Introduction
Surfactant protein A (SP-A), a collagen-containing
C-type collectin, is an important host defense protein that
functions to maintain pulmonary homeostasis and regulate
inflammatory responses (1,2). SP-A is involved in various immune
functions by mediating a variety of processes, including
recognition and opsonization of pathogenic invaders and apoptotic
cells, downregulation of inflammation and modulation of the
expression of Toll-like receptors (TLRs) (3–5).
During acute and chronic lung injury, serum levels of SP-A are
markedly increased and SP-A knockout mice experience more severe
inflammation with increased levels of pro-inflammatory cytokines
(1,6). Although the lung appears to be the
primary site of synthesis (7),
SP-A protein and mRNA are widely expressed in several
extrapulmonary tissues, including the intestine, colon, mucosa and
brain tissue (8–10). Previous studies have demonstrated
that SP-A modulates inflammatory responses in an animal model of
experimental necrotizing enterocolitis (11) and increased expression of SP-A was
observed during chronic rhinosinusitis and allergic rhinitis
(12,13). The potential function of SP-A in
extrapulmonary immunity is an emerging area of research.
Previous studies have revealed that SP-A is
expressed in rat and human central nervous systems (CNS) (10,14).
SP-A exhibits immunoreactivity in rat and human brains, and levels
are lower in the cerebrospinal fluid (CSF) of patients with
autoimmune inflammatory diseases, such as multiple sclerosis (MS),
which indicates that there is an association between the
established functions of SP-A and the pathogenic factors of MS
(10,14). TLR4 is a critical cellular receptor
for SP-A (15). Increased
expression and activation of CNS TLR4 is involved in the activation
and modulation of proinflammatory responses during the progression
of MS (16). TLR4 is widely
expressed in resident immune cells of the CNS, including astrocytes
and microglia, and stimulates the pathway that predominantly
activates the transcription factor nuclear factor-κB (NF-κB)
(17,18). Through this pathway, TLR4 regulates
the production of several inflammatory factors, including tumor
necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) (19,20).
Accordingly, we hypothesized that SP-A may be involved in the
modulation of inflammatory responses in the CNS, and interact with
the lipopolysaccharide (LPS)-TLR4/NF-κB pathway in astrocytes and
microglia. Using the established rat experimental autoimmune
encephalomyelitis (EAE) model of MS, and in vitro
experiments with LPS-stimulated human astrocytes and microglia, the
present study investigated the distribution of SP-A in the rat CNS
and changes in SP-A expression over different courses of EAE
progression. Additionally, the current study focused on the
interactions between SP-A and LPS-induced inflammatory responses,
and TLR4 expression in human astrocyte and microglial cells.
Materials and methods
Animal models
Female Lewis rats (n=40) aged 6–8 weeks old and
weighing ~200 g were obtained from the animal laboratories of China
Medical University (CMU; Shenyang, China). Animals were housed
under a 12 h light/dark cycle and had free access to food and
water. All experimental protocols were performed in accordance with
the Guidelines for the Care and Use of Laboratory Animals of CMU
and National Institutes of Health Publication (NIH publication no.
86-23, revised 1985). EAE model was induced as previously described
(21). Briefly, rats were
immunized with subcutaneous injections of 0.4 ml solution composed
of guinea pig spinal cord homogenate and an equal volume of
Mycobacterium tuberculosis H37Ra (10 mg/ml; Difco; BD
Biosciences, Franklin Lakes, NJ, USA)-enriched complete Freund's
adjuvant (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in both
hind footpads and in the back. The day of immunization was termed
day 0. A preliminary experiment was performed prior to the
detection of SP-A in different stages of EAE. It was observed that
disease onset typically occurred between 9 and 11 days after
immunization, and reached its peak between days 15 and 17
post-immunization. From day 21 onwards, the disease entered
recovery stage. Therefore, the stages in the present study were
designed to consist of initial, peak and recovery stages and the
CNS tissues were obtained on day 10, day 16 and day 21 for
different stages (n=10 per stage). Non-immunized rats (n=10)
received an equal amount of normal saline as a control group. All
rats were sacrificed under anesthesia to obtain CNS tissue.
Preparation of purified SP-A
SP-A protein used in this study was prepared as
described previously (21).
Briefly, bronchoalveolar lavage was obtained from human volunteers
with pulmonary alveolar proteinosis, and SP-A was isolated from
lavage material by pelleting and delipidating with isopropyl and
1-butanol, the purified SP-A protein was donated by the Respiratory
Department of Shenjing Hospital (Shenyang, China). Following
precipitation, SP-A was further purified via column affinity and
purification (~0.8 mg/ml), which was determined by SDS-PAGE.
Endotoxin levels in the SP-A preparation were assessed via
immunoblotting and ranged from undetectable to 0.18 pg/µg.
Human astrocyte and microglial
cultures
Human astrocytes and microglia (cat. nos. 1800 and
1900, respectively; Sciencell Research Laboratories, Carlsbad, CA,
USA) derived from normal human brain were cultured with a mixture
of formulated Dulbecco's modified Eagle's medium (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 10% fetal
bovine serum (Invitrogen; Thermo Fisher Scientific, Inc.) in a
humidified atmosphere of 5% CO2 at 37°C. The culture
medium was renewed every two days. Cells were dissociated with
0.25% EDTA (Invitrogen; Thermo Fisher Scientific, Inc.) once 80%
confluency was reached.
Immunohistochemical staining for SP-A,
glial fibrillary acidic protein (GFAP), allograft inflammatory
factor 1 (Iba-1) and CD11b in the rat CNS
Tissue specimens from rat CNS tissues were fixed in
4% paraformaldehyde (PFA) in PBS (pH 7.4) overnight and
subsequently embedded in paraffin, from which 4 µm thick sections
were prepared. Following deparaffinization, sections were blocked
with 5% normal goat serum (Beyotime Institute of Biotechnology,
Haimen, China) at room temperature for 30 min, and incubated with
rabbit SP-A polyclonal antibody (1:200; cat. no. sc-13977; Santa
Cruz Biotechnology, Inc., Dallas, TX, USA), rabbit GFAP polyclonal
antibody (1:500; cat. no. ab7260; Abcam, Cambridge, UK), rabbit
IBA1 polyclonal antibody (1:500; cat. no. GTX101495; GeneTex, Inc.,
Irvine, CA, USA) or rabbit CD11b polyclonal antibody (1:200; cat.
no. DF2911; Affinity Biosciences, Cincinatti, OH, USA) overnight at
4°C. Subsequently, sections were incubated with goat anti-rabbit
IgG (H+L) secondary antibody (1:1,000; cat. no. A0277; Beyotime
Institute of Biotechnology) at room temperature for 30 min and
stained with 3,3′-diaminobenzidine (DAB) solution (Sigma-Aldrich;
Merck KGaA). When detection of the distribution of SP-A in healthy
rat CNS was performed, lung sections were stained as the positive
control. CNS sections incubated with PBS instead of a primary
antibody served as the negative control. All sections were
counterstained with hematoxylin to identify nuclei. In addition,
conventional hematoxylin and eosin (H&E) staining was
performed. The sections of the cortex and lung were observed at
×200 and ×400 magnification with a light microscope (Olympus
Corporation, Tokyo, Japan).
Immunofluorescence staining for
SP-A
Paraffin sections (3–4 mm) of rat CNS were
deparaffinized for double fluorescence staining for SP-A and GFAP,
or SP-A and Iba-1. Subsequent to blockage with 5% normal donkey
serum (Sigma-Aldrich; Merck KGaA) at room temperature for 2 h,
sections were incubated with a mixture of goat SP-A polyclonal
antibody (1:200; cat. no. sc-7699; Santa Cruz Biotechnology, Inc.)
and rabbit GFAP polyclonal antibody (1:500; cat. no. ab7260;
Abcam), or goat SP-A polyclonal antibody and rabbit Iba-1
polyclonal antibody (1:500; cat. no. GTX101495, GeneTex, Inc.)
overnight at 4°C, followed by a mixture of Alexa Fluor®
488 donkey anti-rabbit IgG (1:200; cat. no. A21206; Invitrogen;
Thermo Fisher Scientific, Inc.) and Alexa Fluor® 594
donkey anti-goat IgG (1:200; cat. no. A11058; Invitrogen; Thermo
Fisher Scientific, Inc.) for 2 h at room temperature. Hoechst 33258
was used for nuclear staining. CNS sections incubated with only
secondary antibody served as negative controls for GFAP, Iba-1 and
SP-A. Brain sections were observed at ×400 magnification with a
fluorescence microscope (Leica Microsystems GmbH, Wetzlar,
Germany).
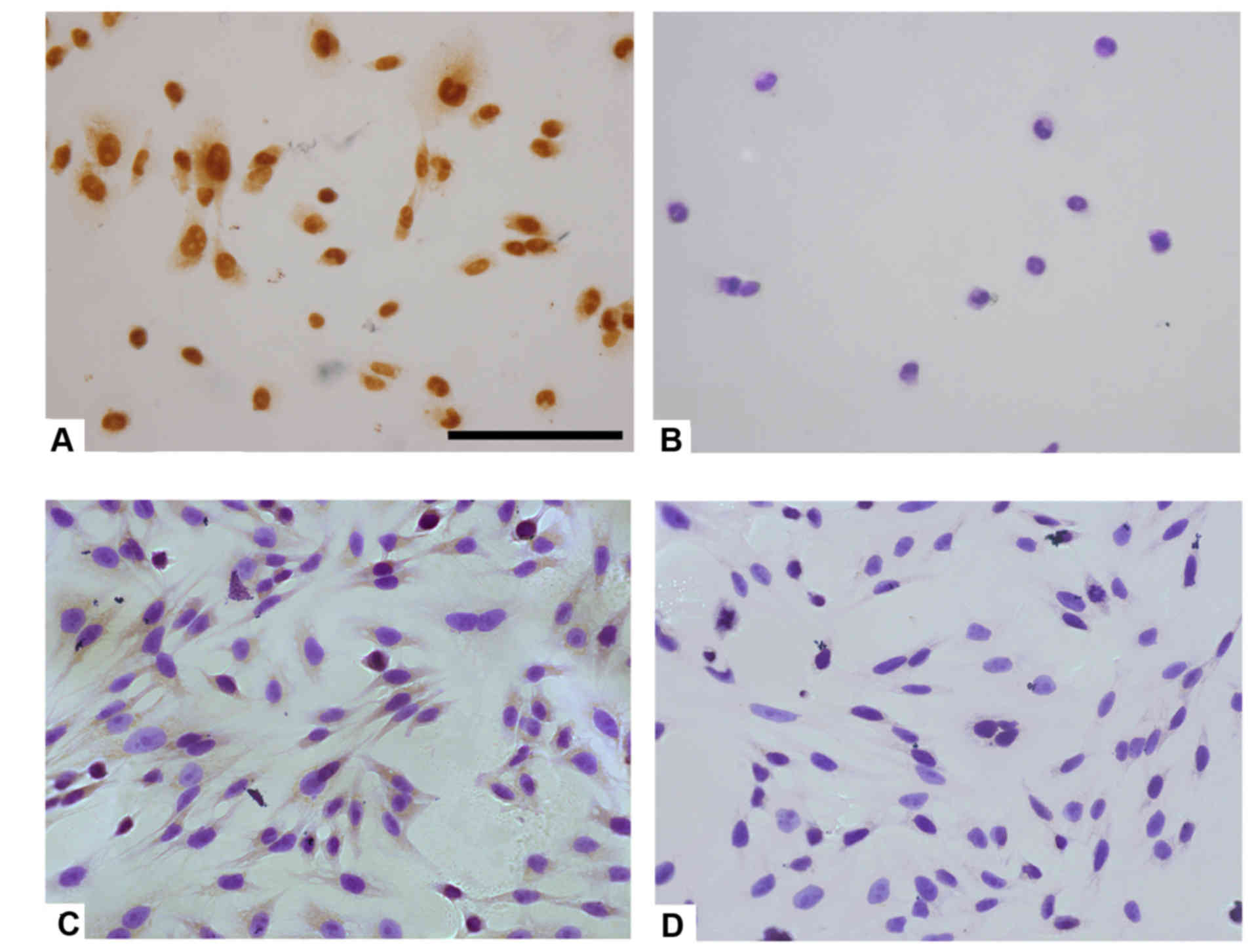
Immunocytochemical staining of
astrocytes and microglia
Immunocytochemical staining for SP-A was performed
in human astrocytes and microglia in vitro. Coverglass
cultured cells were fixed in 4% PFA, blocked with 5% normal goat
serum for 30 min and incubated with rabbit SP-A polyclonal antibody
(1:200; cat. no. sc-13977; Santa Cruz Biotechnology, Inc.)
overnight at 4°C, followed by rabbit IgG secondary antibody at room
temperature for 30 min. Sections were stained with DAB solution
(Sigma-Aldrich; Merck KGaA), followed by counterstaining with
hematoxylin to observe nuclei. Cells incubated only with secondary
antibody served as negative controls.
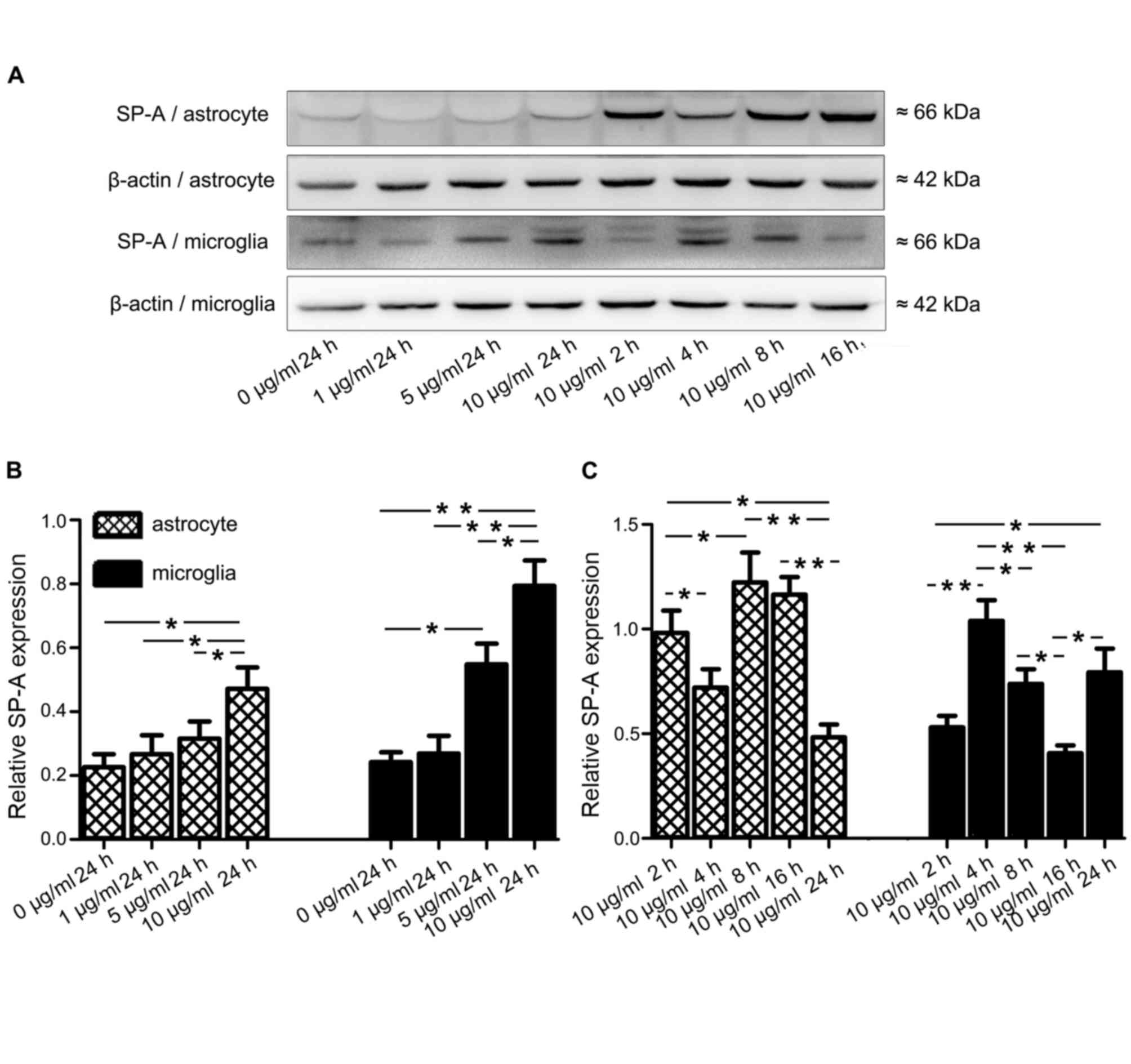
Evaluation of LPS stimulation of SP-A
protein expression in human astrocytes and microglia
Human astrocytes and microglia were incubated with a
range of LPS concentrations from Escherichia coli 0111: B4
(0, 1, 5 and 10 µg/ml; Sigma; St. Louis, MO, USA) for 24 h, or 10
µg/ml LPS for different durations (2, 4, 8, 16 and 24 h). Cells
were collected for analysis of SP-A protein expression by western
blotting.
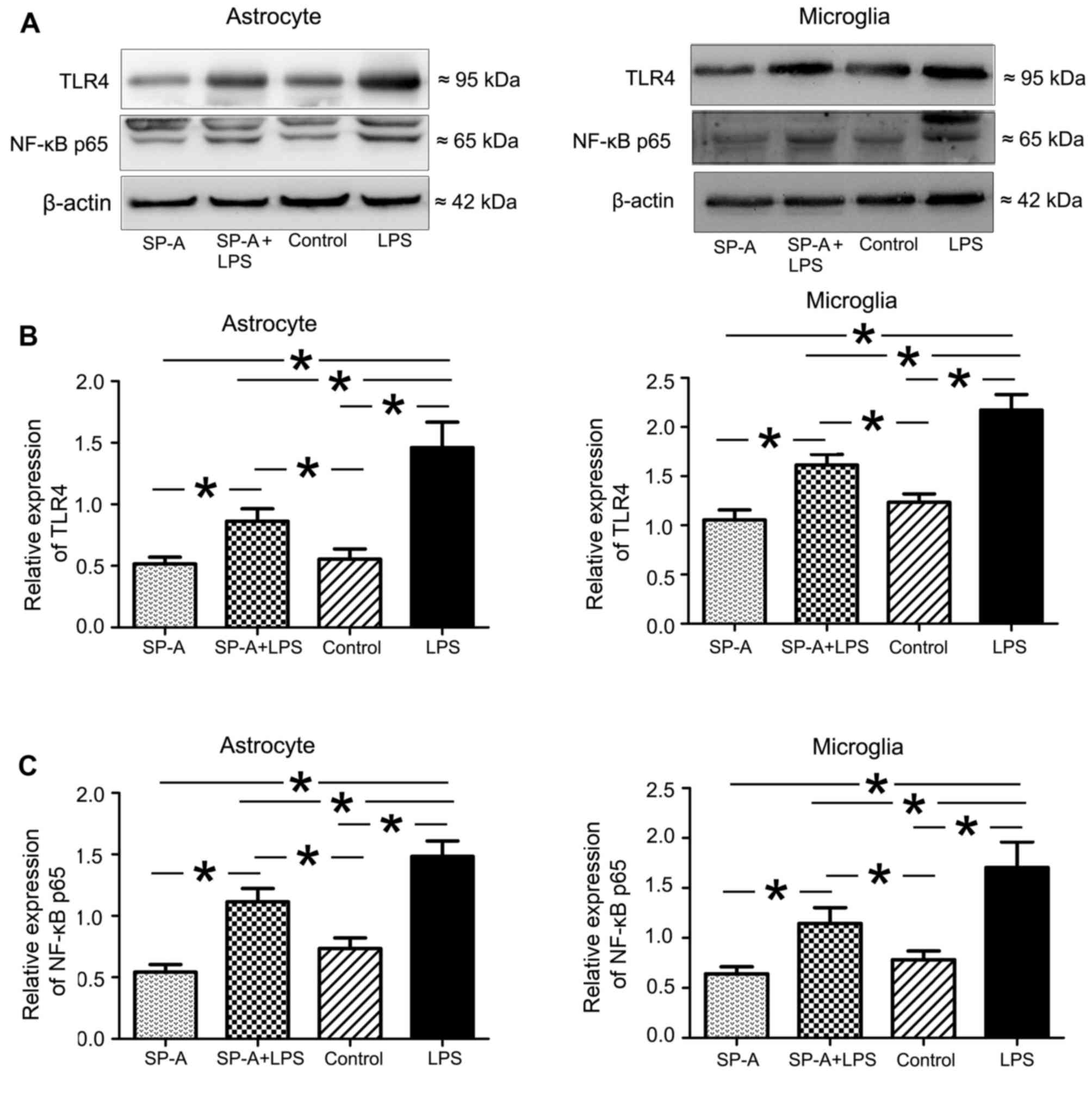
Evaluation of the effect of SP-A on
LPS-induced TLR4/NF-κB expression and TNF-α/IL-1β release
Human astrocytes and microglial cells were divided
into the following four groups: LPS, incubated with 5 µg/ml LPS for
8 h; LPS + SP-A, incubated with 5 µg/ml LPS + 0.5 µg/ml SP-A for 8
h; SP-A, incubated with 0.5 µg/ml SP-A for 8 h; and control,
incubated with normal culture medium. Cells and culture media were
collected separately for the analysis of TLR4/NF-κB proteins by
western blotting, and TNF-α/IL-1β levels by ELISA.
Protein preparation and western
blotting
Proteins from CNS tissues and cell cultures were
extracted using radioimmunoprecipitation assay lysis buffer (cat.
no. sc-24948; Santa Cruz Biotechnology, Inc.) and 10 mg/ml
phenylmethanesulfonyl fluoride (Sigma-Aldrich; Merck KGaA) for 15
min. Homogenates were centrifuged at 15,000 × g for 30 min at 4°C,
and the supernatant was collected. Protein concentrations were
quantified with the BCA assay (Novagen; Merck Millipore). Protein
samples (30–50 µg/well) were subjected to 10% SDS-PAGE and
transferred to PVDF membranes (EMD Millipore, Billerica, MA, USA).
Subsequent to blocking with 5% skimmed milk solution at room
temperature for 2 h, membranes were incubated with rabbit SP-A
polyclonal antibody (1:200; cat. no. sc-13977; Santa Cruz
Biotechnology, Inc.), rabbit TLR4 polyclonal antibody (1:200; cat.
no. sc-10741; Santa Cruz Biotechnology, Inc.), rabbit NF-κB
polyclonal antibody (1:200; cat. no. sc-109, Santa Cruz
Biotechnology, Inc.) or mouse β-actin monoclonal antibody (1:1,000;
cat. no. sc-69879, Santa Cruz Biotechnology, Inc.) overnight at
4°C, followed by peroxidase-conjugated goat anti-rabbit or
anti-mouse secondary antibody (1:2,000; cat. nos. TA130023 and
130004, respectively; OriGene Technologies, Inc., Beijing, China)
at room temperature for 2 h. Immunoproducts were detected by
chemiluminescence with Luminol reagent (cat. no. sc-2048; Santa
Cruz Biotechnology, Inc.) and analyzed using an electrophoresis gel
imaging analysis system (MF-ChemiBIS 3.2; DNR Bio-Imaging Systems,
Ltd., Jerusalem, Israel). Subsequently, densitometry of the bands
was semi-quantitatively analyzed (n=9 per group) using Image-Pro
Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA) and
calculated relative to β-actin control.
ELISA analysis of TNF-α and IL-1β
release
Culture medium from the 4 groups was centrifuged at
15,000 × g for 30 min to remove debris. The supernatant was
collected and used to analyze TNF-α and IL-1β levels using matched
ELISA kits (cat. nos. DTA00C and DLB50, respectively; R&D
systems, Inc., Minneapolis, MN, USA). Concentrations were
quantified relative to standard curves, according to the
manufacturer's instructions.
Statistical analysis
All data were analyzed using GraphPad Prism 6.0
software (GraphPad Software, Inc., La Jolla, CA, USA) and presented
as the mean ± standard deviation. Differences among groups were
analyzed by one-way analysis of variance, followed by the least
significant difference test or Kruskal-Wallis test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Detection of SP-A in healthy rat CNS
tissues
SP-A was detected in brain and spinal cord tissue
sections from normal rats via immunostaining. Immunohistochemical
results demonstrated widespread distribution of SP-A. In the
cortex, SP-A was demonstrated to be expressed on glial cells rather
than neurons and in positively stained cells the staining intensity
was higher in the nuclei than in the cytoplasm (Fig. 1A). SP-A was also expressed in the
cytoplasm and granular structures of hippocampal neurons. Strong
immunoreactivity for SP-A was detected in the cytoplasm of
ependymocytes of the outer layer of choroid plexus. SP-A was also
present in Purkinje cells of the cerebellar cortex. In a vertical
section of brain stem and horizontal section of spinal cord, the
nuclei and cytoplasm of specific cells, presumably glia, exhibited
immunoreactivity for SP-A. Lung tissue sections, used as a positive
control, exhibited positive immunoreactivity, and CNS sections
incubated with only secondary antibody, used as the negative
control, exhibited no immunoreactivity (Fig. 1A). Proteins isolated from spinal
cord, choroid plexus, cerebellum, hippocampus, cortex and brain
stem tissue were analyzed by western blotting to investigate the
protein expression of SP-A, with lung tissue used as the positive
control. SP-A bands from western blot analysis of different rat CNS
samples are presented in Fig. 1B.
The results demonstrated that choroid plexus, hippocampus and brain
stem tissues expressed higher levels of SP-A protein compared with
the other tissues in the rat CNS (Fig.
1C).
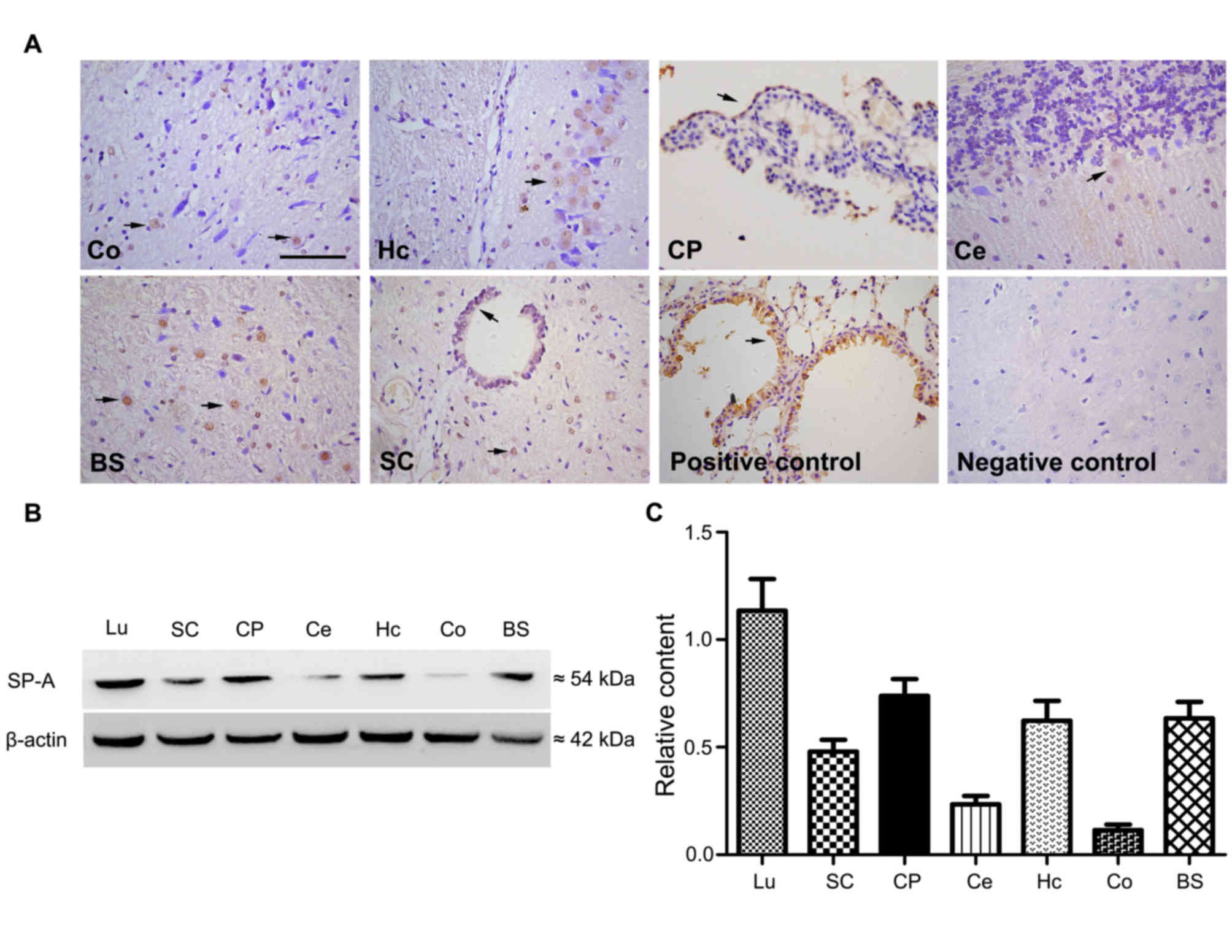 | Figure 1.Detection of SP-A in rat CNS tissues
by immunohistochemistry and western blotting. (A) Expression of
SP-A in normal rat CNS was detected with immunohistochemistry at
magnification ×400. Sections from the Co, Hc, CP, Ce, BS and SC
were analyzed. Lu sections from normal rats were stained as a
positive control and negative control sections of normal rat brains
were stained with secondary antibody only. Arrows indicate the
location of SP-A expressing cells. (B) Proteins isolated from SC,
CP, Hc, Co and BS were detected with western blot analysis, Lu
served as a positive control. (C) Western blot quantitative
analysis was performed and levels were normalized relative to
β-actin. Data is presented as the mean ± standard deviation. Co,
cortex; Hc, hippocampus; CP, choroid plexus; Ce, cerebellum; BS,
brain stem; SC, spinal cord; Lu, lung; SP-A, surfactant protein-A;
CNS, central nervous system. |
H&E staining and changes in the
expression of SP-A, GFAP, Iba-1 and CD11b in the initial, peak and
recovery stages of EAE
As immunoreactivity for SP-A was detected in healthy
rat CNS sections, the present study also investigated SP-A
expression patterns at different stages of EAE. Parallel sections
were stained for inflammation (H&E), astrocytes (GFAP),
microglia and monocytes (Iba-1), and also a general marker for
inflammatory cells (CD11b), which allowed changes in SP-A
expression and the degree of inflammation during EAE to be
analyzed. Rats under normal conditions, with no infiltrating
inflammatory cells in the brain detected by HE staining and normal
microglial/leukocyte CD11b expression, were used as the control.
GFAP-positive astrocytes and Iba-1 positive microglia were evenly
distributed in the brain with normal morphology(Fig. 2A). At the initial stage of EAE
(days 9–11), H&E staining revealed a small number of scattered
infiltrating cells around the vessels. The pattern of GFAP-positive
astrocytes was similar in the initial stage of EAE compared with
the pattern in normal conditions, while Iba-1-positive microglia
exhibited a modified rod shape and increased numbers in the initial
stage of EAE compared with normal conditions, which was associated
with inflammatory cell aggregation. Expression of CD11b was
increased in peri-foci during the initial stage compared with
normal conditions. In the initial stage of EAE, SP-A levels were
marginally increased compared with normal conditions. Notably,
immunoreactive SP-A aggregated to inflammatory foci, which is
evident from immunostaining (Fig.
2A).
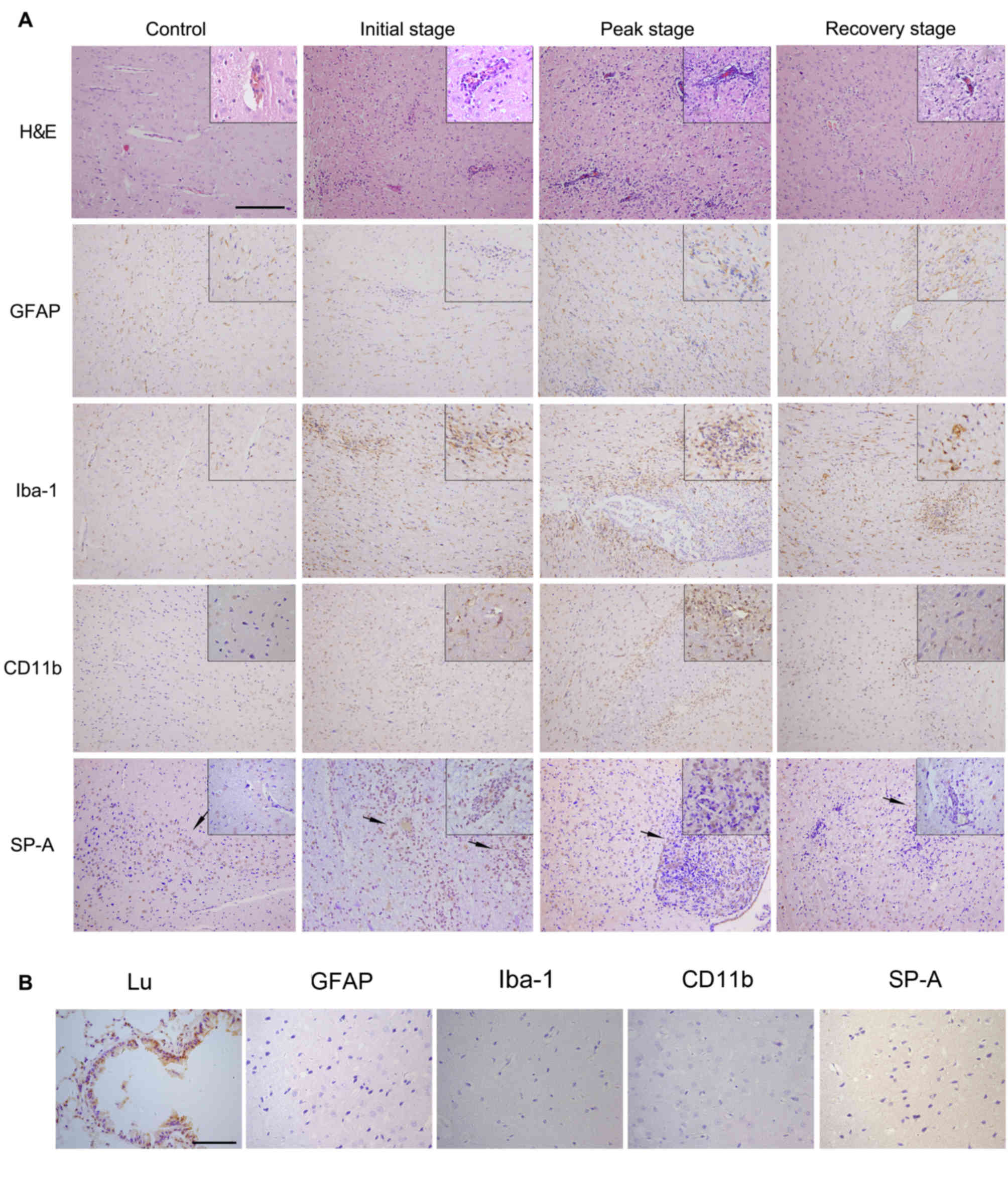 | Figure 2.H&E staining and
immunohistochemical staining of GFAP, Iba-1, CD11b and SP-A in the
rat CNS at different stages of EAE. (A) CNS sections at initial,
peak and recovery stages of EAE were stained for inflammation by
H&E, GFAP-positive astrocytes, Iba-1-postive microglia, CD11b
general marker of inflammatory cells and SP-A. Whole image
magnification, ×200 and corner image magnification, ×400. Section
of rats under normal conditions served as the control. Arrows
indicate the location of SP-A expressing cells. (B) Lu sections of
normal rats were stained as a positive control. Negative control
sections of normal rat CNS were treated with secondary antibody
only and were unstained for GFAP, Iba-1, CD11b and SP-A.
Magnification, ×400. H&E, hematoxylin and eosin; GFAP, glial
fibrillary acidic protein; Iba-1, ionized calcium-binding adapter
molecule 1; SP-A, surfactant protein-A; Lu, lung; CNS, central
nervous system; EAE, experimental autoimmune encephalomyelitis. |
At the peak EAE stage (days 15 and 17), large-scale
inflammatory cell aggregation and numerous perivascular cuffs were
observed in HE-stained sections. Astrocytic reactions occurred
around the EAE lesions with increased staining intensity.
Microglial cells increased in number and were of the activated form
(Fig. 2A). Microglial/leukocyte
CD11b levels were also markedly increased at the peak stage
compared with the initial stage and normal conditions. In CNS
samples in the peak EAE stage, SP-A levels were markedly increased
compared with the initial stage and normal conditions, particularly
within certain severe lesions with huge amounts of inflammatory
cells (Fig. 2A). From day 21
onwards, the recovery stage commenced and the number of
inflammatory cells and EAE lesions observed from H&E staining
was markedly reduced compared with the peak stage. Additionally,
reactive astrocytes and microglia cells decreased in number and
interacted to form glial scars. Positive staining for CD11b was
also decreased, along with SP-A expression in recovery stage CNS
samples compared with peak stage samples. SP-A was distributed far
from lesions, which was also observed in control samples (Fig. 2A). Lung sections of normal rats
were stained as a positive control. Negative control sections of
normal rat CNS were treated with secondary antibody only and were
unstained for GFAP, Iba-1, CD11b and SP-A, as presented in Fig. 2B. Protein levels of the
aforementioned indicators at different EAE stages were assessed by
western blot analysis (Fig. 3A).
Quantification of blots demonstrated gradually increasing
expression of GFAP, Iba-1 and CD11b in the initial and peak stages
compared with control samples, followed by a significant decrease
in expression at the recovery stage compared with the peak stage
(P<0.05; Fig. 3B). Similarly,
SP-A protein expression increased with increasing severity of
inflammation and expression of reactive astrocytes and microglial
cells, and decreased significantly in the recovery stage compared
with initial and peak stages (P<0.05; Fig. 3B).
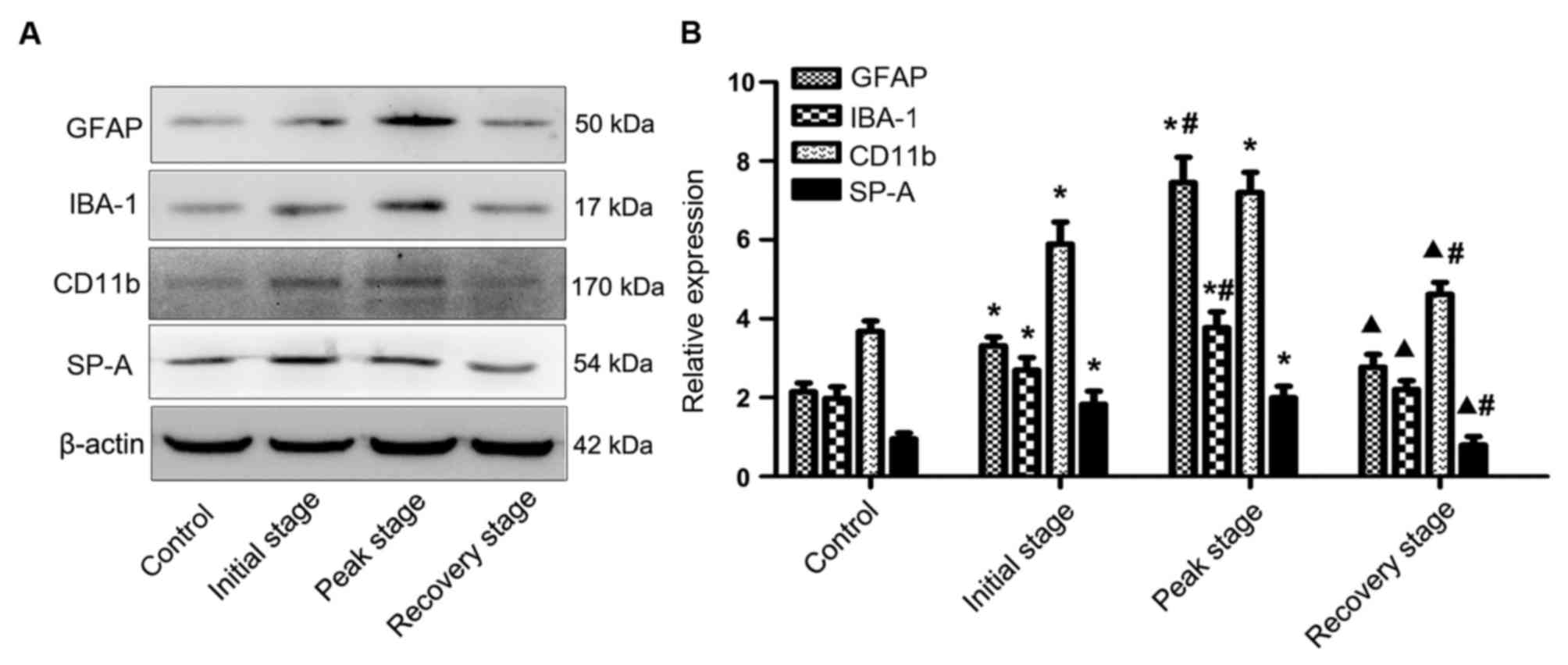 | Figure 3.Western blot analysis of GFAP, Iba-1,
CD11b and SP-A in the rat CNS at different stages of EAE. (A)
Proteins obtained from the rat CNS at the initial, peak and
recovery stages of EAE were detected for GFAP, IBA-1, CD11b and
SP-A via western blotting. (B) Quantitative analysis of western
blots was performed and levels were normalized relative to β-actin.
Results are presented as the mean ± standard deviation. *P<0.05
vs. control group; #P<0.05 vs. initial stage group;
▲P<0.05 vs. peak stage group. GFAP, glial fibrillary
acidic protein; Iba-1, ionized calcium-binding adapter molecule 1;
SP-A, surfactant protein-A; CNS, central nervous system; EAE,
experimental autoimmune encephalomyelitis. |
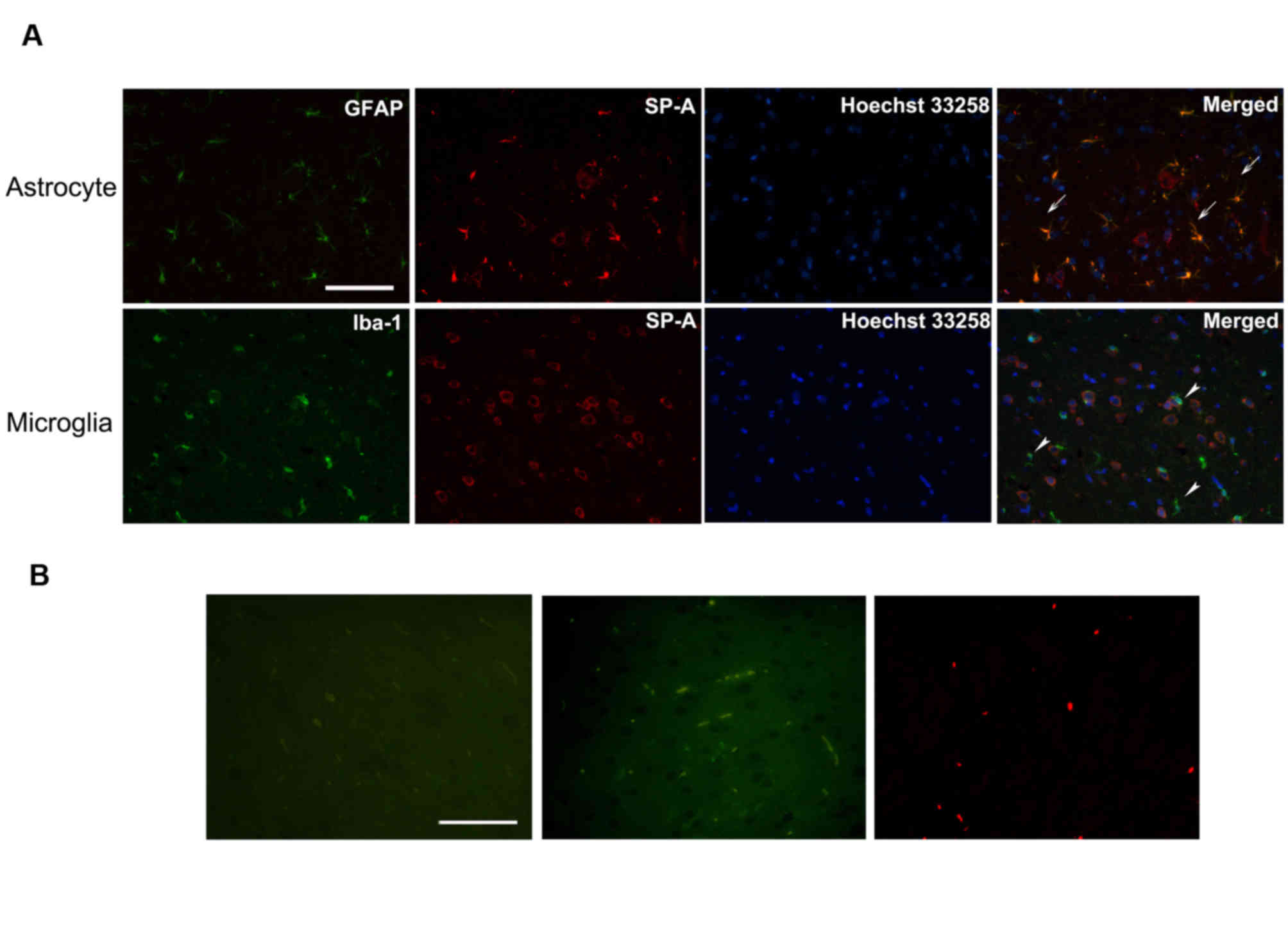
Detection of SP-A in astrocytes and
microglia
To clarify the association between immunoreactive
SP-A, and astrocytes and microglia, the two types of CNS resident
immune cells, the present study detected SP-A expression in
astrocytes and microglia via double immunofluorescence staining of
normal rat CNS sections and immunocytochemical staining of human
astrocyte and microglia cell lines. In rat CNS sections, SP-A was
diffusely distributed in the cytoplasm of astrocytes, but not
microglia (Fig. 4). Additionally,
cytoplasmic and nuclear staining patterns of SP-A in human
astrocytes were observed (Fig.
5A). Fig. 5B presents results
from human astrocytes treated with secondary antibody only as a
negative control. However, immunopositive SP-A was detected in the
cytoplasm of human microglia cells, which contrasted to the results
obtained with rat resident microglia cells (Fig. 5C). Fig. 5D presents results from human
microglia treated with secondary antibody only as a negative
control.
Inflammatory stimuli affect SP-A
protein expression in human astrocytes and microglia
Having demonstrated that SP-A is expressed in human
astrocytes and microglia, the present study subsequently
investigated whether SP-A expression in these cells is affected by
proinflammatory stimuli, such as LPS. Human astrocytes and
microglia were exposed to a range of LPS concentrations, (0–10
µg/ml) for 24 h or 10 µg/ml LPS for 2–24 h, and SP-A protein levels
were investigated by western blotting (Fig. 6A) and quantification was performed
(Fig. 6B). After 24 h of exposure
to 0, 1, 5 or 10 µg/ml LPS, SP-A levels in both cell types
increased in a dose-dependent manner (P<0.05; Fig. 6B). However, application of 10 µg/ml
LPS for 2, 4, 8, 16 and 24 h did not lead to a time-dependent
increase in SP-A levels (Fig.
6C).
SP-A suppresses LPS-induced expression
of TLR4 and NF-κB p65 in human astrocytes and microglia
To determine the potential role of SP-A in the
TLR4/NF-κB signaling pathway, human astrocytes and microglia were
treated with LPS to induce TLR signaling. LPS stimulation for 24 h
induced significantly increased expression of TLR4 and NF-κB p65 in
both cell types compared with the control group (P<0.05;
Fig. 7). SP-A + LPS treatment
induced a significant inhibitory effect on LPS-induced TLR4 and
NF-κB in astrocytes and microglia (P<0.05; Fig. 7). SP-A alone reduced the expression
of TLR4 and NF-κB marginally compared with the control, however
differences were not statistically significant (Fig. 7).
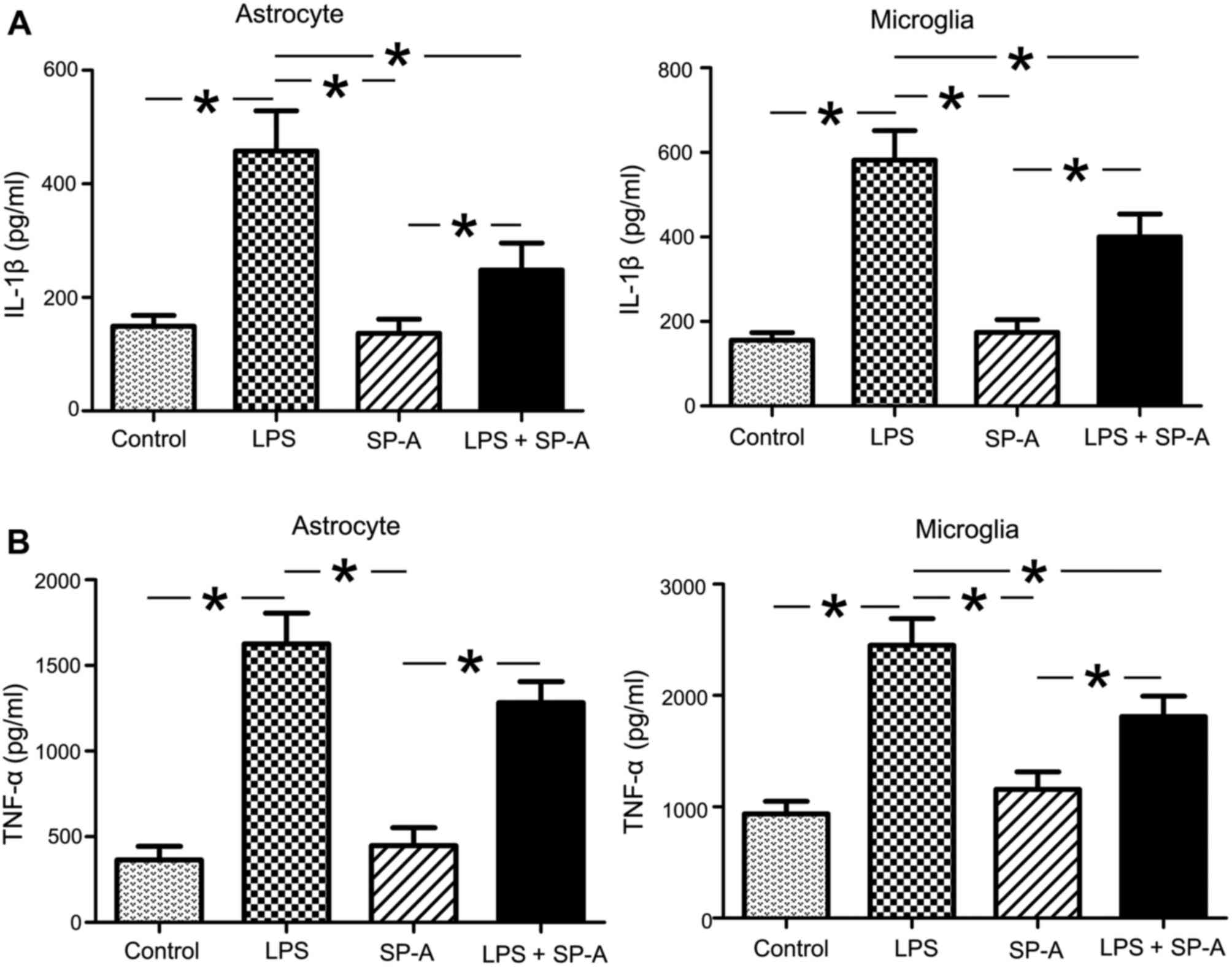
SP-A suppresses the release of
LPS-induced TNF-α and IL-1β
Increased production of proinflammatory cytokines is
associated with the pathogenesis of MS and EAE. Accordingly, the
present study investigated the effects of exogenous SP-A on the
release of TNF-α and IL-1β in LPS-stimulated human astrocytes and
microglia. As presented in Fig. 8,
TNF-α and IL-1β levels in culture medium of both cell types were
significantly increased in LPS groups, compared with the control
groups (P<0.05). SP-A + LPS groups exhibited significantly
decreased TNF-α and IL-1β levels in microglia supernatant, compared
with the LPS groups (P<0.05; Fig.
8). TNF-α and IL-1β levels in the SP-A + LPS group in
astrocytes were reduced compared with the LPS group, however, the
reduction in TNF-α release was not statistically significant
(Fig. 8). SP-A alone did not
significantly reduce the release of TNF-α and IL-1β in astrocytes
and microglia compared with the control (Fig. 8).
Discussion
The present study indicated a potential role for
SP-A in the regulation of inflammatory responses in the CNS. SP-A
was demonstrated to be widely distributed in rat CNS tissues and
was expressed in rat and human astrocytes, and also human
microglia. Changes in the expression of SP-A at different stages of
EAE were also observed. The results of the current study
demonstrated that LPS enhances SP-A protein expression in human
astrocytes and microglial cells in a dose-dependent manner.
Notably, SP-A inhibited LPS-induced expression of TLR4 and NF-κB,
and LPS-induced release of TNF-α and IL-1β. These results indicate
that SP-A may function in the modulation of neuroinflammation,
potentially through its inhibitory effects on TLR4/NF-κB activation
and proinflammatory cytokine release.
The SP-A protein is a hydrophilic,
collagen-containing lectin with a hexameric structure formed by six
trimers of glycoprotein monomers. Characterization studies have
suggested that this potent innate immune molecule performs a
diverse range of functions, including anti-microbial activity,
clearance of apoptotic and necrotic cells and control of
inflammation triggered by self or non-self (22). Initially identified in the alveolar
surface of the lung, SP-A was subsequently demonstrated to be
widely distributed in other tissues (23), such as brain tissue (10). Schob et al (10) were the first to report an
association between SP-A and CNS pathology by examining the
expression patterns of SP-A in human brains, which revealed
increased SP-A expression in the CSF of patients with autoimmune
infectious diseases and indicated a potential role for SP-A in MS
development. MS is a chronic autoimmune-mediated inflammatory
disease of CNS. A primary mechanism underlying the pathogenesis of
MS is that peripherally activated immune cells infiltrate the CNS,
where they trigger inflammatory reactions and cause myelin damage
(24). Accordingly, the present
study investigated the expression of SP-A in different stages of
EAE, the most widely used animal model of MS. In normal rats, SP-A
was widely distributed in the CNS and expressed on various resident
cells, including hippocampal neurons, cerebellum Purkinje cells,
ependymal cells and certain glial cells, which indicates potential
interactions between the peripheral innate and CNS immune systems.
SP-A protein expression in different stages of the EAE model was
subsequently examined. SP-A expression was increased as EAE
progressed, and expression moved towards the inflammatory foci, as
with reactive microglia cells and astrocytes. As CNS resident
immune cells, astrocytes and microglia are involved in the
modulation of immuno-inflammatory responses in EAE. In the present
study, microglial cells responded to inflammation earlier than
astrocytes. However, levels of SP-A were not markedly increased
until the peak stage, consistent with the changes in expression
observed in astrocytes. To further investigate the association
between SP-A and astrocytes and microglia, expression of SP-A was
detected in rat brain astrocytes and microglia in vivo. SP-A
was demonstrated to be expressed on astrocytes but not microglia in
rat brains. Notably, similar examination of SP-A expression in
human astrocytes and microglia in vitro revealed that both
cell lines endogenously produced and expressed SP-A protein.
Furthermore, human astrocytes and microglia responded to
inflammatory stimulation with LPS through dose-dependent increased
SP-A expression.
In EAE, invading immune cells produce endotoxin and
proinflammatory mediators, which are important triggers of CNS
infection. The severity of EAE at different stages partially
depends on the levels of these pathogenic substances (25). Thus, changes in SP-A expression in
human astrocytes and microglia depend on the amount of LPS, but not
time of exposure to LPS, which is consistent with in vivo
results. Activation of astrocytes and microglia is reported to
cause severe inflammatory insult driven by activation of the TLR4
signaling pathway via release of proinflammatory cytokines
(26–28). As SP-A is an endogenous ligand for
TLR4 (3), we hypothesized that the
protein modulates inflammation through effects on TLR4/NF-κB
signaling in astrocytes and microglia. Consistent with this theory,
SP-A exerted a significant inhibitory effect on LPS-induced
overexpression of TLR4 and NF-κB in both cell types. In human
lungs, SP-A also regulates TLR4 activation and expression in
macrophages and dendritic cells (15,29).
Furthermore, SP-A has been previously reported to inhibit TLR4
signaling, decrease IκBα phosphorylation and nuclear translocation
of p65, which are critical for NF-κB activation, and lead to
diminished TLR ligand-induced TNF-α secretion (15). This effect of SP-A on human
astrocytes and microglia was confirmed in the current study, which
demonstrated that LPS-induced secretion of TNF-α and IL-1β was
suppressed in the presence of SP-A. Based on these results, LPS may
promote the expression of SP-A, which may subsequently participate
in the regulation of LPS-induced TLR4/NF-κB activation and
proinflammatory cytokine release. The ability of SP-A to reduce
levels of proinflammatory cytokines has been demonstrated in
various mammalian diseases. Quintanilla et al (11) demonstrated that SP-A treatment of
experimental necrotizing enterocolitis significantly suppressed
TLR4 expression and reduced TNF-α and IL-1β levels. Retinal SP-A is
also reported to localize on GFAP-reactive astrocytes in mice and
TLR4 ligands upregulate SP-A expression and myeloid differentiation
primary response 88 protein essential for NF-κB signaling (30). Similarly, SP-A has been
demonstrated to significantly inhibit TLR ligand-induced expression
of proinflammatory mediators, including TNF-α and IL-1β, in an
inflammation-induced preterm delivery model (31). The functional mechanism of
SP-A-mediated regulation of CNS inflammation may be complex as SP-A
has been reported to bind LPS and modulate TLR4. In the present
study, SP-A potentially interacts with TLR4 directly to inhibit its
overexpression and activation, alternatively, SP-A may interact
with LPS to prevent it binding to TLR4 (32). Further in vitro studies
using blockers or inhibitors are required to clarify this. SP-A has
also been identified as an endogenous ligand for one or more
receptors, including TLR2, CD14 and glycoprotein 340, each of which
is thought to trigger a proinflammatory signal transduction cascade
(3,33,34).
Thus, LPS-induced increased expression of SP-A in astrocytes and
microglia may partially act on the feedback mechanism to inhibit
activation of inflammatory signaling pathways induced by these
receptors. Notably, these results are consistent with results from
the present study. The involvement of a pulmonary innate immune
protein in the regulation of CNS inflammatory pathology is not
surprising. However, the contribution of SP-A in EAE is remains
unclear and it is important to note that species differences exist
between rats and humans with regard to the immune system. Further
studies of SP-A gene deletion or overexpression in vivo are
therefore required to ascertain the effects of SP-A on the
regulation of CNS inflammation and also MS pathogenesis, including
effects on the clearance of pathogenic immune cells and regulation
of T-cell polarization.
In conclusion, the present study provides
preliminary evidence of SP-A expression in normal and EAE rat CNS,
and human astrocytes and microglia. The results indicate that LPS
induces increased expression of SP-A protein in human astrocytes
and microglia in a dose-dependent manner. Furthermore, SP-A may
regulate inflammation in these cell lines at least partially by
reducing the expression of TLR4 protein and inflammatory cytokine
release, indicating that the immunomodulatory properties of SP-A
may function in neuroinflammatory disease.
Acknowledgements
The study was partially financially supported by
grants from the Liaoning Province Science and Technology
Project-Animal Scientific Research and Clinical Application for
Major Disease of Liaoning Province (grant no. 2012225021), the
Program of Basic and Clinical Research Platform of China Medical
University (grant no. CMU-201406) and the Technology Projects of
Liaoning Province (grant no. 2009225010-2) awarded to Dr Juan
Feng.
Glossary
Abbreviations
Abbreviations:
|
CNS
|
central nervous system
|
|
SP-A
|
surfactant protein-A
|
|
TLR4
|
Toll-like receptor 4
|
|
EAE
|
experimental autoimmune
encephalomyelitis
|
References
|
1
|
Wright JR: Immunoregulatory functions of
surfactant proteins. Nat Rev Immunol. 5:58–68. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chroneos ZC, Midde K, Sever-Chroneos Z and
Jagannath C: Pulmonary surfactant and tuberculosis. Tuberculosis
(Edinb). 89 Suppl 1:S10–S14. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kishore U, Greenhough TJ, Waters P, Shrive
AK, Ghai R, Kamran MF, Bernal AL, Reid KB, Madan T and Chakraborty
T: Surfactant proteins SP-A and SP-D: Structure, function and
receptors. Mol Immunol. 43:1293–1315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schaub B, Westlake RM, He H, Arestides R,
Haley KJ, Campo M, Velasco G, Bellou A, Hawgood S, Poulain FR, et
al: Surfactant protein D deficiency influences allergic immune
responses. Clin Exp Allergy. 34:1819–1826. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hartshorn KL, Crouch E, White MR,
Colamussi ML, Kakkanatt A, Tauber B, Shepherd V and Sastry KN:
Pulmonary surfactant proteins A and D enhance neutrophil uptake of
bacteria. Am J Physiol. 274:L958–L969. 1998.PubMed/NCBI
|
|
6
|
Chuang CY, Chen TL and Chen RM: Molecular
mechanisms of lipopolysaccharide-caused induction of surfactant
protein-A gene expression in human alveolar epithelial A549 cells.
Toxicol Lett. 191:132–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yuan HT, Gowan S, Kelly FJ and Bingle CD:
Cloning of guinea pig surfactant protein A defines a distinct
cellular distribution pattern within the lung. Am J Physiol.
273:L900–L906. 1997.PubMed/NCBI
|
|
8
|
Rubio S, Lacaze-Masmonteil T, Chailley-Heu
B, Kahn A, Bourbon JR and Ducroc R: Pulmonary surfactant protein A
(SP-A) is expressed by epithelial cells of small and large
intestine. J Biol Chem. 270:12162–12169. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Snyder GD, Oberley-Deegan RE, Goss KL,
Romig-Martin SA, Stoll LL, Snyder JM and Weintraub NL: Surfactant
protein D is expressed and modulates inflammatory responses in
human coronary artery smooth muscle cells. Am J Physiol Heart Circ
Physiol. 294:H2053–H2059. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schob S, Schicht M, Sel S, Stiller D,
Kekulé AS, Paulsen F, Maronde E and Bräuer L: The detection of
surfactant proteins a, b, c and d in the human brain and their
regulation in cerebral infarction, autoimmune conditions and
infections of the CNS. PLoS One. 8:e744122013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quintanilla HD, Liu Y, Fatheree NY, Atkins
CL, Hashmi SS, Floros J, McCormack FX, Rhoads JM and Alcorn JL:
Oral administration of surfactant protein-a reduces pathology in an
experimental model of necrotizing enterocolitis. J Pediatr
Gastroenterol Nutr. 60:613–620. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee HM, Kang HJ, Woo JS, Chae SW, Lee SH
and Hwang SJ: Upregulation of surfactant protein a in chronic
rhinosinusitis. Laryngoscope. 116:328–330. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wootten CT, Labadie RF, Chen A and Lane
KF: Differential expression of surfactant protein A in the nasal
mucosa of patients with allergy symptoms. Arch Otolaryngol Head
Neck Surg. 132:1001–1007. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Luo JM, Wan YS, Liu ZQ, Wang GR, Floros J
and Zhou HH: Regularity of distribution of immunoreactive pulmonary
surfactant protein A in rat tissues. Int J Mol Med. 14:343–351.
2004.PubMed/NCBI
|
|
15
|
Henning LN, Azad AK, Parsa KV, Crowther
JE, Tridandapani S and Schlesinger LS: Pulmonary surfactant protein
a regulates TLR expression and activity in human macrophages. J
Immunol. 180:7847–7858. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Duperray A, Barbe D, Raguenez G, Weksler
BB, Romero IA, Couraud PO, Perron H and Marche PN: Inflammatory
response of endothelial cells to an endogenous retrovirus
associated to MS is mediated by TLR4. Int Immunol. 27:545–553.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parajuli B, Sonobe Y, Kawanokuchi J, Doi
Y, Noda M, Takeuchi H, Mizuno T and Suzumura A: GM-CSF increases
LPS-induced production of proinflammatory mediators via
upregulation of TLR4 and CD14 in murine microglia. J
Neuroinflammation. 9:2682012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu Y, Li W and Zhou C, Lu F, Gao T, Liu Y,
Cao J, Zhang Y, Zhang Y and Zhou C: Ketamine inhibits
lipopolysaccharide-induced astrocytes activation by suppressing
TLR4/NF-ĸB pathway. Cell Physiol Biochem. 30:609–617. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur
C and Ling EA: Toll-like receptor 4 mediates microglial activation
and production of inflammatory mediators in neonatal rat brain
following hypoxia: Role of TLR4 in hypoxic microglia. J
Neuroinflammation. 10:232013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blanco AM, Vallés SL, Pascual M and Guerri
C: Involvement of TLR4/type IIL-1 receptor signaling in the
induction of inflammatory mediators and cell death induced by
ethanol in cultured astrocytes. J Immunol. 175:6893–6899. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mohajeri M, Sadeghizadeh M and Javan M:
Pertussis toxin promotes relapsing-remitting experimental
autoimmune encephalomyelitis in Lewis rats. J Neuroinflammation.
289:105–110. 2015.
|
|
22
|
Nayak A, Dodagatta-Marri E, Tsolaki AG and
Kishore U: An insight into the diverse roles of surfactant
proteins, SP-A and SP-D in innate and adaptive immunity. Frontiers
in immunology. 3:1312012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Hu F, Wang G, Zhou Q and Ding G:
Lipopolysaccharide- induced expression of surfactant proteins A1
and A2 in human renal tubular epithelial cells. J Inflamm (Lond).
10:22013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Furlan R, Cuomo C and Martino G: Animal
models of multiple sclerosis. Methods Mol Biol. 549:157–173. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Files DK, Jausurawong T, Katrajian R and
Danoff R: Multiple Sclerosis. Primary Care. 42:159–175. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsumoto Y, Ohmori K and Fujiwara M:
Microglial and astroglial reactions to inflammatory lesions of
experimental autoimmune encephalomyelitis in the rat central
nervous system. J Neuroimmunol. 37:23–33. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Napoli I and Neumann H: Protective effects
of microglia in multiple sclerosis. Exp Neurol. 225:24–28. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lundgaard I, Osório MJ, Kress BT,
Sanggaard S and Nedergaard M: White matter astrocytes in health and
disease. Neuroscience. 276:161–173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Awasthi S, Madhusoodhanan R and Wolf R:
Surfactant protein-A and toll-like receptor-4 modulate immune
functions of preterm baboon lung dendritic cell precursor cells.
Cell Immunol. 268:87–96. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhatti F, Ball G, Hobbs R, Linens A,
Munzar S, Akram R, Barber AJ, Anderson M, Elliott M and Edwards M:
Pulmonary surfactant protein a is expressed in mouse retina by
muller cells and impacts neovascularization in oxygen-induced
retinopathy. Invest Ophthalmol Vis Sci. 56:232–242. 2015.
View Article : Google Scholar :
|
|
31
|
Agrawal V, Smart K, Jilling T and Hirsch
E: Surfactant Protein (SP)-A suppresses preterm delivery and
inflammation via TLR2. PLoS One. 8:e6399902013. View Article : Google Scholar
|
|
32
|
Kawai T and Akira S: Pathogen recognition
with Toll-like receptors. Curr Opin Immunol. 17:338–344. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Holmskov U, Lawson P, Teisner B, Tornoe I,
Willis AC, Morgan C, Koch C and Reid KB: Isolation and
characterization of a new member of the scavenger receptor
superfamily, glycoprotein-340 (gp-340), as a lung surfactant
protein-D binding molecule. J Biol Chem. 272:13743–13749. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sano H, Sohma H, Muta T, Nomura S, Voelker
DR and Kuroki Y: Pulmonary surfactant protein A modulates the
cellular response to smooth and rough lipopolysaccharides by
interaction with CD14. J Immunol. 163:387–395. 1999.PubMed/NCBI
|