Introduction
Sepsis is associated with a high level of mortality
worldwide and is frequently associated with the functional failure
of one or more organs (1,2). In the cardiovascular system, it is
well known that sepsis induces myocardial dysfunction and heart
failure, which is characterized by a significant decrease in
cardiac output (1). In a clinical
setting, cardiac dysfunction induced by sepsis accounts for ~15% of
all cases of mortality, thus it is closely associated with the
prognosis of sepsis (3,4). Therefore, it is critical to improve
cardiac function in order to increase survival rates during sepsis,
particularly at the early stages. Clinical studies have indicated
that sepsis leads to functional depression of the heart without
severe structural damage (2,5),
which suggests that sepsis-associated myocardial dysfunction may be
reversible. Despite previous studies investigating sepsis-induced
pathological alterations in human and animal models (1,6). the
precise mechanisms by which sepsis induces myocardial depression
during the early stages remains unknown. However, a number of
molecules and signaling pathways, including those involved in
oxidative stress, mitochondrial damage and energy metabolism
disorders, have been proposed to be involved (4).
Calcineurin, a Ca2+-calmodulin-activated
serine-threonine phosphatase, serves an important role in
cardiovascular physiology and pathophysiology (7). Overexpression of calcineurin in
cardiomyocytes regulated by the myosin heavy chain promoter has
been demonstrated to promote cardiac hypertrophy (8), which is reversible (9). In addition, inhibition of calcineurin
by specific inhibitors, such as cyclosporine and FK506, protected
the heart from pressure overload-induced cardiac hypertrophy
(10). A previous study indicated
that the activity of calcineurin was increased in cardiomyocytes
upon exposure to lipopolysaccharides, one of the predominant
molecule types released following sepsis (11). These molecules promote pathological
alterations in sepsis-associated myocardial dysfunction (12), indicating a potential role for
calcineurin in the myocardial depression induced by sepsis.
Cyclosporine A (CSA), an immunosuppressant and calcineurin
inhibitor, has been widely used in the clinic to suppress immune
responses against transplanted organs. However, to date, a
relatively small number of studies have been performed to examine
the efficacy of CSA in the treatment of sepsis, and the results
generated have been controversial. One study in particular
demonstrated that CSA served a protective role in rats suffering
from sepsis-induced acute kidney injury (13), however, it was later reported to
exacerbate mortality in a murine model of septic shock (14). Previous studies have revealed that
calcineurin is involved in sepsis-associated myocardial dysfunction
(15), and that this dysfunction
is improved by CSA treatment in animal models (16). However, the mechanisms underlying
efficient CSA treatment are not fully understood.
During sepsis, free fatty acid metabolic disorders,
such as those involving non-esterified free fatty acids (NEFA), and
the misregulation of hormones and cytokines have been observed to
occur (17). The AMP-activated
protein kinase (AMPK)-acetyl CoA carboxylase (ACC)-carnitine
palmitoyl transferase 1 (CPT1) signaling pathway is one of the
pathways involved in mediating myocardial energy metabolism
(18,19). The AMPK-ACC-CPT1 pathway is
activated in response to hormone signaling and stressors, including
low blood glucose levels, hypoxia and ischemia (20). A previous study demonstrated that
elevated calcineurin activity interferes with AMPK-ACC-CPT1 pathway
function, whereas CSA ameliorated fatty acid metabolism by
inhibiting calcineurin activity in rodent hippocampi (3). However, whether the AMPK-ACC-CPT1
signaling pathway contributes to the sepsis-induced alterations in
the lipid metabolism of the heart remains unclear.
The aim of the present study was to determine
whether CSA might be an effective treatment for myocardial
repression during the early stages of the septic process using a
rodent model of sepsis. In addition, the present study investigated
whether the AMPK-ACC-CPT1 pathway may be involved in CSA-mediated
protection against sepsis-associated functional damage of the
myocardium.
Materials and methods
Animals
A total of 135 male Wistar rats (age, 4–6 weeks;
weight, 250–300 g). were purchased from the Experimental Animal
Center of China Medical University (Shenyang, China). The rats, 3
per cage, were maintained at temperature at 22°C, 45% humidity, and
12 h light/dark cycle, having free access to food and water.
All animal experiments were approved by the
Institutional Animal Care and Use Committee of the Medical Ethics
Committee of The First Hospital of China Medical University
(Beijing, China).
Materials
CSA (Pharmaceuticals AG, Rothreuz ZG, Switzerland),
a calcineurin activity measurement kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China), a free fatty acid assay
kit (Nanjing Jiancheng Bioengineering Institute), a bicinchoninic
acid (BCA) assay kit (Beyotime Institute of Biotechnology, Haimen,
China), and primary antibodies against AMPK (cat. no. sc-25792),
phosphorylated (p)-AMPK (cat. no. sc-33524), ACC-β (cat. no.
sc-26822), p-ACC-β (cat. no. sc-30446-R), CPT1 (cat. no. sc-139482)
and β-actin (cat. no. sc-47778, all from Santa Cruz Biotechnology
Inc., Dallas, TX, USA) were used in the present study. In addition,
10% chloral hydrate, phosphate-buffered saline, TBS solution and
Tween-20 were procured from Sinopharm Chemical Reagent Co., Ltd.
(Shanghai, China).
Generation of the septic animal model
using the cecal ligation puncture (CLP) procedure
Rats were anesthetized with a subcutaneous injection
of 10% chloral hydrate (4 ml/kg), fixed on a 22°C warm pad and a
section of abdominal skin was sterilized. A ventral midline
incision of ~1.5 cm was made, followed by cecal ligation at one
third of the distance from the end of the cecum. The cecum was
pierced twice with a needle (no. 18) and fecal overflow was
promoted by squeezing. The cecum was then ligated and placed back
in the abdominal cavity before the opening was closed. A total of 5
ml saline was immediately injected into the abdomen to prevent
shock.
Animal grouping
The rats were randomly divided into the following 3
groups: the control group (sham) where an abdominal incision was
performed without cecal ligation; the sepsis group (CLP group),
whereby abdominal incision and cecal ligation were performed; the
CSA intervention group (CSA group), whereby 10 mg/kg CSA was
administered by intraperitoneal injection at 30 min following
anesthesia, which was followed by abdominal incision and cecal
ligation. Each group was observed at 2, 6, 12, 24 and 72 h
following the operation. Rats that did not survive over course of
the study were excluded (control, 0; CLP, 23; CSA, 19) from the
sample collection and data analysis.
Determination of cardiac function
Cardiac function was measured via catheterization.
Briefly, each rat was anesthetized with 10% chloral hydrate (4
ml/kg, i.p.), fixed with the strip wrapped around the limbs on a
supine position on a small operation table, and the right common
carotid artery was exposed. The proximal end of the catheter, which
was connected to a computer monitored by a Biopac polygraph (Biopac
UK Ltd., Pershore, UK), was inserted into the left ventricle via
the proximal end of the right common carotid artery. The full
procedure was guided by the computer-aided imaging system
AcqKnowledge 4.0 (Biopac UK Ltd.). Following several min of
stabilization, the left ventricular end-diastolic pressure, the
maximal rate of left ventricle systolic pressure rise
(+dP/dtmax) and the maximal rate of the left ventricle
diastolic pressure decrease (−dP/dtmax) were measured
using the Biopac polygraph. Rat cardiac function was measured at 2,
6, 12, 24 and 72 h following the operation.
Blood sample collection
After the rats were anesthetized, blood samples were
collected from the carotid artery using an arterial clamp at 2, 6,
12, 24 and 72 h following the operation. The collected blood (5 ml)
was centrifuged at 1,566 × g for 10 min at room temperature
and the supernatant was immediately frozen at −80°C for future
use.
Determination of the plasma and
cardiac tissue homogenate index
Following blood sample collection, the rat chest was
immediately opened, the heart was isolated and the left ventricle
was removed and immediately frozen at −80°C. The cardiac
calcineurin activity assay, NEFA concentration determination, and
the BCA protein quantification assay were performed based on the
protocols provided by the manufacturers. Briefly, left ventricles
from rats were collected and snap-frozen at −80°C. For protein
purification, the heart tissues were homogenized in RIPA buffer (25
mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate,
0.1% SDS) and protease inhibitors PMSF (ST505, Beyotime Institute
of Biotechnology, Haimen, China were added prior to the
purification. Protein concentrations were determined with a BCA kit
(Beyotime Institute of Biotechnology). cardiac calcineurin activity
assays were performed with a calcineurin activity assay kit
(according to the protocol provided by the manufacturers. NEFA
concentrations were determined with a commercial kit.
Oil red O staining
Oil red O staining was performed as previously
described (21,22) with some modifications. Briefly,
frozen myocardial tissues were sectioned to produce 8-µm-thick
tissue slices. The sections were fixed in 4% formaldehyde for 10
min at room temperature. The slides were then rinsed with distilled
water and dried, before they were by incubated with Oil red O dye
(Oil red, 0.5 g; dissolved in 60% isopropronol) at 37°C for 20 min.
The slides were subsequently submersed in 60% isopropanol for 10 s,
washed with water, and stained with 0.5% hematoxylin for 30 sec at
room temperature. The slides were mounted in glycerogelatin and
sealed.
Western blot analysis
Western blotting was performed to determine the
expression levels of AMPK, p-AMPK, ACC, p-ACC and CPT1 in the
myocardium. Briefly, proteins were extracted from the myocardial
tissues using a protein extraction reagent (Beyotime Institute of
Biotechnology) and protein concentration was measured using the BCA
Assay kit (Beyotime Institute of Biotechnology). Heat-denatured
protein (60 µg) was loaded onto a 10% SDS-PAGE gel, followed by
protein transfer to a nitrocellulose membrane. The membrane was
subsequently blocked with 5% non-fat dried milk for 1 h at room
temperature, before it was probed overnight with the diluted
primary antibody of interest (AMPK, 1:500; p-AMPK, 1:500; ACC,
1:1,000; p-ACC, 1:1,000; CPT1, 1:500) at 4°C. The PVDF membrane was
then washed with TBS-Tween solution and incubated with horseradish
peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse
immunoglobulin G (1:10,000; OriGene Technologies, Beijing, China)
at room temperature for 1 h. Actin expression levels were used as a
control. Protein bands were visualized using an enhanced
chemiluminescence developer (MicroChemi, DNR, Israel) and relative
densitometry was performed using ImageJ version 1.63 (imagej.nih.gov/ij/).
Statistical analysis
Data are expressed as the mean ± standard deviation.
Statistical significance was analyzed using SPSS statistical
software (version, 19.0; IBM SPSS, Armonk, NY, USA). Independent
sample t-tests were used for comparing the statistical
significance between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
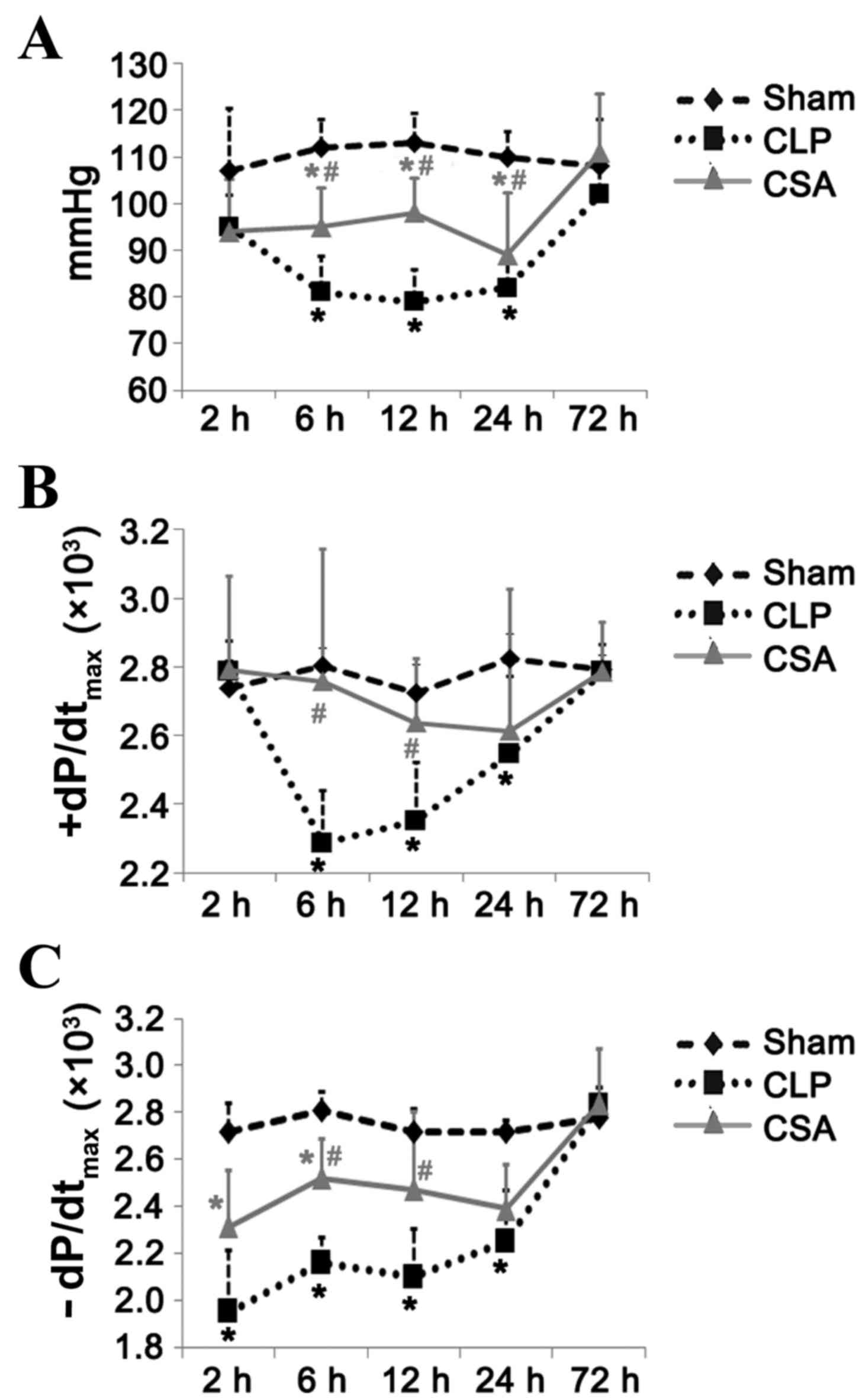
CSA significantly attenuated the
sepsis-induced decrease in blood pressure and cardiac function
As expected, sepsis induced a significant decrease
in blood pressure at 6, 12 and 24 h in the CSA group, respectively,
when compared with the CLP group (P<0.05; Fig. 1A). At 72 h following the induction
of sepsis, the blood pressure of the CLP rats recovered to the same
level as that observed in the sham group, suggesting that this
decrease in blood pressure occurred at an early stage in the septic
model. CSA treatment significantly attenuated this decrease at 6
and 12 h; however, not at 24 h when compared with the CLP group
(P<0.05; Fig. 1A).
Sepsis-induced cardiac dysfunction was evident at 6 h and persisted
for up to 24 h, as demonstrated by the decreased
+dP/dtmax and -dP/dtmax values in the CLP
group when compared with the sham group (Fig. 1B and C). The CSA group demonstrated
significantly improved cardiac function when compared with the CLP
group at 6 and 12 h following the operation (+dP/dtmax,
P<0.05 at both time points; -dP/dtmax, P<0.05 at
both time points; Fig. 1B and C).
At 72 h following the induction of sepsis, no significant
difference was observed between the +dP/dtmax and
-dP/dtmax values of the CLP and CSA groups when compared
with those of the sham group (Fig. 1B
and C). Therefore, this sepsis model may facilitate the study
of the effects of sepsis on myocardial function during the early
stages of sepsis. The results demonstrated that CSA administration
efficiently attenuated the decrease in blood pressure and cardiac
function typically induced by sepsis.
CSA significantly inhibited the
increased activity of calcineurin by sepsis in the myocardium
As CSA is a specific inhibitor of calcineurin, and
calcineurin has been implicated in the maintenance of cardiac
homeostasis (23), the present
study investigated whether sepsis may alter the activity of
calcineurin in the myocardium. As shown in Table I, calcineurin activity was
significantly higher in the CLP group when compared with the sham
group at 6, 12, 24 and 72 h following induction of sepsis
(P<0.05, CLP group vs. the sham group). However, CSA treatment
significantly inhibited the increase in calcineurin activity at 6,
12 and 24 h when compared with the CSP group (P<0.05; Table I). No significant difference in
calcineurin levels were observed in the CSA group when compared
with the CLP group at 72 h (Table
I). These results indicated that sepsis is associated with
elevated calcineurin activity in the myocardium, which may be
suppressed by CSA treatment.
 | Table I.CSA significantly inhibited the
sepsis-induced increase in calcineurin activity in the
myocardium. |
Table I.
CSA significantly inhibited the
sepsis-induced increase in calcineurin activity in the
myocardium.
| Group | 2 h | 6 h | 12 h | 24 h | 72 h |
|---|
| Sham (unit/mg
protein) | 0.67±0.042 | 0.57±0.037 | 0.37±0.058 | 0.5±0.046 | 0.52±0.074 |
| CLP (unit/mg
protein) | 0.77±0.096 |
0.93±0.076a |
1.38±0.077a |
1.6±0.085a |
1.12±0.102a |
| CSA (unit/mg
protein) | 0.61±0.069 |
0.59±0.056b |
0.87±0.059b |
1.04±0.072b | 1.03±0.06 |
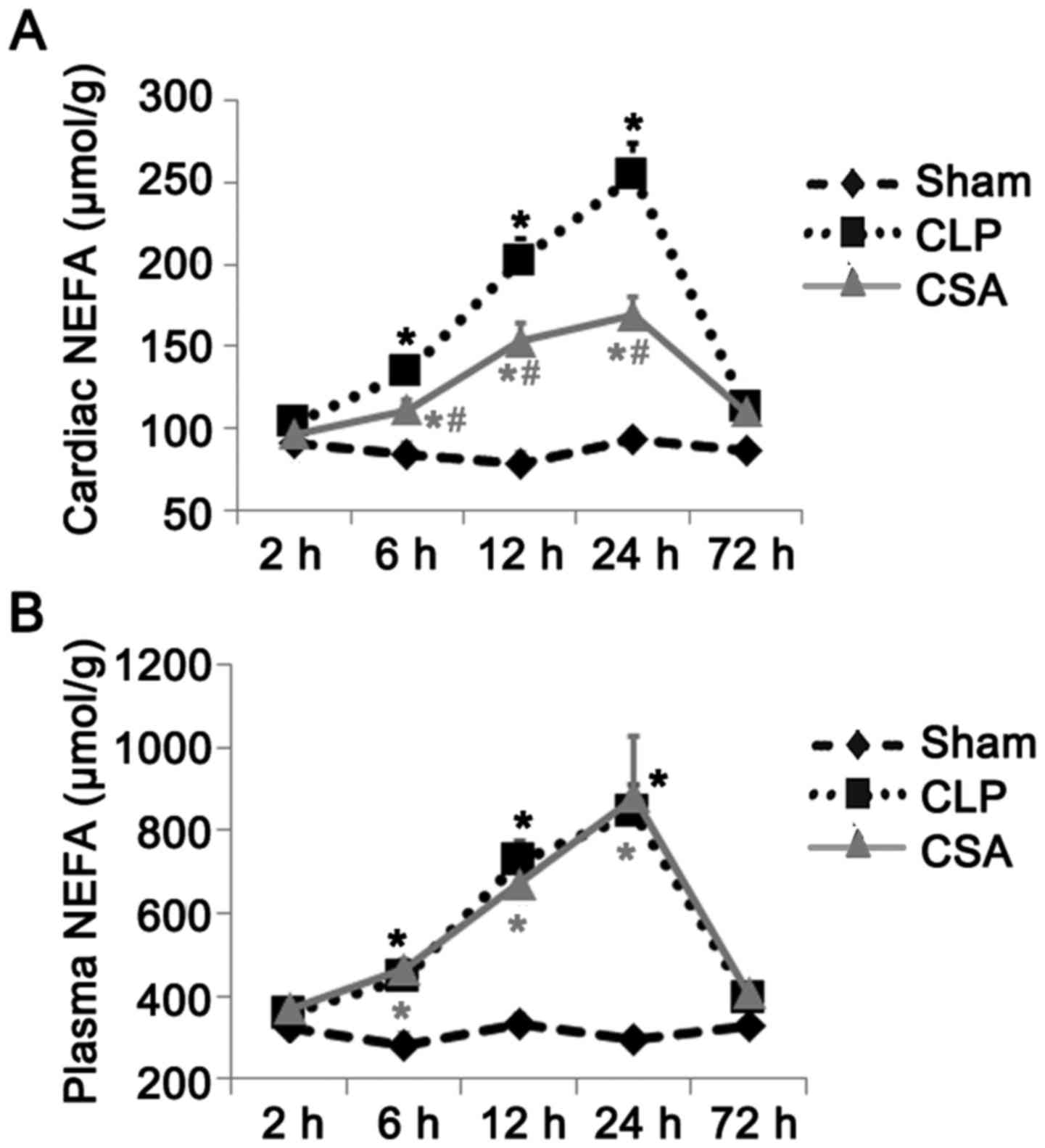
CSA significantly decreased the
sepsis-induced increase in NEFA levels in the myocardium but not in
the serum
It has been previously reported that sepsis alters
energy metabolism in tissues and organs (5). Therefore, the authors of the present
study investigated whether cardiac lipid metabolism was altered in
the rat model of sepsis. As the levels of NEFA are a useful
indicator of altered lipid oxidation, the concentration of NEFA in
the myocardium and plasma of rats with and without sepsis was
measured. As shown in Fig. 2, the
levels of NEFA were significantly elevated in the plasma and
myocardium at 6, 12 and 24 h in the CLP group when compared with
those in the sham group (P<0.05). Although CSA treatment was
associated with a significant increase in NEFA concentration in the
myocardium at 6, 12 and 24 h when compared with the sham group, no
significant difference in plasma NEFA concentrations was observed
in the CSA group when compared with the CLP group at any time point
(Fig. 2). Thus, CSA treatment
substantially diminished the levels of NEFA in the myocardium but
not in the serum of rats with sepsis.
To confirm the altered levels of NEFA in the
myocardium of rats in the three experimental groups, the myocardia
were stained using Oil red O. The sham group exhibited no obvious
Oil red O staining at any of the time points examined (Fig. 3A and B, and data not shown). As
early as 2 h following sepsis induction, the myocardia in the CLP
group exhibited marked red staining, which peaked at 24 h and
persisted for at least 72 h (Fig.
3C-G). Despite positive red staining in the myocardia of the
CSA group, the intensity of red staining was lower when compared
with that of the CLA group at each time point examined (Fig. 3H-L). Oil red O staining reflects
free fatty acid accumulation in the myocardium. Therefore, these
results indicate that CSA may facilitate free fatty acid metabolism
in the myocardium, thus reducing the accumulation induced by
sepsis.
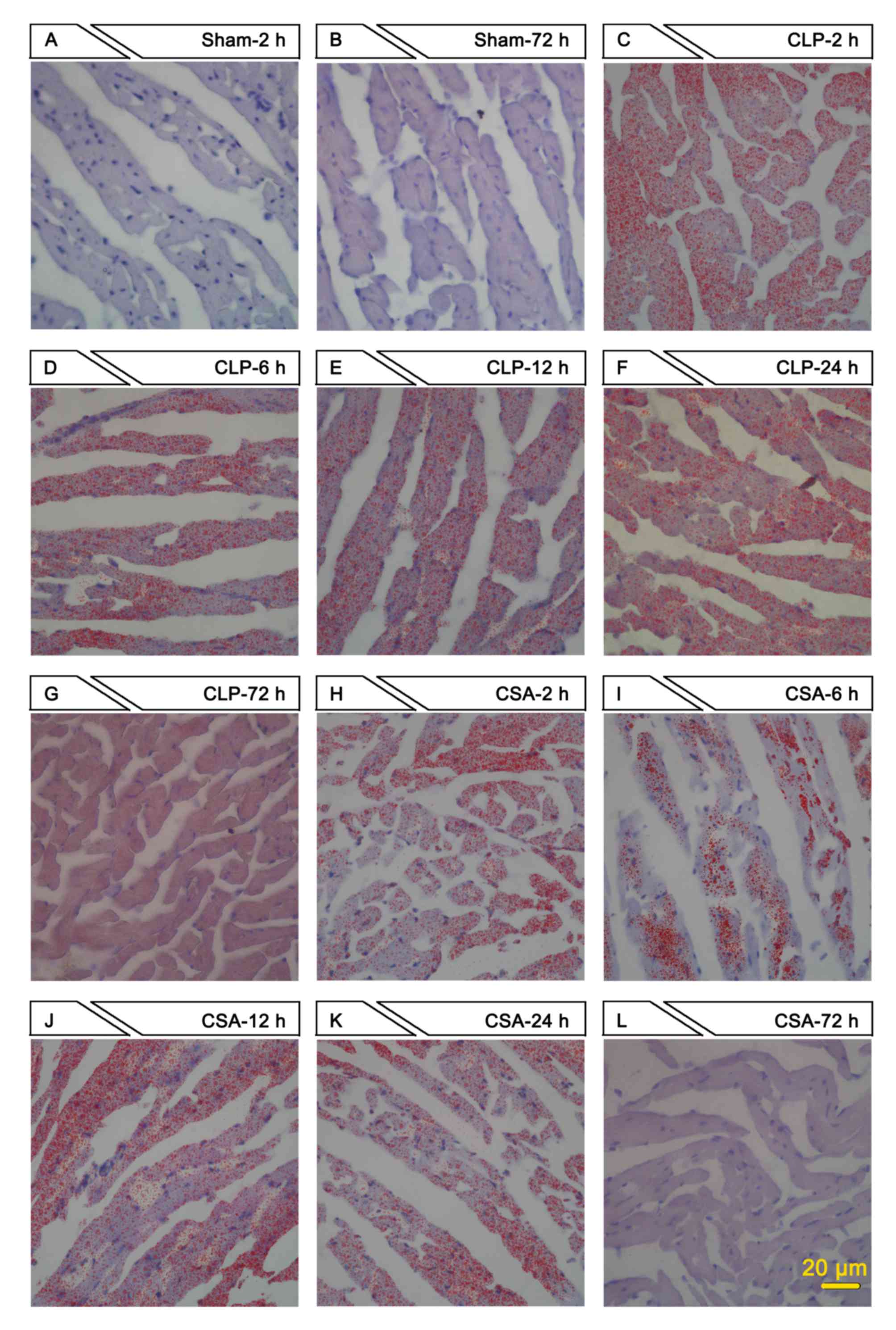 | Figure 3.CSA significantly decreased NEFA
accumulation in the myocardium. Oil red O staining was performed on
heart tissue sections prepared from rats in sham, CLP and CSA
groups at different time points (2, 6, 12, 24 and 72 h).
Representative images are shown (scale bar, 20 µm; magnification,
×400). Sham group at (A) 2 h and (B) 72 h; CLP group at (C) 2 h,
(D) 6 h, (E) 12 h, (F) 24 h and (G) 72 h; CSA group at (H) 2 h, (I)
6 h, (J) 12 h, (K) 24 h and (L) 72 h. NEFA, non-esterified fatty
acids; sham, sham-operated rats; CLP, cecal ligation puncture
procedure sepsis model group; CSA, cyclosporine A-treated sepsis
model group. |
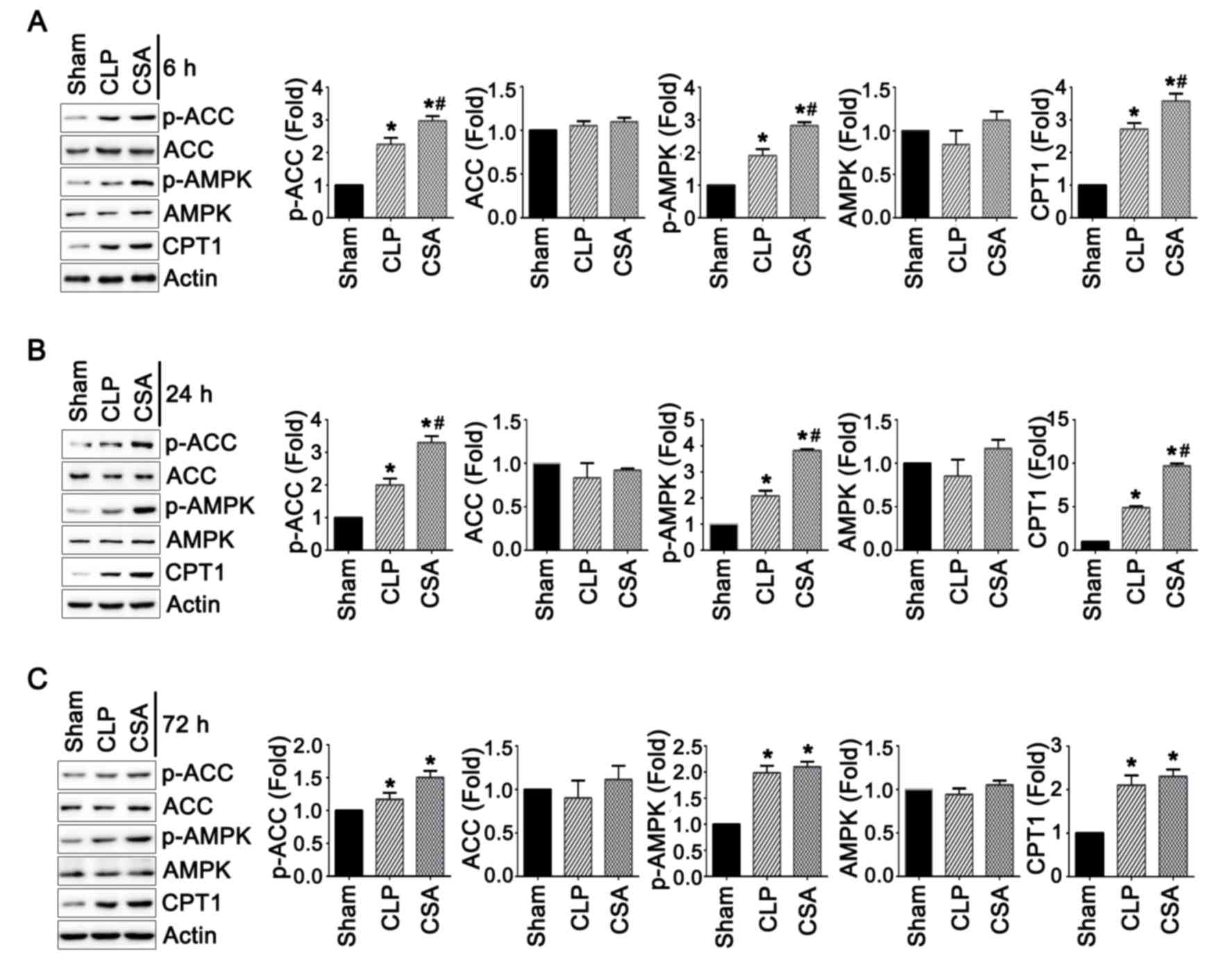
CSA treatment activated the
AMPK-ACC-CPT1 signaling pathway
In order to investigate whether sepsis induced
alterations in lipid metabolism, the present study investigated
whether the AMPK-ACC-CPT1 signaling pathway may be involved. The
AMPK-ACC-CPT1 pathway has been proposed to be an important
regulator of lipid metabolism in the heart (24). Therefore, the expression levels of
AMPK, ACC and CPT1 in the heart tissues from rats in all three
experimental groups were analyzed by western blotting. As shown in
Fig. 4, the protein expression
levels of AMPK and ACC did not exhibit any significant differences
among the three groups at 6, 24 and 72 h time points. However, the
active forms of the two proteins, p-AMPK and p-ACC, were
significantly elevated in the CLP group when compared with the sham
group at all time points (P<0.05; Fig. 4). In addition, CPT1 expression was
elevated in the CLP group when compared with the sham group at all
time points (P<0.05; Fig. 4).
CSA treatment increased the activity of p-AMPK, p-ACC and CPT1 at
6, and 24 h, when compared with the CLP and sham groups (Fig. 4, P<0.05). Collectively, these
data suggest that sepsis may increase the activity of the
AMPK-ACC-CPT1 signaling pathway, and CSA may further enhance the
activation of this pathway by increasing the levels of the active,
phosphorylated forms or the total protein levels of these
components.
Discussion
Hemodynamic alterations serve a crucial role in the
occurrence and development of shock and multiple organ dysfunction
syndrome in the pathophysiology of sepsis (25). In particular, left ventricular
function is an important contributor to hemodynamic alterations,
and significantly affects the prognosis of sepsis (26). A previous study demonstrated that
early sepsis may lead to decreased heart function, which is
attributable to myocardial ischemia and hypoxia as well as altered
myocardial energy metabolism (27).
The present study investigated how early stage
sepsis affected cardiac function and whether CSA may be able to
exert any significant protective effects. The sepsis model induced
a decrease in blood pressure and impaired cardiac function; the
latter of which was demonstrated by decreased +dP/dtmax
and -dP/dtmax values at 6 and 24 h following sepsis
induction. However, these functional indices returned to levels
comparable to those of the sham group, suggesting that the sepsis
model applied in the current study may be a short-term model that
is only appropriate for studying the effects of early-stage sepsis
on the heart. In addition, CSA treatment was observed to
substantially improve blood pressure and cardiac function at 6 and
12 h following sepsis induction. Thus, CSA may be an effective drug
in facilitating the treatment of sepsis at the early stages of
infection, in order to ameliorate cardiac function.
Previously, CSA has been widely used as an
immunosuppressant to repress immune responses against transplanted
organs in the clinic (28).
However, previous studies have indicated that CSA is a specific
inhibitor of calcineurin; a calcium-sensitive phosphatase, which
serves a well-defined role in cardiac physiology and
pathophysiology (12,15). In addition, calcineurin may be
involved in sepsis-induced cardiac dysfunction (12,15).
The present study demonstrated that calcineurin activity was
elevated following induction of sepsis. This elevated activity of
calcineurin was repressed by CSA treatment. In the CLP group,
calcineurin activity was significantly increased between 6 and 72 h
following sepsis induction, and CSA treatment efficiently
suppressed this increase during the 6–24 h period. This coincided
with cardiac functional recovery at 6 to 24 h by CSA treatment. The
results indicated that CSA treatment may alleviate the cardiac
dysfunction induced by sepsis, in part, through suppression of
calcineurin activity. Cardiac function was normal in the CLP group
at 72 h. However, at this time point, calcineurin activity remained
significantly higher in the CLP group when compared with the sham
group. Therefore, it is possible that additional mechanisms, other
than calcineurin activity, may be involved in mediating myocardial
repression during the early stages of sepsis. Alternatively, it is
possible that the elevated calcineurin levels at 72 h may not have
been sufficient to induce cardiac dysfunction.
Sepsis-induced alterations in myocardial energy
metabolism serve an important role in the development of cardiac
dysfunction (29). Altered
myocardial energy metabolism is characterized by impaired
mitochondrial function, reduced lipid metabolism and glucose
metabolism disorders in patients and mouse models (20). Myocardial NEFA accumulation is an
important manifestation of myocardial metabolic dysregulation
(20). Under physiological
conditions, cardiomyocytes do not synthesize and store a
significant quantity of NEFA; instead, myocardial cells take up
NEFA from the circulating serum. Following uptake, NEFA is
predominantly transported to the mitochondria to provide energy,
and to participate in triglyceride and lysophospholipid synthesis.
Myocardial energy is primarily obtained via mitochondrial oxidative
metabolism, where fatty acid oxidation contributes 60–80% of the
energy generated, whereas pyruvate oxidation generates 10–40% and
glycolysis contributes the remainder (30,31).
However, during the development of sepsis, glycolysis, instead of
free fatty acid oxidation (using NEFA), serves as the major source
of energy, which leads to the accumulation of NEFA in
cardiomyocytes. Excessive NEFA accumulation in the heart induces
the inhibition of mitochondrial oxidative phosphorylation and
adenosine triphosphate (ATP)-to-inorganic phosphate conversion,
leading to heart damage (32,33).
However, how NEFA metabolism is regulated in the myocardium of rats
in the septic model is unknown. In the present study, the CLP group
exhibited elevated concentrations of NEFA in the plasma and an
increased accumulation of NEFA in cardiomyocytes at 6–24 h
following sepsis induction when compared with the sham group.
Notably, CSA treatment decreased NEFA accumulation in
cardiomyocytes at 6, 12 and 24 h. However, no significant effects
on plasma NEFA concentrations were observed at any of the time
points examined. In cardiomyocytes, a decrease in NEFA accumulation
was observed alongside the recovery of cardiac function achieved by
CSA treatment. These findings indicated that the CSA-induced
improvement in cardiac function in the rat sepsis model may be
partially reduced following NEFA storage in cardiomyocytes. In
addition, the increase in the calcineurin activity in plasma
observed in rats with sepsis may have been induced via different
mechanisms compared with calcineurin induction in the heart and
were therefore CSA-independent. As CSA efficiently decreased NEFA
accumulation in cardiomyocytes but not in plasma, and CSA is a
specific inhibitor for calcineurin, it is possible that increased
NEFA accumulation in cardiomyocytes may involve
calcineurin-associated signaling pathways. However, further
investigation is required to evaluate this hypothesis.
In the present study, the AMPK-ACC-CPT1
calcineurin-mediated signaling pathway, which is a key pathway for
the mediation of energy metabolism (NEFA oxidation in particular)
(34), was investigated.
Activation of the AMPK pathway in the heart may facilitate NEFA
oxidation, thereby improving the energy supply and enhancing
cardiac function (35). By
contrast, inhibition of the AMPK pathway has been observed to
reduce NEFA oxidation, thereby increasing NEFA accumulation and the
associated toxicity to cardiomyocytes (36). In addition, NEFA accumulation may
induce cardiac endoplasmic reticulum stress, together with
decreased ATP production, thereby inducing heart failure (21,37–39).
Calcineurin is one of the factors that regulate the AMPK-ACC-CPT1
pathway (40). Cardiac injuries
induced by external stimuli, such as
H2O2-activated calcineurin and repression of
the AMPK-ACC-CPT1 pathway, lead to altered lipid metabolism and the
accumulation of NEFA (3,40). By contrast, inhibition of
calcineurin through the use of inhibitors, such as CSA and FK506,
has been observed to improve the activity of AMPK-ACC-CPT1
signaling and alleviate myocardial injury (3,40).
The present study demonstrated that, during the early stages of
sepsis, the total protein levels of AMPK and ACC were not
significantly altered. However, the levels of their activated
forms, p-AMPK and p-ACC, were significantly increased in the
initial 72 h-period following sepsis induction. In addition, a
similar increase in the protein levels of CPT1, was observed.
However, the sepsis-induced activation of the AMPK-ACC-CPT1 pathway
was not sufficient to inhibit NEFA accumulation. By contrast, CSA
treatment increased the activity of this pathway, which was
associated with a significant reduction in NEFA accumulation.
Therefore, sepsis may alter lipid metabolism in the heart, thereby
activating calcineurin and the AMPK-ACC-CPT1 pathway
simultaneously. However, in the presence of activated calcineurin
activity, AMPK-ACC-CPT1 signaling may have been unable to
facilitate the efficient mobilization of NEFA metabolism in the
myocardium. CSA treatment may have partially diminished NEFA
accumulation by further upregulating the activity of the
AMPK-ACC-CPT1 pathway. Of particular note, the myocardial function
in rats with sepsis was comparable to rats in the sham group at 72
h, whereas calcineurin activity and NEFA concentration in the
myocardium of rats in the CLP group remained significantly higher
at the same time point, which suggests that there may be
alternative mechanisms involved in mediating myocardial function at
the early stages of sepsis progression.
In conclusion, the results of the present study
indicate that sepsis may induce cardiac dysfunction, calcineurin
activation and abnormal lipid metabolism. Improved cardiac function
by CSA was evidenced by the observed decrease in calcineurin
activity and reduced NEFA accumulation in cardiomyocytes.
Calcineurin and AMPK signaling are involved in altered lipid
metabolism, and CSA reduced NEFA accumulation in the myocardium
potentially via partial suppression of calcineurin activity and
increased AMPK signaling. Therefore, the results of the present
study provide a greater insight into the mechanisms by which CSA is
able to alleviate myocardial repression induced by sepsis. In
addition, the results present a potential therapeutic treatment for
sepsis-induced cardiac dysfunction, at least at the early stages of
infection.
References
|
1
|
Iskander KN, Osuchowski MF,
Stearns-Kurosawa DJ, Kurosawa S, Stepien D, Valentine C and Remick
DG: Sepsis: Multiple abnormalities, heterogeneous responses, and
evolving understanding. Physiol Rev. 93:1247–1288. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (Sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Park HG, Yi H, Kim SH, Yu HS, Ahn YM, Lee
YH, Roh MS and Kim YS: The effect of cyclosporine A on the
phosphorylation of the AMPK pathway in the rat hippocampus. Prog
Neuropsychopharmacol Biol Psychiatry. 35:1933–1937. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Parrillo JE: The cardiovascular
pathophysiology of sepsis. Annu Rev Med. 40:469–485. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
dos Santos CC, Gattas DJ, Tsoporis JN,
Smeding L, Kabir G, Masoom H, Akram A, Plotz F, Slutsky AS, Husain
M, et al: Sepsis-induced myocardial depression is associated with
transcriptional changes in energy metabolism and contractile
related genes: A physiological and gene expression-based approach.
Crit Care Med. 38:894–902. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Remick DG: Pathophysiology of sepsis. Am J
Pathol. 170:1435–1444. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bueno OF, Van Rooij E, Molkentin JD,
Doevendans PA and De Windt LJ: Calcineurin and hypertrophic heart
disease: Novel insights and remaining questions. Cardiovasc Res.
53:806–821. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Molkentin JD, Lu JR, Antos CL, Markham B,
Richardson J, Robbins J, Grant SR and Olson EN: A
calcineurin-dependent transcriptional pathway for cardiac
hypertrophy. Cell. 93:215–228. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Berry JM, Le V, Rotter D, Battiprolu PK,
Grinsfelder B, Tannous P, Burchfield JS, Czubryt M, Backs J, Olson
EN, et al: Reversibility of adverse, calcineurin-dependent cardiac
remodeling. Circ Res. 109:407–417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sussman MA, Lim HW, Gude N, Taigen T,
Olson EN, Robbins J, Colbert MC, Gualberto A, Wieczorek DF and
Molkentin JD: Prevention of cardiac hypertrophy in mice by
calcineurin inhibition. Science. 281:1690–1693. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Suzuki J, Bayna E, Li HL, Molle ED and Lew
WY: Lipopolysaccharide activates calcineurin in ventricular
myocytes. J Am Coll Cardiol. 49:491–499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Larche J, Lancel S, Hassoun SM, Favory R,
Decoster B, Marchetti P, Chopin C and Neviere R: Inhibition of
mitochondrial permeability transition prevents sepsis-induced
myocardial dysfunction and mortality. J Am Coll Cardiol.
48:377–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Iwamura H, Sato M and Wakitani K:
Comparative study of glucocorticoids, cyclosporine A, and JTE-607
[(−)-Ethyl-N[3,5-dichloro-2-hydroxy-4-[2-(4-methylpiperazin-1-yl)ethoxy]benzoyl]-L-phenylalaninate
dihydrochloride] in a mouse septic shock model. J Pharmacol Exp
Ther. 311:1256–1263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Joshi MS, Julian MW, Huff JE, Bauer JA,
Xia Y and Crouser ED: Calcineurin regulates myocardial function
during acute endotoxemia. Am J Respir Crit Care Med. 173:999–1007.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fauvel H, Marchetti P, Obert G, Joulain O,
Chopin C, Formstecher P and Nevière R: Protective effects of
cyclosporin A from endotoxin-induced myocardial dysfunction and
apoptosis in rats. Am J Respir Crit Care Med. 165:449–455. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zu L, He J, Jiang H, Xu C, Pu S and Xu G:
Bacterial endotoxin stimulates adipose lipolysis via toll-like
receptor 4 and extracellular signal-regulated kinase pathway. J
Biol Chem. 284:5915–5926. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heidrich F, Schotola H, Popov AF, Sohns C,
Schuenemann J, Friedrich M, Coskun KO, von Lewinski D, Hinz J,
Bauer M, et al: AMPK-activated protein kinase and its role in
energy metabolism of the heart. Curr Cardiol Rev. 6:337–342. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fillmore N and Lopaschuk GD: Targeting
mitochondrial oxidative metabolism as an approach to treat heart
failure. Biochim Biophys Acta. 1833:857–865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kiuchi S, Matsuo N, Takeyama N and Tanaka
T: Accelerated hepatic lipid synthesis in fasted septic rats. Eur
Surg Res. 25:146–154. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Borradaile NM, Han X, Harp JD, Gale SE,
Ory DS and Schaffer JE: Disruption of endoplasmic reticulum
structure and integrity in lipotoxic cell death. J Lipid Res.
47:2726–2737. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Son NH, Park TS, Yamashita H, Yokoyama M,
Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS and Goldberg
IJ: Cardiomyocyte expression of PPARgamma leads to cardiac
dysfunction in mice. J Clin Invest. 117:2791–2801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Torac E, Gaman L and Atanasiu V: The
regulator of calcineurin (RCAN1. an important factor involved in
atherosclerosis and cardiovascular diseases development. J Med
Life. 7:481–487. 2014.PubMed/NCBI
|
|
24
|
Kuwabara Y, Horie T, Baba O, Watanabe S,
Nishiga M, Usami S, Izuhara M, Nakao T, Nishino T, Otsu K, et al:
MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and
high-fat diet-induced cardiac hypertrophy in mice through
suppression of the LKB1/AMPK pathway. Circ Res. 116:279–288. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahrens T: Hemodynamics in sepsis. AACN Adv
Crit Care. 17:435–445. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merx MW and Weber C: Sepsis and the heart.
Circulation. 116:793–802. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zaky A, Deem S, Bendjelid K and Treggiari
MM: Characterization of cardiac dysfunction in sepsis: An ongoing
challenge. Shock. 41:12–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Molnar AO, Fergusson D, Tsampalieros AK,
Bennett A, Fergusson N, Ramsay T and Knoll GA: Generic
immunosuppression in solid organ transplantation: Systematic review
and meta-analysis. BMJ. 350:h31632015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kakihana Y, Ito T, Nakahara M, Yamaguchi K
and Yasuda T: Sepsis-induced myocardial dysfunction:
Pathophysiology and management. J Intensive Care. 4:222016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stanley WC, Lopaschuk GD, Hall JL and
McCormack JG: Regulation of myocardial carbohydrate metabolism
under normal and ischaemic conditions. Potential for
pharmacological interventions. Cardiovasc Res. 33:243–257. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van der Vusse GJ, Glatz JF, Stam HC and
Reneman RS: Fatty acid homeostasis in the normoxic and ischemic
heart. Physiol Rev. 72:881–940. 1992.PubMed/NCBI
|
|
32
|
Ebong IA, Goff DC Jr, Rodriguez CJ, Chen H
and Bertoni AG: Mechanisms of heart failure in obesity. Obes Res
Clin Pract. 8:e540–e548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Haffar T, Bérubé-Simard FA and Bousette N:
Cardiomyocyte lipotoxicity is mediated by Il-6 and causes
down-regulation of PPARs. Biochem Biophys Res Commun. 459:54–59.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Srivastava RA, Pinkosky SL, Filippov S,
Hanselman JC, Cramer CT and Newton RS: AMP-activated protein
kinase: An emerging drug target to regulate imbalances in lipid and
carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid
Res. 53:2490–2514. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Viollet B and Andreelli F: AMP-activated
protein kinase and metabolic control. Handb Exp Pharmacol. 303–330.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park TS, Hu Y, Noh HL, Drosatos K, Okajima
K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED and Goldberg
IJ: Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid
Res. 49:2101–2112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Borradaile NM, Buhman KK, Listenberger LL,
Magee CJ, Morimoto ET, Ory DS and Schaffer JE: A critical role for
eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol
Cell. 17:770–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Song XJ, Yang CY, Liu B, Wei Q, Korkor MT,
Liu JY and Yang P: Atorvastatin inhibits myocardial cell apoptosis
in a rat model with post-myocardial infarction heart failure by
downregulating ER stress response. Int J Med Sci. 8:564–572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Turner MD: Fatty acyl CoA-mediated
inhibition of endoplasmic reticulum assembly. Biochim Biophys Acta.
1693:1–4. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
He H, Liu X, Lv L, Liang H, Leng B, Zhao
D, Zhang Y, Du Z, Chen X, Li S, et al: Calcineurin suppresses
AMPK-dependent cytoprotective autophagy in cardiomyocytes under
oxidative stress. Cell Death Dis. 5:e9972014. View Article : Google Scholar : PubMed/NCBI
|