Introduction
Loss of elastic recoil, perpetual destruction of
alveolar structure and airspace enlargement are the pathological
characteristics of patients with chronic obstructive pulmonary
disease (COPD) (1). Senescence and
apoptosis of alveolar epithelial cells results in the destruction
of the alveolar structure, which may contribute to the pathogenesis
of COPD (2–4). However, there is no effective
treatment to prevent the destruction of the alveolar structure in
patients with COPD. As progenitors of type I alveolar epithelial
cells (AECI) in mammals, type II alveolar epithelial cells (AECII)
are involved in synthesizing and secreting pulmonary surfactant
proteins, maintaining alveolar homeostasis, reducing surface
tension of the alveoli, improving lung tissue repair and gas
exchange (5,6). Although stem cells are involved in
lung repair (7,8), there still is no effective method to
prevent AECII senescence and damage.
As a nicotinamide adenine dinucleotide
(NAD)+-dependent class III histone deacetylase, sirtuin
1 (SIRT1) is an important member of the sirtuin protein family
(9,10). SIRT1 deacetylate H1, H3 and H4
histones, forkhead box O 3a (FoxO3a), p53 and nuclear factor-κB,
which are involved in the regulation of signaling pathways of
stress resistance, inflammation, cellular senescence, apoptosis and
proliferation (11–13). FoxO3a is an important member of the
forkhead box O subfamily and is a non-histone substrate of SIRT1.
p53, another non-histone substrate of SIRT1, activates the
transcription of p21 and inhibit the activity of cyclin-dependent
kinases, which resulted in accelerating cellular senescence
(14).
Long non-coding RNAs (lncRNAs) are
non-protein-coding RNAs that are longer than 200 nucleotides
(15), which are involved in
regulating gene transcription and thus processes including
embryonic development, cell differentiation and cellular senescence
(16,17). Previous studies have demonstrated
that lncRNAs directly regulate cell functions, and are associated
with multiple diseases (18).
Abdelmohsen et al (19)
indicated that the expression levels of senescence-associated
lncRNA1 (SAL-RNA1) and SIRT1 were downregulated, but SAL-RNA2,
SAL-RNA3, p53 and p21 were upregulated in senescent fetal
lung-derived WI-38 human diploid fibroblasts (WI-38 HDFs).
Furthermore, inhibiting SAL-RNA1 levels enhances the phenotypic
characteristics of senescent WI-38 HDFs, which increases
senescence-associated β-galactosidase (SA-β-gal) activity and p53
expression.
However, it remains unknown whether SAL-RNA-mediated
SIRT1/p53 and the FoxO3a signaling pathways are involved in
regulating AECII senescence in the pathogenesis of COPD. The
present study investigated whether AECII senescence in lung tissues
promoted the pathogenesis of COPD.
Materials and methods
Patients and specimens
A total of 34 patients who underwent lung surgery at
Zhejiang Provincial People's Hospital (Hangzhou, China) from
January 2014 to April 2015 were enrolled in the present study.
These patients included 28 men and 6 women, ranging from 41–81
years old, with a median age of 66.5 years. None of the patients
received preoperative chemotherapy, radiotherapy or intravenous
corticosteroid therapy, and these patients did not exhibit failure
of the heart, lung, brain or kidney, or any hematological diseases.
Patients diagnosed with respiratory diseases other than COPD,
including idiopathic pulmonary fibrosis and asthma, were excluded.
According to the global strategy for the diagnosis of chronic
obstructive pulmonary disease (1),
these patients were sorted into 2 groups as follows: A control
group (22 patients) and a COPD group (12 patients, exposed to
cigarette smoke for >20 years). Lung function of each patient
was examined by spirometry, which was performed by a trained
technician according to the American Thoracic Society/European
Respiratory Society guidelines (20). The lung tissues, taken from a
distance of 5 cm from the negative margin of cancer tissues, were
collected immediately following surgical resection and frozen
instantly in liquid nitrogen or immersed in paraformaldehyde
solution. The present study was approved by the Ethics Committee of
Zhejiang Provincial People's Hospital (Hangzhou, China). Informed
consent was obtained from all patients.
Histological examinations
Lungs were immersed in paraformaldehyde for 24 h,
then embedded in paraffin and cut into 4 µm thick sections. A total
of 3 discontinuous paraffin-embedded sections from each lung tissue
sample were stained with hematoxylin and eosin (H&E) in order
to assess the morphological changes in the lungs. A total of 5
fields of view from each of the 3 sections from each lung sample
were assessed using a light microscope (Olympus Corporation, Tokyo,
Japan). The mean linear intercept (MLI), the mean alveolar airspace
(MAA) and the number of alveolar counted per unit area (MAN) were
obtained. MLI indicates the average distance between opposing walls
of a single alveolus and is a measure of pulmonary airspace
enlargement. MAA is also a measure of pulmonary airspace
enlargement, while MAN is a measure of pulmonary airspace
density.
Senescence-associated-β-galactosidase
(SA-β-gal) activity assay
SA-β-gal activity was assessed using an in
situ β-galactosidase staining kit (Beyotime Institute of
Biotechnology, Shanghai, China) according to the manufacturer's
protocol. Lung tissues were fixed in β-galactosidase stationary
solution for 15 min, then washed 3 times for 10 min each in PBS.
Sections were then incubated with 1 ml staining solution mixture
(10 µl staining solution A, 10 µl staining solution B, 930 µl
staining solution C and 50 µl X-gal solution) for 2 h at 37°C.
Following 3 washes with PBS, 5 fields of view from each of the 3
sections from each lung sample were examined using a light
microscope (Olympus Corporation).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from each lung sample using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) according to the manufacturer's protocol. The
final RNA purity and concentrations were determined using a
spectrophotometer. cDNA was synthesized from the total RNA using
the PrimeScript™ RT reagent kit with gDNA eraser (Takara Bio, Inc.,
Otsu, Japan) according to the manufacturer's protocol. qPCR
analysis was performed using SYBR-Green qPCR kit (Takara Bio, Inc.)
and the Agilent MX3000P qPCR system (Agilent Technologies, Inc.,
Santa Clara, CA, USA). The thermocycling conditions were as
follows: Initial denaturation at 95°C for 15 min, followed by 45
cycles of denaturation at 95°C for 15 sec, annealing at 58°C for 15
sec, and elongation at 72°C for 1 min. The following primers were
used (Invitrogen; Thermo Fisher Scientific, Inc.): SAL-RNA1,
forward 5′-AGGCTGCCATCTCACCCTCATAC-3′ and reverse
5′-TCCTTACCTCTTCCCTTCCACCC-3′; SAL-RNA2, forward
5′-GGATGCTGTGAGCTTTGTGA-3′ and reverse 5′-GAAACCCCCAGAGCTGAGAC-3′;
SAL-RNA3, forward 5′-ACTGCTGGGATAACGGTGAC-3′ and reverse
5′-TCTGTGCTCAGCTCTGCAAT-3′; SIRT1, forward
5′-GCAGATTAGTAGGCGGCTTG-3′ and reverse 5′-ACTTTCATCCTCCATGGGTTC-3′;
p53, forward 5′-ACCACCATCCACTACAACTACAT-3′ and reverse
5′-CAGGACAGGCACAAACACG-3′; FOXO3a, forward
5′-TGGCAAGCACAGAGTTGGATGA-3′ and reverse
5′-TGGCGGGAGCGTGATGTTAT-3′; p21, forward
5′-ACTTTGATTAGCAGCGGAACA-3′ and reverse
5′-GAAAACAGTCCAGGCCAGTATG-3′; and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH), forward 5′-TGAAGGTCGGAGTCAACGG-3′ and
reverse 5′-CTGGAAGATGGTGATGGGATT-3′. All analyses were performed in
triplicate. GADPH was used as a reference gene. The results were
quantified using the 2−ΔΔCq method (21).
Western blotting
Proteins were extracted from each lung sample by
homogenizing the samples in ice-cold lysis buffer [50 mmol/l Tris
(pH 7.4), 150 mmol/l NaCl, 0.1% sodium dodecyl sulfate, 1 mmol/l
EDTA, 1% sodium deoxycholate, and 1% Triton X-100; Invitrogen;
Thermo Fisher Scientific, Inc.] containing a protease inhibitor
cocktail (1 mmol/l phenylmethylsulfonyl fluoride, 1 mg/l leupeptin
and 1 mg/l aprotinin; Beyotime Institute of Biotechnology). The
protein concentration in the samples was determined using a micro
bicinchoninic acid protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. Next,
equal amounts of protein (20 µg) from each sample were heated at
100°C for 5 min and then separated by electrophoresis on 8% sodium
dodecyl sulfate-polyacrylamide gels. The separated proteins were
electrotransferred to nitrocellulose membranes and blocked with
Tris-buffered saline containing 0.1% Tween-20 (TBS-T) and 5% bovine
serum albumin (BSA; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
for 2 h at room temperature. The membranes were then incubated with
primary antibodies against p53 (#9282; 1:1,000 in TBS-T; Cell
Signaling Technology, Inc., Danvers, MA, USA), p21 (sc-397; 1:500
in TBS-T; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), FoxO3a
(#12829; 1:1,000 in TBS-T; Cell Signaling Technology, Inc.) SIRT1
(#2493; 1:1,000 in TBS-T; Cell Signaling Technology) and β-actin
(ab8227; 1:1,000; Abcam, Cambridge, UK) overnight at 4°C on an
orbital shaker. Following washing 3 times for 10 min each in TBS-T,
the membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit immunoglobulin G (#35552; 1:5,000 in TBS-T;
Pierce; Thermo Fisher Scientific, Inc.) for 1 h at room temperature
on an orbital shaker. Following 5 washes with TBS-T, immunoreactive
bands on the membrane were detected using an enhanced
chemiluminescence solution (GE Healthcare Life Sciences, Chalfont,
UK), visualized by means of X-ray-film exposure, and analyzed using
an UVP-GDS8000 gel-analysis system (Ultra-Violet Products, Ltd.,
Cambridge, UK). Protein expression levels were analyzed by
densitometry and the values were normalized relative to those
measured for β-actin, which was used as the loading control.
SIRT1 deacetylase activity assay
SIRT1 deacetylase activity was measured using the
SIRT1 fluorimetric activity assay kit (Enzo Life Sciences, Inc.,
Farmingdale, NY, USA) according to the manufacturer's protocol.
SIRT1 was immunoprecipitated as described previously (22). Following washing with PBS, Fluor de
Lys substrate and NAD+ were added to the
SIRT1-conjugated beads and incubated for 90 min at 37°C. Then, the
substrate-SIRT1 mixture was placed on a white 96-well plate, and
the Fluor de Lys developer II reagent was added to the 96 wells for
30 min at 37°C. Finally, the plate was analyzed by a microplate
reading fluorimeter (M200; Tecan Group, Ltd., Männedorf,
Switzerland) at 405 nm.
Statistical analyses
Statistical analyses were performed using SPSS 17.0
software (SPSS Inc., Chicago, IL, USA). Data were expressed as the
mean ± standard deviation. The significance of difference between
groups was determined by means of independent-sample Student's
t-tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
Lung function examination
The lung function of each patient was examined by
spirometry. There were no significant differences between patients
from the control group and the COPD group in forced vital capacity
(FVC; P>0.05; Table I).
However, the ratio of forced expiratory ventilation in 1 sec to FVC
(FEV1/FVC) and %FEV1 in patients with COPD
was significantly reduced compared with the control group
(P<0.001; Table I).
 | Table I.Lung function examination in patients
with COPD. |
Table I.
Lung function examination in patients
with COPD.
| Variable | Control | COPD | P-value |
|---|
| FVC (L) | 2.47±0.59 | 2.44±0.63 | 0.789 |
| FEV1/FVC
(%) | 82.39±5.59 | 64.13±6.83 | <0.001 |
| FEV1, %
predicted | 92.15±10.11 | 63.28±14.09 | <0.001 |
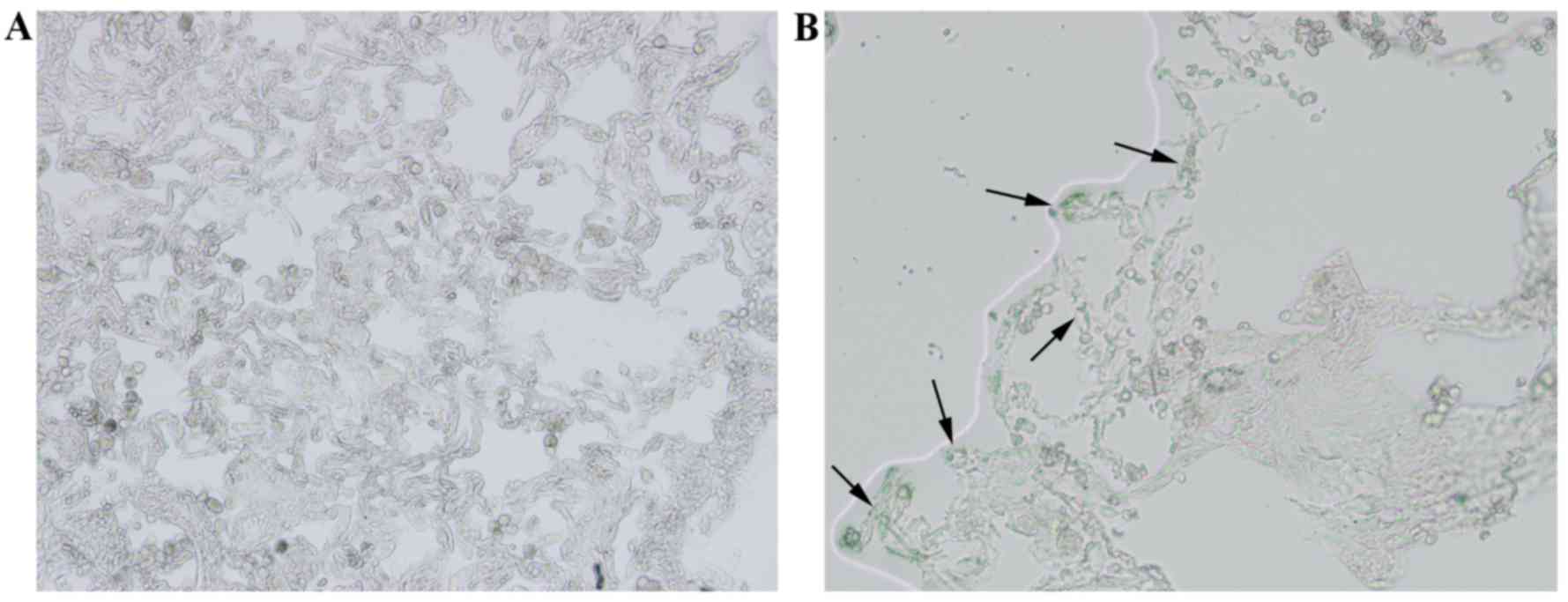
Lung histological characteristics
The histological characteristics of lung tissues
from the control group and the COPD group were analyzed by H&E
staining. Compared with the control group (Fig. 1A), the airspace was visibly
enlarged and the number of alveoli was decreased in lung tissues of
the COPD group (Fig. 1B). The lung
samples from the COPD group also exhibited merged alveoli and the
formation of bullae, which was in accord with the pathological
characteristics of COPD (Fig. 1B).
Quantitative analyses of lung histomorphology revealed that the MLI
and MAA of the COPD group was significantly higher compared with
the control group (P<0.05; Table
II) and the MAN was significantly decreased in the COPD group
compared with the control group (P<0.05; Table II).
| Table II.Quantitative analyses of lung
histomorphology in patients with COPD. |
Table II.
Quantitative analyses of lung
histomorphology in patients with COPD.
| Group | MLI (µM) | MAA
(µm2) | MAN
(mm2) |
|---|
| Control | 35.26±2.47 | 3166.57±127.32 | 264.76±25.81 |
| COPD |
189.16±15.22a |
20681.16±768.86a |
50.44±4.85a |
SA-β-gal activity
SA-β-gal activity was assayed in lung samples to
analyze whether cellular senescence was exhibited in lung tissues
of the patients with COPD. The cells in lung tissues with blue
color were considered SA-β-gal positive, and so senescent. Compared
with the control group (Fig. 2A),
SA-β-gal activity was visibly increased in the COPD group (Fig. 2B) Most SA-β-gal-positive cells were
located at the edges of alveoli (Fig.
2B).
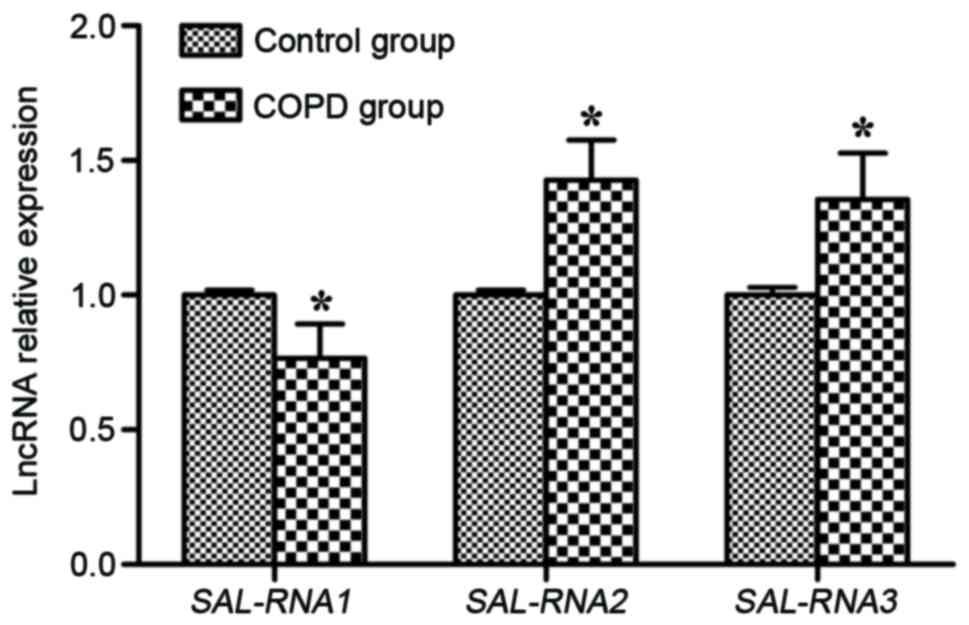
LncRNA expression of SAL-RNAs
The lncRNA expression of SAL-RNA1, SAL-RNA2 and
SAL-RNA3 was measured by RT-qPCR. SAL-RNA2 and SAL-RNA3 mRNA
expression levels were significantly increased in the lung tissues
of the COPD group compared with the control group (P<0.05;
Fig. 3). However, the SAL-RNA1
level in the COPD group was significantly decreased compared with
the control group (P<0.05; Fig.
3).
mRNA expression of SIRT1, FoxO3a, p53
and p21
p53 and p21 mRNA expression levels were
significantly increased in lung tissues in the patients with COPD
compared with the control group (P<0.05; Fig. 4). By contrast, SIRT1 and FoxO3a
mRNA expression levels in the COPD group were significantly
decreased compared with the control group (P<0.05; Fig. 4).
Protein expression and SIRT1
deacetylase activity
SIRT1, FoxO3a, p53 and p21 protein expression levels
were visualized by western blotting. SIRT1 and FoxO3a protein
levels were visibly reduced in the COPD group compared with the
control group, while p53 and p21 protein levels were visibly
upregulated in the COPD group compared with the control group
(Fig. 5A). Accompanying the
downregulation in SIRT1 protein levels, there was a significant
reduction of SIRT1 activity in the COPD group compared with the
control group (P<0.05; Fig.
5B).
Discussion
COPD is the fourth leading cause of death globally
(1). Cigarette smoking is a major
risk factor in the development of COPD. AECII are involved in the
maintenance and repair of lung tissue and alveolar homeostasis
(5,6). Cellular senescence and apoptosis of
alveolar epithelial cells in lung tissues are important
characteristics of COPD pathogenesis (2,23,24).
SA-β-gal is considered an important biomarker for cellular
senescence (25). In the present
study, the airspace was visibly enlarged and the number of alveoli
decreased in the lung tissues of patients with COPD. The results
also revealed that the MLI, MAA and SA-β-gal activity were
significantly increased and MAN was significantly decreased in the
COPD group compared to the control group. Cigarette smoke exposure
induces cellular senescence and apoptosis of alveolar cells, and
results in air space enlargement (23,26).
Previous studies have demonstrated that the
expression of SIRT1 is significantly decreased in the lung tissues
of COPD patients as well as in lung samples of rats with emphysema
(27–29). SIRT1 is associated with cellular
senescence in the development of emphysema, and the activation of
SIRT1 may be a target for COPD/emphysema treatment (30). Activation of SIRT1 increases SIRT1
expression, which is associated with the upregulation of FoxO3,
downregulation of p53 and inhibition of the AECII apoptosis
(29). Previous studies have
demonstrated that SIRT1 is an antiaging protein, and is reduced in
the lung tissues of patients with COPD (27,28).
Cellular senescence mediated by SIRT1 participates in the
progression of COPD, but the positive effects of SIRT1 on AECII
senescence in patients with COPD remain elusive.
p53 is a tumor suppressor protein, that possesses
the ability to induce cellular senescence by activating p21
expression. SIRT1 possesses the biochemical potential to
deacetylate lysine residues of p53 or regulate the gene promoter of
p53, which inhibits p53 activity and reduces cellular senescence
(31). FoxO3a is an important
member of the forkhead box O subfamily, which is a non-histone
substrate of SIRT1. Protein complexes with the combination of SIRT1
and FoxO3a may regulate apoptosis and cellular senescence (32). SIRT1/FoxO3a and SIRT1/p53 pathways
may regulate the cell cycle inhibitors (p16 and p21) to accelerate
cellular senescence (33).
LncRNAs are associated with multiple diseases,
including Alzheimer's disease (34), coronary artery disease (35), prostate cancer (36) and lung cancer (37), but few lncRNA are associated with
COPD. A previous study (19)
revealed that high levels of SAL-RNA1 are expressed in
early-passage WI-38 HDFs, but the expression levels of SAL-RNA2 and
SAL-RNA3 are low. Following 52 population doublings SAL-RNA1 and
SIRT1 expression levels are downregulated, but SAL-RNA2, SAL-RNA3,
p53 and p21 expression levels are upregulated in the senescent
WI-38 HDFs. Furthermore, inhibiting SAL-RNA1 levels may enhance the
phenotypic characteristics of senescent WI-38 HDFs, which may
increase SA-β-gal activity and p53 expression. In the present
study, SAL-RNA1, SIRT1 and FoxO3a expression was downregulated, but
SAL-RNA2, SAL-RNA3, p53 and p21 expression was upregulated in lung
tissues of the COPD group compared with the control group. Thus,
the results of the present study indicated that the SIRT1/FoxO3a
and SIRT1/p53 pathways were mediated by SAL-RNA1, SAL-RNA2 and
SAL-RNA3, which may regulate AECII senescence in the pathogenesis
of COPD, and may provide novel targets for COPD therapy.
Acknowledgements
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81470241,
81000016 and 81470109) and the Foundation of Science and Technology
Department of Zhejiang Province (grant no. 2014C37022).
References
|
1
|
Global Initiative for Chronic Obstructive
Lung Disease: Global strategy for the diagnosis, management and
prevention of chronic obstructive pulmonary disease. Seattle: GOLD;
http://www.goldcopd.com/2015
|
|
2
|
Tsuji T, Aoshiba K and Nagai A: Alveolar
cell senescence exacerbates pulmonary inflammation in patients with
chronic obstructive pulmonary disease. Respiration. 80:59–70. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siganaki M, Koutsopoulos AV, Neofytou E,
Vlachaki E, Psarrou M, Soulitzis N, Pentilas N, Schiza S, Siafakas
NM and Tzortzaki EG: Deregulation of apoptosis mediators' p53 and
bcl2 in lung tissue of COPD patients. Respir Res. 11:462010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao CZ, Fang XC, Wang D, Tang FD and Wang
XD: Involvement of type II pneumocytes in the pathogenesis of
chronic obstructive pulmonary disease. Respir Med. 104:1391–1395.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whitsett JA, Wert SE and Weaver TE:
Alveolar surfactant homeostasis and the pathogenesis of pulmonary
disease. Annu Rev Med. 61:105–119. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hoffman AM and Ingenito EP: Alveolar
epithelial stem and progenitor cells: Emerging evidence for their
role in lung regeneration. Curr Med Chem. 19:6003–6008. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Y, Xu W, Yan J, Xia Y, Gu C, Ma Y and
Tao H: Differentiation of human amniotic fluid-derived mesenchymal
stem cells into type II alveolar epithelial cells in vitro. Int J
Mol Med. 33:1507–1513. 2014.PubMed/NCBI
|
|
8
|
Li Y, Gu C, Xu W, Yan J, Xia Y, Ma Y, Chen
C, He X and Tao H: Therapeutic effects of amniotic fluid-derived
mesenchymal stromal cells on lung injury in rats with emphysema.
Respir Res. 15:1202014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Milne JC and Denu JM: The sirtuin family:
Therapeutic targets to treat diseases of aging. Curr Opin Chem
Biol. 12:11–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bordo D: Structure and evolution of human
sirtuins. Curr Drug Targets. 14:662–665. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lavu S, Boss O, Elliott PJ and Lambert PD:
Sirtuins--novel therapeutic targets to treat age-associated
diseases. Nat Rev Drug Discov. 7:841–853. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Finkel T, Deng CX and Mostoslavsky R:
Recent progress in the biology and physiology of sirtuins. Nature.
460:587–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Satoh A, Stein L and Imai S: The role of
mammalian sirtuins in the regulation of metabolism, aging and
longevity. Handb Exp Pharmacol. 206:125–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guterres FA, Martinez GR, Rocha ME and
Winnischofer SM: Simvastatin rises reactive oxygen species levels
and induces senescence in human melanoma cells by activation of
p53/p21 pathway. Exp Cell Res. 319:2977–2988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kung JT, Colognori D and Lee JT: Long
noncoding RNAs: Past, present and future. Genetics. 193:651–669.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Batista PJ and Chang HY: Long noncoding
RNAs: Cellular address codes in development and disease. Cell.
152:1298–1307. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ohsawa R, Seol JH and Tyler JK: At the
intersection of non-coding transcription, DNA repair, chromatin
structure and cellular senescence. Front Genet. 4:1362013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Abdelmohsen K, Panda A, Kang MJ, Xu J,
Selimyan R, Yoon JH, Martindale JL, De S, Wood WH III, Becker KG
and Gorospe M: Senescence-associated lncRNAs: Senescence-associated
long noncoding RNAs. Aging Cell. 12:890–900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miller MR, Hankinson J, Brusasco V, Burgos
F, Casaburi R, Coates A, Crapo R, Enright P, van der Grinten CP,
Gustafsson P, et al: Standardisation of spirometry. Eur Respir J.
26:319–338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Caito S, Rajendrasozhan S, Cook S, Chung
S, Yao H, Friedman AE, Brookes PS and Rahman I: SIRT1 is a
redox-sensitive deacetylase that is post-translationally modified
by oxidants and carbonyl stress. FASEB J. 24:3145–3159. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Farkas L, Farkas D, Warburton D, Gauldie
J, Shi W, Stampfli MR, Voelkel NF and Kolb M: Cigarette smoke
exposure aggravates air space enlargement and alveolar cell
apoptosis in Smad3 knockout mice. Am J Physiol Lung Cell Mol
Physiol. 301:L391–L401. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mimae T, Hagiyama M, Inoue T, Yoneshige A,
Kato T, Okada M, Murakami Y and Ito A: Increased ectodomain
shedding of lung epithelial cell adhesion molecule 1 as a cause of
increased alveolar cell apoptosis in emphysema. Thorax. 69:223–231.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gary RK and Kindell SM: Quantitative assay
of senescence-associated beta-galactosidase activity in mammalian
cell extracts. Anal Biochem. 343:329–334. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hara H, Araya J, Takasaka N, Fujii S,
Kojima J, Yumino Y, Shimizu K, Ishikawa T, Numata T, Kawaishi M, et
al: Involvement of creatine kinase B in cigarette smoke-induced
bronchial epithelial cell senescence. Am J Respir Cell Mol Biol.
46:306–312. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rajendrasozhan S, Yang SR, Kinnula VL and
Rahman I: SIRT1, an antiinflammatory and antiaging protein, is
decreased in lungs of patients with chronic obstructive pulmonary
disease. Am J Respir Crit Care Med. 177:861–870. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nakamaru Y, Vuppusetty C, Wada H, Milne
JC, Ito M, Rossios C, Elliot M, Hogg J, Kharitonov S, Goto H, et
al: A protein deacetylase SIRT1 is a negative regulator of
metalloproteinase-9. FASEB J. 23:2810–2819. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu C, Li Y, Xu WL, Yan JP, Xia YJ, Ma YY,
Chen C, Wang HJ and Tao HQ: Sirtuin 1 activator SRT1720 protects
against lung injury via reduction of type II alveolar epithelial
cells apoptosis in emphysema. COPD. 12:444–452. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yao H, Chung S, Hwang JW, Rajendrasozhan
S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, et
al: SIRT1 protects against emphysema via FOXO3-mediated reduction
of premature senescence in mice. J Clin Invest. 122:2032–2045.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Arunachalam G, Samuel SM, Marei I, Ding H
and Triggle CR: Metformin modulates hyperglycaemia-induced
endothelial senescence and apoptosis through SIRT1. Br J Pharmacol.
171:523–535. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ganesan S, Unger BL, Comstock AT, Angel
KA, Mancuso P, Martinez FJ and Sajjan US: Aberrantly activated EGFR
contributes to enhanced IL-8 expression in COPD airways epithelial
cells via regulation of nuclear FoxO3A. Thorax. 68:131–141. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Furukawa A, Tada-Oikawa S, Kawanishi S and
Oikawa S: H2O2 accelerates cellular senescence by accumulation of
acetylated p53 via decrease in the function of SIRT1 by
NAD+ depletion. Cell Physiol Biochem. 20:45–54. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faghihi MA, Modarresi F, Khalil AM, Wood
DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G III, Kenny PJ and
Wahlestedt C: Expression of a noncoding RNA is elevated in
Alzheimer's disease and drives rapid feed-forward regulation of
beta-secretase. Nat Med. 14:723–730. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Visel A, Zhu Y, May D, Afzal V, Gong E,
Attanasio C, Blow MJ, Cohen JC, Rubin EM and Pennacchio LA:
Targeted deletion of the 9p21 non-coding coronary artery disease
risk interval in mice. Nature. 464:409–412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chung S, Nakagawa H, Uemura M, Piao L,
Ashikawa K, Hosono N, Takata R, Akamatsu S, Kawaguchi T, Morizono
T, et al: Association of a novel long non-coding RNA in 8q24 with
prostate cancer susceptibility. Cancer Sci. 102:245–252. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schmidt LH, Spieker T, Koschmieder S,
Schäffers S, Humberg J, Jungen D, Bulk E, Hascher A, Wittmer D,
Marra A, et al: The long noncoding MALAT-1 RNA indicates a poor
prognosis in non-small cell lung cancer and induces migration and
tumor growth. J Thorac Oncol. 6:1984–1992. 2011. View Article : Google Scholar : PubMed/NCBI
|