
that H. pylori infection activated the TLR4-mediated and
MyD88-dependent signaling pathway, activated NF-κB, and promoted
inflammatory reactions. Pathak et al (20) reported that H. pylori 0175
proteins come directly in contact with the extracellular domain of
TLR4 and that they activate the MAPK and NF-κB signaling pathways,
thus leading to IL-6 secretion from macrophages. Maeda et al
(21) reported that H.
pylori activated the NF-κB signaling pathway and increased
TNF-α release from human monocytic THP-1 cells but failed to
activate the NF-κB signaling pathway in macrophages derived from
TLR4 mutant mice. Mandell et al (22) reported that H. pylori
infection increases the secretion of TNF-α and IL-6 from THP-1
cells in a concentration-dependent manner. However, macrophages
lacking TLR4 expression do not respond to H. pylori LPS
stimulation. In summary, TLR4 signaling pathway activation in
macrophages is important in the abnormal inflammatory reaction that
is caused by H. pylori infection, but the exact regulatory
mechanism remains unclear.
Tim-3 was the first, and is presently the only,
surface molecule shown to specifically identify Th1 cells in mice
and humans (23). Numerous studies
have demonstrated that Tim-3 is also expressed in innate immune
cells, including dendritic cells, macrophages and natural killer
cells, but the effect of Tim-3 on immune cells is different than on
other cells (5,23). Tim-3 has been demonstrated to be
important in innate immune cells, including macrophages/monocytes
(24). The present study revealed
that H. pylori infection upregulated the mRNA and protein
expression of Tim-3 in macrophages in a concentration-dependent
manner. In a previous study by this group, it was demonstrated that
following 12 h of H. pylori stimulation, there was a marked
increase in Tim-3 production by mouse spleen lymphocytes (25). Tim-3 expression was also confirmed
in vivo to be higher in the gastric mucosa of H.
pylori-infected mice than of uninfected mice (25). However, the function of Tim-3 in
H. pylori infection remains unclear.
TLR4 and Tim-3 are commonly expressed in various
immune cells, and their interaction is closely related to the
development of many diseases (10,11).
However, no report to date has examined the effect of Tim-3
overexpression on the activation of the TLR4 signaling pathway and
the H. pylori-induced inflammatory reactions in macrophages.
The present study revealed that mRNA expression of TLR4 and MyD88,
and secretion of IL-6, IFN-γ and TNF-α, in Tim-3-overexpressing
macrophages infected with H. pylori were significantly lower
than control macrophages. These results suggest that Tim-3
inhibited the activation of macrophages by negatively regulating
the TLR4 signaling pathway. A previous study from this group
(26) demonstrated that blocking
Tim-3 with an inhibitory antibody upregulated TLR4 and MyD88
expression, promoted NF-κB activation, decreased the number of
CD4+CD25+Foxp3+ Treg cells, and
upregulated Th1 immune responses, resulting in intensified
inflammation of the gastric mucosa of H. pylori-infected
mice. Frisancho et al (11)
reported that, in a mouse model of inflammatory heart disease,
blocking Tim-3 significantly increases TLR4 expression and cardiac
inflammation, indicating that Tim-3 is a negative regulator of the
TLR4 signaling pathway. The present study also demonstrated that
following H. pylori infection, Tim-3 overexpression in
macrophages inhibited the TLR4 signaling pathway and reduced
pro-inflammatory cytokine secretion. However, these results were
not statistically significant in the absence of H. pylori
infection, indicating that Tim-3 strongly and negatively regulated
TLR4 signaling only in the presence of pathology, and not under
normal physiological conditions. The mechanism by which Tim-3
negatively regulated the TLR4 signaling pathway remains unclear.
Yang et al (9) reported
that Tim-3 overexpression in macrophages significantly suppresses
TLR-mediated pro-inflammatory cytokine production. Li et al
(27) reported that the H.
pylori cytotoxin-associated gene A activates the
phosphatidylinositol 3-kinase (PI3K)/AKT serine/threonine kinase 1
(Akt1) pathway, which inhibits interleukin 8 release in H.
pylori-infected gastric carcinoma AGS cells. However, further
studies are needed to determine how H. pylori infection
induces Tim-3 overexpression and downregulates TLR4-mediated
inflammatory cytokine production through the PI3K-Akt pathway. The
present study also demonstrated that following H. pylori
infection, Tim-3 overexpression in macrophages not only inhibited
the release of IL-6, IFN-γ and TNF-α, but also suppressed the
release of IL-10. Zhang et al (24) reported that blocking or silencing
Tim-3 in macrophages not only increased the release of IL-12 and
IL-6, but also increased the release of IL-10. Therefore, the role
of Tim-3 in regulating inflammatory and immune responses may be
influenced not only by Tim-3 expression levels but also by the
state of the macrophages and the molecular balance between
inhibition and stimulation. Anderson et al (28) reported that Tim-3, by virtue of its
differential expression on cells of the innate and adaptive immune
systems, can both promote inflammation and terminate Th1 immunity.
Therefore, Tim-3 serves opposing roles, influenced by multiple
factors, including variations in stimuli, cell type and immune
activation state. Further research will focus on the mechanism by
which Tim-3 downregulates the TLR4 signaling pathway during H.
pylori infection.
In conclusion, H. pylori infection in
RAW264.7 macrophages activated the TLR4 signaling pathway,
upregulated Tim-3 expression and increased secretion of
pro-inflammatory cytokines (TNF-α, IL-6 and IFN-γ), while
decreasing secretion of an anti-inflammatory cytokine (IL-10).
Under conditions of H. pylori infection, however, Tim-3
overexpression inhibited the TLR4 pathway activation and the
secretion of pro-inflammatory cytokines. Taken together, the
present study indicated that Tim-3 may represent a novel
therapeutic target for treating H. pylori infection-related
diseases.
Acknowledgements
The present study was supported by the Major
Research Program Foundation of China (Prevention and control of
major chronic non-communicable diseases, grant no. 2016YFC1302201)
and the National Natural Science Foundation of China (grant no.
81260076), and was edited for proper English language, grammar,
punctuation, spelling, and overall style by American Journal
Experts.
References
|
1
|
Koch M, Meyer TF and Moss SF:
Inflammation, immunity, vaccines for Helicobacter pylori infection.
Helicobacter. 18:(Suppl 1). S18–S23. 2013. View Article : Google Scholar
|
|
2
|
Demiray E and Bekmen N: Helicobacter
pylori infection and phagocytosis. Mikrobiyol Bul. 42:177–184.
2008.(In Turkish). PubMed/NCBI
|
|
3
|
Duque Arango G and Descoteaux A:
Macrophage cytokines: Involvement in immunity and infectious
diseases. Front Immunol. 5:4912014.PubMed/NCBI
|
|
4
|
Rad R, Ballhorn W, Voland P, Eisenächer K,
Mages J, Rad L, Ferstl R, Lang R, Wagner H, Schmid RM, et al:
Extracellular and intracellular pattern recognition receptors
cooperate in the recognition of Helicobacter pylori.
Gastroenterology. 136:2247–2257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Freeman GJ, Casasnovas JM, Umetsu DT and
DeKruyff RH: TIM genes: A family of cell surface phosphatidylserine
receptors that regulate innate and adaptive immunity. Immunol Rev.
235:172–189. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mariat C, Sánchez-Fueyo A, Alexopoulos SP,
Kenny J, Strom TB and Zheng XX: Regulation of T cell dependent
immune responses by TIM family members. Philos Trans R Soc Lond B
Biol Sci. 360:1681–1685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Monney L, Sabatos CA, Gaglia JL, Ryu A,
Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel
RA, et al: Th1-specific cell surface protein Tim-3 regulates
macrophage activation and severity of an autoimmune disease.
Nature. 415:536–541. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Khademi M, Illés Z, Gielen AW, Marta M,
Takazawa N, Baecher-Allan C, Brundin L, Hannerz J, Martin C, Harris
RA, et al: T Cell Ig- and mucin-domain-containing molecule-3
(TIM-3) and TIM-1 molecules are differentially expressed on human
Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear
cells in multiple sclerosis. J Immunol. 172:7169–7176. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang X, Jiang X, Chen G, Xiao Y, Geng S,
Kang C, Zhou T, Li Y, Guo X, Xiao H, et al: T cell Ig mucin-3
promotes homeostasis of sepsis by negatively regulating the TLR
response. J Immunol. 190:2068–2079. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Uchida Y, Ke B, Freitas MC, Yagita H,
Akiba H, Busuttil RW, Najafian N and Kupiec-Weglinski JW: T-cell
immunoglobulin mucin-3 determines severity of liver
ischemia/reperfusion injury in mice in a TLR4-dependent manner.
Gastroenterology. 139:2195–2206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Frisancho-Kiss S, Davis SE, Nyland JF,
Frisancho JA, Cihakova D, Barrett MA, Rose NR and Fairweather D:
Cutting edge: Cross-regulation by TLR4 and T cell Ig mucin-3
determines sex differences in inflammatory heart disease. J
Immunol. 178:6710–6714. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schumacher MA, Donnelly JM, Engevik AC,
Xiao C, Yang L, Kenny S, Varro A, Hollande F, Samuelson LC and
Zavros Y: Gastric sonic hedgehog acts as a macrophage
chemoattractant during the immune response to Helicobacter pylori.
Gastroenterology. 142:1150–1159.e6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deen NS, Gong L, Naderer T, Devenish RJ
and Kwok T: Analysis of the relative contribution of phagocytosis,
LC3-associated phagocytosis, and canonical autophagy during
helicobacter pylori infection of macrophages. Helicobacter.
20:449–459. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Lu Y, Ma L, Cao X, Xiao J, Chen
J, Jiao S, Gao Y, Liu C, Duan Z, et al: Activation of vascular
endothelial growth factor receptor-3 in macrophages restrains
TLR4-NF-κB signaling and protects against endotoxin shock.
Immunity. 40:501–514. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Byun EB, Sung NY, Byun EH, Song DS, Kim
JK, Park JH, Song BS, Park SH, Lee JW, Byun MW and Kim JH: The
procyanidin trimer C1 inhibits LPS-induced MAPK and NF-κB signaling
through TLR4 in macrophages. Int Immunopharmacol. 15:450–456. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Käbisch R, Mejias-Luque R, Gerhard M and
Prinz C: Involvement of toll-like receptors on Helicobacter
pylori-induced immunity. PLoS One. 9:e1048042014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Su YL, Yang JC, Lee H, Sheu F, Hsu CH, Lin
SL and Chow LP: The C-terminal disulfide bonds of Helicobacter
pylori GroES are critical for IL-8 secretion via the TLR4-dependent
pathway in gastric epithelial cells. J Immunol. 194:3997–4007.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fujihara M, Muroi M, Tanamoto K, Suzuki T,
Azuma H and Ikeda H: Molecular mechanisms of macrophage activation
and deactivation by lipopolysaccharide: Roles of the receptor
complex. Pharmacol Ther. 100:171–194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pathak SK, Basu S, Bhattacharyya A, Pathak
S, Banerjee A, Basu J and Kundu M: TLR4-dependent NF-kappaB
activation and mitogen- and stress-activated protein kinase
1-triggered phosphorylation events are central to Helicobacter
pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated
induction of IL-6 release from macrophages. J Immunol.
177:7950–7958. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maeda S, Akanuma M, Mitsuno Y, Hirata Y,
Ogura K, Yoshida H, Shiratori Y and Omata M: Distinct mechanism of
Helicobacter pylori-mediated NF-kappa B activation between gastric
cancer cells and monocytic cells. J Biol Chem. 276:44856–44864.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mandell L, Moran AP, Cocchiarella A,
Houghton J, Taylor N, Fox JG, Wang TC and Kurt-Jones EA: Intact
gram-negative Helicobacter pylori, Helicobacter felis, and
Helicobacter hepaticus bacteria activate innate immunity via
toll-like receptor 2 but not toll-like receptor 4. Infect Immun.
72:6446–6454. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anderson DE: TIM-3 as a therapeutic target
in human inflammatory diseases. Expert Opin Ther Targets.
11:1005–1009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang Y, Ma CJ, Wang JM, Ji XJ, Wu XY,
Moorman JP and Yao ZQ: Tim-3 regulates pro- and anti-inflammatory
cytokine expression in human CD14+ monocytes. J Leukoc Biol.
91:189–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu S, Xie Y, Zhou N, Jin L, Tan Y, Liu D,
Gong Y, Liu L, Liu J, Liu W, et al: Expression of T-cell
immunoglobulin- and mucin-domain-containing molecules-1 and −3
(Tim-1 and Tim-3) in Helicobacter pylori infection. Helicobacter.
16:373–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gong Y, Tao L, Wang F, Liu W, Jing L, Liu
D, Hu S, Xie Y and Zhou N: Chitosan as an adjuvant for a
Helicobacter pylori therapeutic vaccine. Mol Med Rep. 12:4123–4132.
2015.v. PubMed/NCBI
|
|
27
|
Li SP, Chen XJ, Sun AH, Zhao JF and Yan J:
CagA(+) H. pylori induces Akt1 phosphorylation and inhibits
transcription of p21(WAF1/CIP1) and p27(KIP1) via PI3K/Akt1
pathway. Biomed Environ Sci. 23:273–278. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anderson AC, Anderson DE, Bregoli L,
Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW,
Hirashima M, et al: Promotion of tissue inflammation by the immune
receptor Tim-3 expressed on innate immune cells. Science.
318:1141–1143. 2007. View Article : Google Scholar : PubMed/NCBI
|