Introduction
Systemic vasculitis represents a heterogeneous group
of disorders characterized by inflammation and necrosis in the
blood vessel wall involving multiple organs (1). Clinical manifestations of systemic
vasculitis are nonspecific and may vary from mild disorders to
life-threatening multisystem conditions (2). Cases of vasculitis exhibit
differences in pathology, type of inflammation, vessel and organ
involvement, and demographics (3).
Therefore, the diagnosis and treatment of systemic vasculitis is
challenging, and improvements in therapy and the understanding of
the etiopathogenesis are required. Although the etiology of
systemic vasculitis is not fully understood, there are a
combination of genetic, immunological and environmental factors
that may be responsible for certain cases. Accumulating evidence
has demonstrated that genetic factors contribute to the
susceptibility to vasculitis (4).
Anti-neutrophil cytoplasmic antibody-associated vasculitis (AAV) is
a necrotizing group of disorders characterized by autoimmune
inflammation that predominantly affects small to medium-sized
vessels, which can lead to vessel occlusion and systemic organ
damage (5). Ordonez et al
(6) reported that
CD45RClow CD4 T cells were significantly increased in
patients with AAV compared with healthy controls, which may
contribute to the susceptibility to AAV. Additionally, Kobayashi
et al (7) demonstrated that
putative gene markers, particularly early growth response 1 and
G0/G1 switch gene 2, may be useful for diagnosing vasculitis, and
monocytes expressing these vasculitis-upregulated genes may be
involved in the pathogenesis of vasculitis. However, the molecular
mechanism underlying the development and progression of systemic
vasculitis remains unclear. Continued investigation and
identification of the genetic factors involved in the pathogenesis
of systemic vasculitis is necessary.
Dual-color cDNA microarray data was used in the
present study to identify differentially expressed genes (DEGs) in
samples from patients with systemic vasculitis compared with
healthy controls. Comprehensive bioinformatics were conducted to
analyze the significant gene ontology (GO) terms and pathways that
the DEGs were involved in. This was followed by the construction of
a protein-protein interaction (PPI) network and significant
functional interaction (FI) module selection. Furthermore,
transcriptional factors (TFs) among the DEGs were predicted and,
subsequently, a transcriptional regulation network was constructed.
The current study aimed to identify the involvement of critical
genes in systemic vasculitis, to obtain an improved understanding
of the molecular circuity in systemic vasculitis and to investigate
novel potential gene targets for systemic vasculitis treatment.
Materials and methods
Data source
The dual-color cDNA microarray data GSE16945
(8), was downloaded from the Gene
Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) based on the
platform of GPL4133 Agilent-014850 Whole Human Genome Microarray
4×44K G4112F (feature number version). This dual-color microarray
dataset consisted of details of 13 microarray chips. Each chip
included a cyanine (Cy) 3-labelled channel derived from mixed
peripheral mononuclear blood cells (PMBCs) from 16 healthy
volunteers as controls and a Cy5-labelled channel derived from
PMBCs from a patient with systemic vasculitis. A total of 13
patients with vasculitis were included in the present study.
Data preprocessing and DEG
screening
Raw data were imported into BRB ArrayTools
(http://linus.nci.nih.gov/BRB-ArrayTools.html; version
4.3.1) developed by Simon et al (9). The preprocessing step included local
background subtraction, averaging of intensities of duplicated
probes and quantile normalization across multiple arrays. DEG
screening was performed via the BRB ArrayTools package (9). Genes were excluded when <50% of
the expression data had >1.5-fold change in either direction
from the median value of the gene. Genes were also excluded when
the percentage of data filtered out or missing was >50%. The
threshold for a DEG was P<0.05 using multivariate permutation
tests as previously described (10).
Functional and pathway enrichment
analysis
The clusterProfiler package is implemented in R,
which is an open-source programming environment (11) and this package automates the
process of biological-term enrichment analysis of gene clusters
(12). In the present study, the
clusterProfiler package version 3.4 (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
was applied to perform GO analysis (including cellular composition,
molecular function and biological process terms) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analysis. False discovery rate (FDR) (13) was performed to adjust P-values
using the Benjamini and Hochberg method (14). An FDR <0.05 was selected as the
cutoff criterion.
PPI network construction
A PPI network was constructed in the present study
using the Search Tool for the Retrieval of Interacting Genes
database (15). Interacting pairs
of DEGs (confidence score >0.4) (16) were selected for PPI network
construction. Additionally, Cytoscape software version 2.8
(17) was used to provide
interactive visualization for the PPI network. The node degrees in
the PPI network were calculated with the igraph package version
0.5.3 (18) in R and the nodes
with higher degrees were considered to be hub proteins.
FI network construction and modules
selection
ReactomeFIViz (19), a Cytoscape plugin, integrates
constructed human protein FI networks by combining curated and
non-curated data sources, and the gene expression matrix, to
calculate the Pearson coefficient of genes (20) in an FI network. In the FI network,
the Pearson coefficient of each pair of genes were considered as
edge weights. In addition, ReactomeFIViz further divides the FI
network into modules using a popular algorithm, Markov Clustering
(MCL) (21). In the current study,
a gene FI network was constructed using ReactomeFIViz (19) and module division was performed.
Furthermore, Gene Cluster with Literature Profiles (GenCLiP)
software version 2.0, which was developed by Huang et al
(22) to cluster gene lists by
literature profiling and to construct gene co-occurrence networks
associated with custom keywords, was used to analyze the biological
behavior of genes identified in the selected modules.
Prediction of TFs and transcriptional
regulation network construction
TRANSFAC is a database on TFs, which contains the
genomic binding sites of TFs and DNA-binding profiles, and is an
integrated system for gene expression regulation (23). TFs among the identified DEGs were
screened for using data derived from TRANSFAC database in the
present study. Candidate binding sites for the identified TFs in
the promoter region were also identified through sequence matching
of the position weight matrix (24) by using the MotifDb R package
(25). The location of the TF
binding site (TFBS) within the promoter region of each DEG was
predicted using the position weight matrix algorithm, in which a
minimum score for a match was set at 85%. Subsequently, a
transcriptional regulation network was constructed that included
the identified TFs and other DEGs.
Results
Screening of DEGs
A total of 266 DEGs were identified in PMBCs from
patients with systemic vasculitis compared with controls, including
173 up- and 93 downregulated genes. The results demonstrated that
there were more upregulated genes than downregulated genes in
patients with vasculitis compared with controls.
GO and pathway enrichment
analysis
GO and pathway analysis demonstrated that up- and
downregulated genes were associated with different GO terms and
pathways. The top five GO terms in each category of the up- and
downregulated genes are presented in Table I. Additionally, the top five KEGG
pathways associated with the up- and downregulated genes are
presented in Table II. The
results of the present study demonstrated that upregulated DEGs
were primarily involved in biological processes associated with
defense response and response to stress or stimulus. However,
downregulated DEGs were enriched in biological processes associated
with immune responses, such as the innate immune response.
 | Table I.Top five enriched BP, CC, and MF GO
terms for up- and downregulated genes in patients with systemic
vasculitis. |
Table I.
Top five enriched BP, CC, and MF GO
terms for up- and downregulated genes in patients with systemic
vasculitis.
| GO ID | Description | P-value | False discovery
rate | Count |
|---|
| Upregulated |
|
|
|
|
| GO_BP
0006950 | Response to
stress | 2.35E-12 | 2.40E-09 | 65 |
| GO_BP
0002376 | Immune system
process | 6.88E-12 | 2.90E-09 | 51 |
| GO_BP
0006952 | Defense
response | 8.52E-12 | 2.90E-09 | 40 |
| GO_BP
0006954 | Inflammatory
response | 3.24E-10 | 8.28E-08 | 23 |
| GO_BP
0050896 | Response to
stimulus | 9.57E-10 | 1.96E-07 | 101 |
| GO_CC
0031982 | Vesicle | 7.96E-09 | 6.77E-07 | 58 |
| GO_CC
0044421 | extracellular
region part | 1.32E-08 | 6.77E-07 | 59 |
| GO_CC
0005833 | Hemoglobin
complex | 1.51E-08 | 6.77E-07 |
5 |
| GO_CC
0005615 | Extracellular
space | 3.96E-08 | 1.33E-06 | 30 |
| GO_CC
0031988 | Membrane-bounded
vesicle | 1.69E-07 | 3.80E-06 | 54 |
| GO_MF
0003674 | Molecular
function | 3.42E-10 | 4.82E-08 | 146 |
| GO_MF
0005515 | Protein
binding | 3.00E-08 | 1.33E-06 | 100 |
| GO_MF
0005488 | binding | 3.11E-08 | 1.33E-06 | 129 |
| GO_MF
0005344 | Oxygen transporter
activity | 3.76E-08 | 1.33E-06 |
5 |
| GO_MF
0005506 | Iron ion
binding | 8.50E-06 | 0.000212228 |
9 |
| Downregulated |
|
|
|
|
| GO_BP
0006952 | Defense
response | 2.30E-16 | 8.58E-14 | 34 |
| GO_BP
0006955 | Immune
response | 2.46E-16 | 8.58E-14 | 33 |
| GO_BP
0045087 | Innate immune
response | 4.35E-14 | 1.01E-11 | 25 |
| GO_BP
0002376 | Immune system
process | 7.59E-14 | 1.32E-11 | 38 |
| GO_BP
0051607 | Defense response to
virus | 4.92E-13 | 6.86E-11 | 14 |
| GO_CC
0044444 | Cytoplasmic
part | 1.86E-06 | 0.000161279 | 55 |
| GO_CC
0005737 | Cytoplasm | 2.56E-06 | 0.000161279 | 66 |
| GO_CC
0042611 | MHC protein
complex | 7.12E-06 | 0.000298999 |
4 |
| GO_CC
0071556 | Integral component
of lumenal side of endoplasmic reticulum membrane | 0.000294636 | 0.005946701 |
3 |
| GO_CC
0098553 | Lumenal side of
endoplasmic reticulum membrane | 0.000294636 | 0.005946701 |
3 |
| GO_MF
0003674 | Molecular
function | 3.66E-06 | 0.00019454 | 84 |
| GO_MF
0003823 | Antigen
binding | 4.42E-06 | 0.00019454 |
6 |
| GO_MF
0005515 | Protein
binding | 1.59E-05 | 0.000466985 | 58 |
| GO_MF
0003924 | GTPase
activity | 6.22E-05 | 0.001368961 |
7 |
| GO_MF
0005525 | GTP binding | 0.000282526 | 0.003941692 |
8 |
| Table II.Enriched KEGG pathways for up- and
downregulated genes in patients with systemic vasculitis. |
Table II.
Enriched KEGG pathways for up- and
downregulated genes in patients with systemic vasculitis.
| KEGG ID | Description | P-value | False discovery
rate | Count |
|---|
| Upregulated |
|
|
|
|
|
hsa05130 | Pathogenic
Escherichia coli infection | 0.000614223 | 0.013512902 | 5 |
|
hsa03010 | Ribosome | 0.004807379 | 0.035254116 | 5 |
|
hsa05323 | Rheumatoid
arthritis | 0.004807379 | 0.035254116 | 5 |
|
hsa05143 | African
trypanosomiasis | 0.008320256 | 0.038297864 | 3 |
|
hsa05146 | Amoebiasis | 0.008704060 | 0.038297864 | 5 |
| Downregulated |
|
|
|
|
|
|
hsa05330 | Allograft
rejection | 0.000139043 | 0.001547821 | 4 |
|
hsa05332 | Graft-versus-host
disease | 0.000204470 | 0.001547821 | 4 |
|
hsa04940 | Type I diabetes
mellitus | 0.000244393 | 0.001547821 | 4 |
|
hsa05320 | Autoimmune thyroid
disease | 0.000495821 | 0.002355147 | 4 |
|
hsa04145 | Phagosome | 0.000652198 | 0.002478353 | 6 |
PPI network construction and
analysis
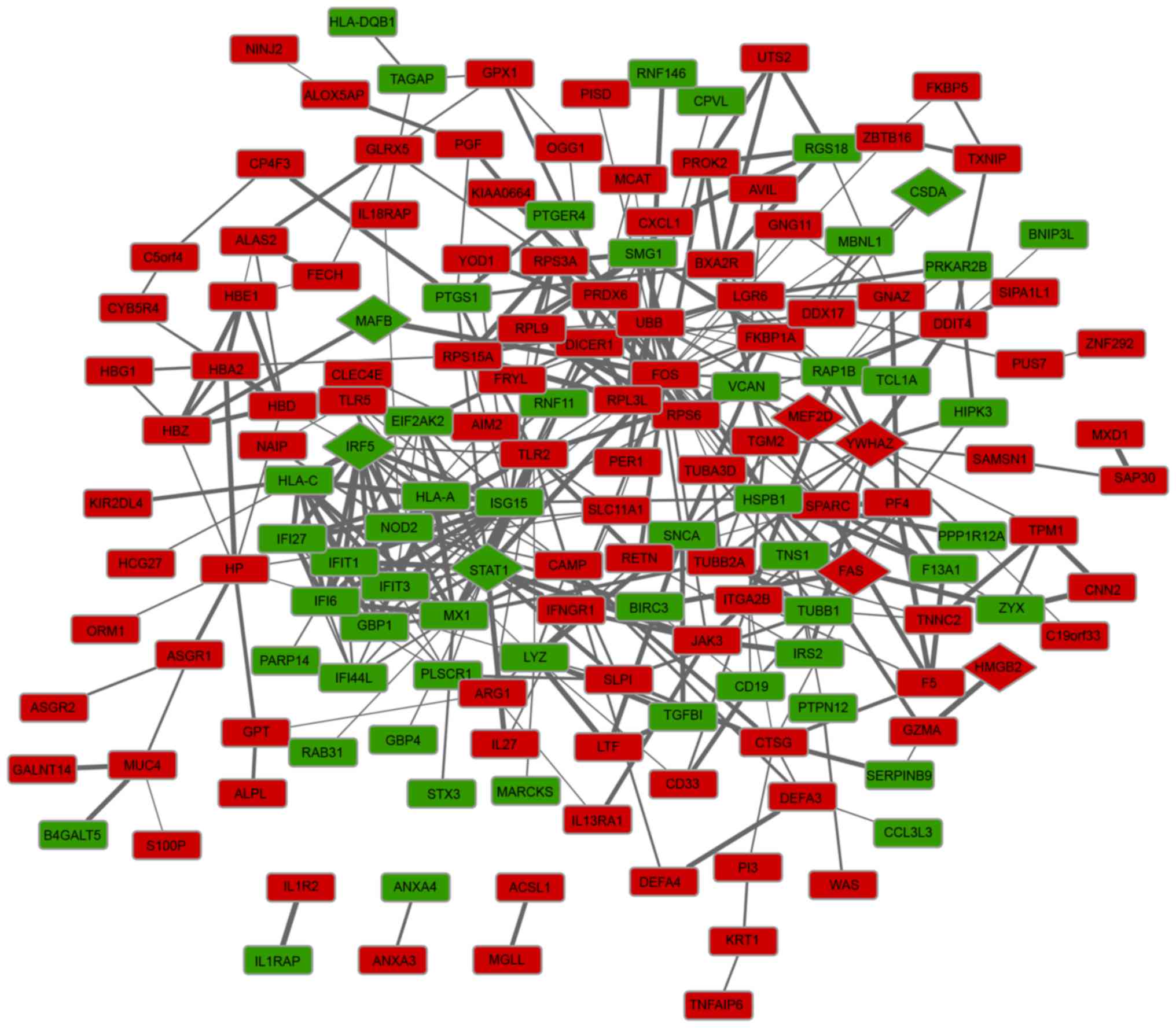
The PPI network was constructed and is presented in
Fig. 1. The PPI network included
163 nodes and 449 interactions, involving 107 up- and 56
downregulated genes. Following the calculation of node degrees, the
present study identified that the top five DEGs in the PPI network,
with higher node degrees, were FBJ murine osteosarcoma viral
oncogene homolog (FOS; degree, 23), ubiquitin B (UBB;
degree, 22), ISG15 ubiquitin-like modifier (degree, 22), signal
transducer and activator of transcription 1 (STAT1; degree,
21) and MX dynamin-like GTPase 1 (MX1; degree, 17).
Furthermore, a total of nine TFs that were involved in the PPI
network were predicted, including cold shock domain protein A, Fas
cell surface death receptor, high mobility group box 2, interferon
regulatory factor 5, v-maf avian musculoaponeurotic
fibrosarcoma oncogene homolog B (MAFB), myocyte enhancer
factor 2D (MEF2D), POU class 2 associating factor 1,
STAT1, tyrosine 3-monooxygenase/tryptophan 5-monooxygenase
activation protein Z (YWHAZ).
Module selection in the FI network and
function analysis of DEGs in the modules
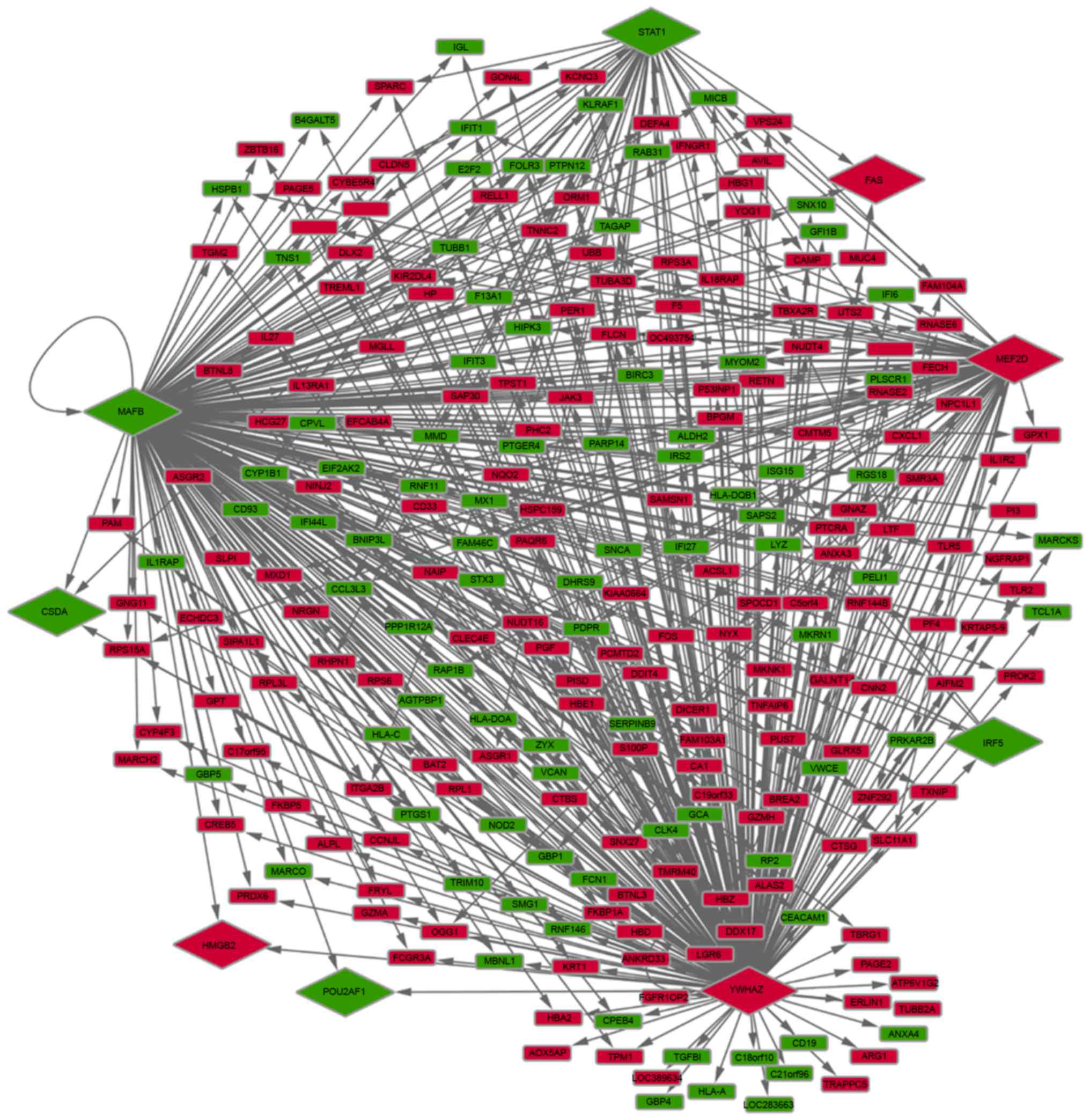
The FI network of DEGs was constructed using the
ReactomeFIViz plugin. Furthermore, three significant modules were
identified using the MCL algorithm (Fig. 2). UBB, FOS and
STAT1 were identified as hub proteins for one of the three
identified modules. In addition, these three DEGs interacted with
each other, and FOS and STAT1 were identified as TFs
following literature profiling. As presented in Fig. 2, there were seven, nine and seven
DEGs enriched in modules 1, 2 and 3, respectively. Additionally,
according to the clustered gene lists by literature profiling, it
was identified that the upregulated DEG, UBB, which was the
hub protein in module 1, was associated with cell death and signal
transduction (Fig. 3). While the
hub protein in module 2, FOS (upregulated), as an important
TF, was associated with inflammatory response, signal transduction
and cell migration. Downregulated STAT1, which was the hub
protein in module 3, was identified to be a TF that was associated
with the immune response, inflammatory response, cell death and
Toll-like receptors (TLRs).
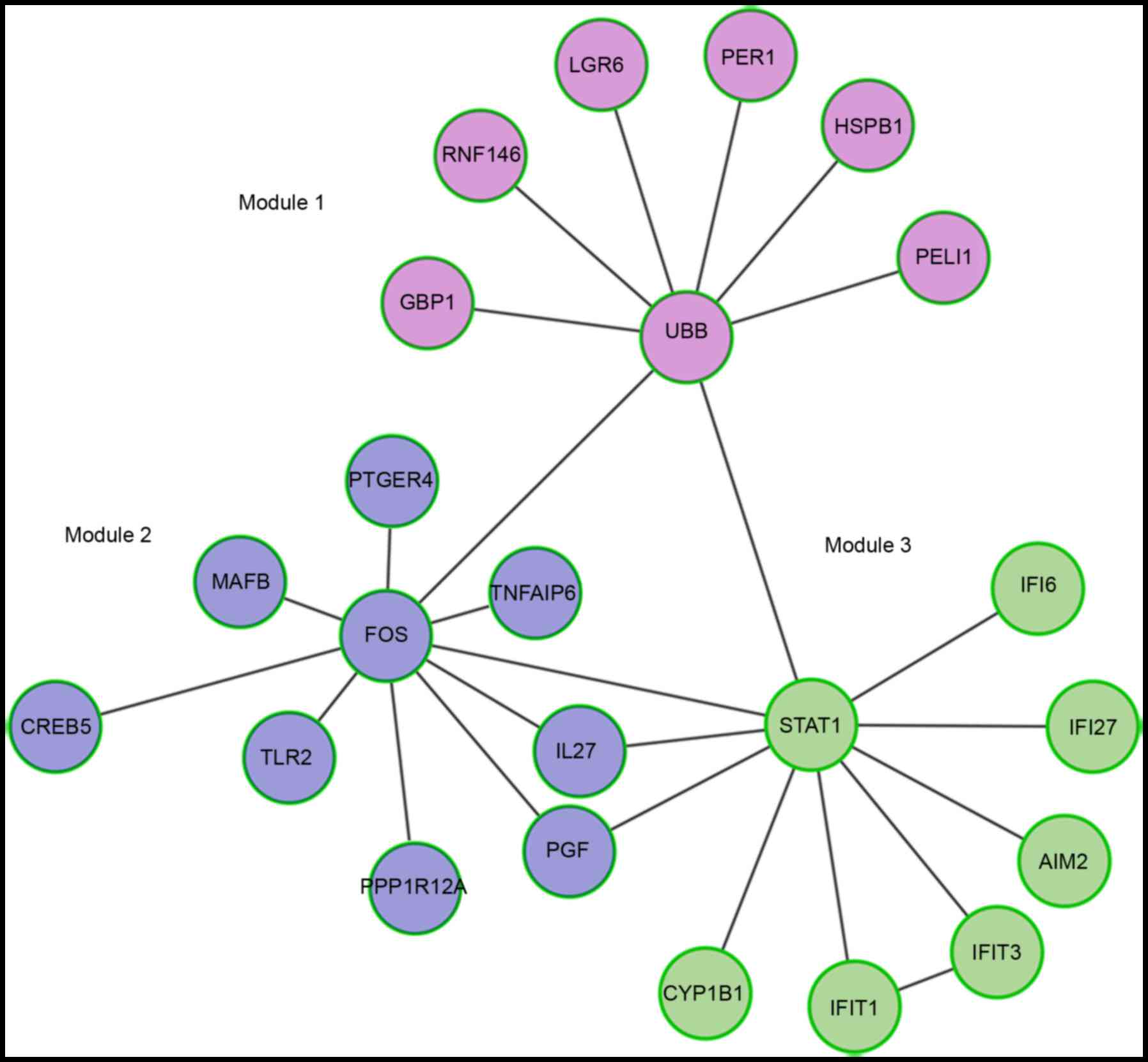 | Figure 2.Functional interaction modules of
differentially expressed genes in patients with systemic vasculitis
compared with controls. GBP1, guanylate binding protein 1; RNF146,
ring finger protein 146; LGR6, leucine rich repeat containing G
protein-coupled receptor 6; PER1, period circadian clock 1; HSPB1,
heat shock protein family B (small) member 1; PELI1, pellino E3
ubiquitin protein ligase 1; UBB, ubiquitin B; PTGER4, prostaglandin
E receptor 4; MAFB, v-maf avian musculoaponeurotic
fibrosarcoma oncogene homolog B; CREB5, cAMP responsive element
binding protein 5; TNFAIP6, TNF α induced protein 6; IL27,
interleukin 27; PGF, placental growth factor; PPP1R12A, protein
phosphatase 1 regulatory subunit 12A; TLR2, Toll-like receptor 2;
FOS, FBJ murine osteosarcoma viral oncogene homolog; IFI,
interferon α inducible protein; AIM2, absent in melanoma 2; IFIT,
interferon induced protein with tetratricopeptide repeats; CYP1B1,
cytochrome P450 family 1 subfamily B member 1; STAT1, signal
transducer and activator of transcription 1. |
 | Figure 3.Gene function in 3 functional
interaction modules identified with the Markov Clustering
algorithm. Green indicates corresponding gene-term association
positively reported. Black represents corresponding gene-term
association not yet reported. UBB, ubiquitin B; HSPB1, heat shock
protein family B (small) member 1; GBP1, guanylate binding protein
1; PER1, period circadian clock 1; LGR6, leucine rich repeat
containing G protein-coupled receptor 6; RNF146, ring finger
protein 146; PELI1, pellino E3 ubiquitin protein ligase 1; CYP1B1,
cytochrome P450 family 1 subfamily B member 1; AIM2, absent in
melanoma 2; IFIT, interferon induced protein with tetratricopeptide
repeats; STAT1, signal transducer and activator of transcription 1;
IFI6, interferon α inducible protein 6; IL27, interleukin 27;
PTGER4, prostaglandin E receptor 4; PPP1R12A, protein phosphatase 1
regulatory subunit 12A; PGF, placental growth factor; FOS, FBJ
murine osteosarcoma viral oncogene homolog; TLR2, Toll-like
receptor 2; TNFAIP6, TNF α induced protein 6; CREB5, cAMP
responsive element binding protein 5; MAFB, v-maf avian
musculoaponeurotic fibrosarcoma oncogene homolog B. |
Construction and analysis of the
transcriptional regulation network
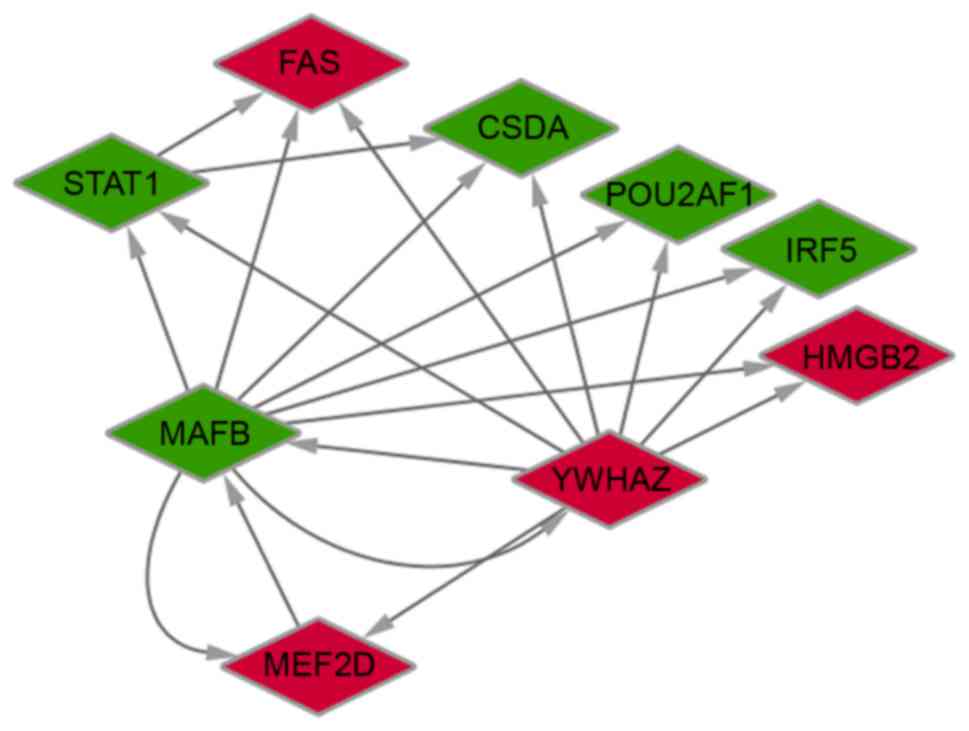
As mentioned above, a total of nine DEGs were
predicted to be TFs based on data derived from the TRANSFAC
database. The present study subsequently predicted all of the
potential target genes, which were also identified as DEGs, of
these nine TFs. The results demonstrated that five TFs potentially
regulated 257 DEGs (167 up- and 90 downregulated genes). In
particular, it was identified that STAT1, MEF2D,
MAFB and YWHAZ potentially regulated further DEGs
(Fig. 4). TFBS were also
identified in pairs of TFs. The interactions among the nine
identified TFs are presented in Fig.
5. Furthermore, genes that were coregulated by STAT1,
MEF2D, MAFB, and YWHAZ were analyzed. The
results indicated that TFs STAT1 and MAFB may
coregulate 39 target genes, while MEF2D and YWHAZ may
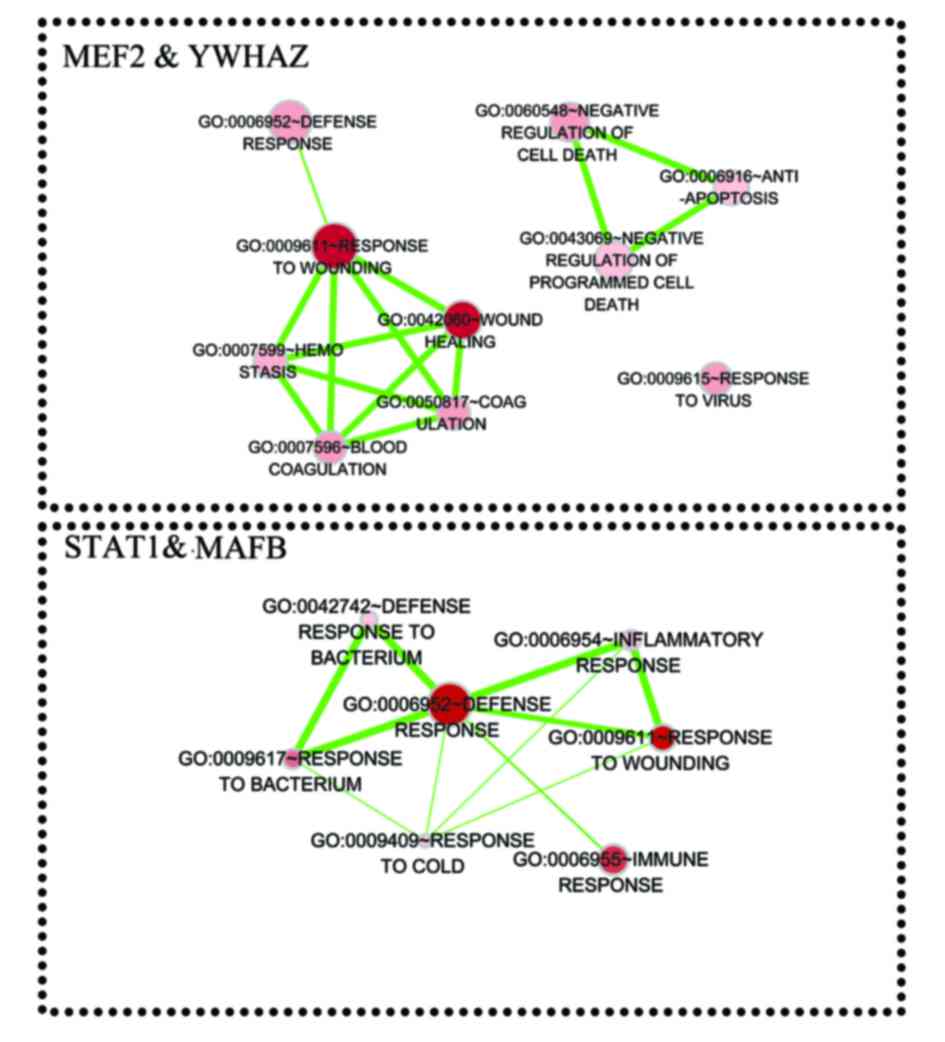
coregulate 70 target genes. These target genes were enriched in
biological processes associated with response to environmental
stimulus and immune response (Fig.
6).
Discussion
Gene expression profiling using DNA microarrays is a
tool for investigating systemic vasculitis at a molecular level
(4,7). In the current study, a total of 173
up- and 93 downregulated genes were identified in PMBCs from
patients with systemic vasculitis compared with controls. GO and
pathway enrichment analysis demonstrated that DEGs were primarily
associated with immune response. FOS, UBB,
STAT1 and MX1 were identified as hub proteins in the
PPI network. Furthermore, UBB, FOS and STAT1
were hub proteins in one of the three identified FI modules. A
total of nine TFs were predicted among the identified DEGs. Of
those nine TFs, STAT1, MAFB and YWHAZ, which
exhibited interactions among each other, were indicated to regulate
further DEGs as target genes in the regulation network. The target
genes of the TFs were primarily associated with response to
environmental stimulus and immune response.
The FOS gene family encodes leucine zipper
proteins, which dimerize with proteins of the JUN family,
forming the TF complex activator protein-1 (AP-1) to regulate gene
expression (5). Previous studies
have demonstrated that the signaling of TLRs, which results in
cytokine production, is integrated by adapter molecules that
activate AP-1 (FOS/JUN) (26–28).
In addition, Tadema et al (29) demonstrated that monocytes and
natural killer cells exhibited increased TLR expression in AAV.
Consistent with the previous study, the results of the current
study revealed that FOS was upregulated in PMBCs from
patients with systemic vasculitis compared with controls, and
FOS was a hub protein in the PPI network and also in the FI
module 2. In this context, we hypothesized that FOS may have
a crucial role in the TLR signaling involved in the inflammatory
response in systemic vasculitis, and, thus, FOS may be a
potential gene target for vasculitis treatment.
UBB encodes ubiquitin, which is one of the
most conserved proteins and has a major role in targeting cellular
proteins for degradation by the 26S proteasome (30). It is apparent that ubiquitination
of various components of the Notch signaling pathway functions in
shaping and orchestrating the Notch signaling pathway (31). Additionally, Piggott et al
(32) revealed that blocking the
Notch pathway inhibited vascular inflammation in large-vessel
vasculitis and modulating the Notch signaling cascade may be a
promising novel method for immunosuppressive therapy of
large-vessel vasculitis. Furthermore, the present study identified
that UBB was upregulated in PMBCs from patients with
systemic vasculitis compared with controls, and UBB was a
hub protein in the PPI network and in the identified FI module 1.
Combined, these results indicate that upregulated UBB may
function in the progression of systemic vasculitis via
participation in the Notch signaling pathway. Therefore, UBB
may be a potential therapeutic target for systemic vasculitis,
which requires further investigation.
The results of the present study also identified
that MX1 was another hub protein in the PPI network.
MX1 encodes a GTP-metabolizing protein, which is induced by
type I and type II interferons and participates in the cellular
antiviral response by antagonizing the replication process of
several viruses (33). Evidence
has demonstrated that the most common vasculitic syndrome
associated with hepatitis C virus (HCV) infection is an immune
complex-mediated type of systemic vasculitis, preferentially
involving the small vessels (34).
MX1 may be associated with interferon signaling and have an
essential role in the cellular antiviral response that is involved
in systemic vasculitis, which requires further investigation.
Furthermore, the present study demonstrated that the
predicted TFs, STAT1, MAFB, and YWHAZ, which
interacted with each other, regulated further DEGs as target genes
in the regulation network, indicating the importance of them. The
protein encoded by STAT1 is a member of the STAT protein
family, which are phosphorylated by receptor associated kinases,
and subsequently act as transcriptional activators. Previous
studies have demonstrated that STAT acts as a signal transducer and
transcriptional activator, and mediates cellular responses to
interferons, other cytokines and growth factors (35–37).
Lin et al (38)
demonstrated that HCV suppressed interferon signaling by degrading
STAT1. Chan et al (39) reported that type I interferons,
when used to treat resistant Churg-Strauss syndrome, a type of
systemic vasculitis, led to complete remission in 25% of a small
case series. In a previous study, it was observed that MAFB
suppressed acute inflammatory responses in
lipopolysaccharide-stimulated lung injury (40). Additionally, it was demonstrated
that MAFB modulated the efficiency of interferon production.
On the other hand, YWHAZ belongs to the 14-3-3 family of
proteins, which mediate signal transduction and gene regulation
events through binding to phosphoserine-containing proteins
(41). Nishimura et al
(41) demonstrated that
overexpression of YWHAZ may inhibit cell apoptosis in breast
cancer cell lines. In addition, the activation of STAT1 was
demonstrated to induce apoptosis (42). Furthermore, Jamin et al
(43) revealed that apoptosis of
endothelial cells was induced by the binding of anti-endothelial
cell antibodies to heat shock protein family D (Hsp60) member 1 in
vasculitis-associated systemic autoimmune diseases. We hypothesized
that STAT1 may be involved in interferon signaling
transduction via interaction with MAFB, and STAT1 may
participate in cell apoptosis through interaction with YWHAZ
in systemic vasculitis.
In conclusion, the critical genes involved in
systemic vasculitis have been investigated based on the microarray
data used in this study. FOS may function in TLR signaling
that is involved in the inflammatory response and UBB may
function in the progression of systemic vasculitis via
participation in the Notch signaling pathway. In addition,
MX1 may be associated with interferon signaling and have an
essential role in the cellular antiviral response in systemic
vasculitis. Furthermore, STAT1 may be involved in interferon
signaling transduction via interaction with MAFB, and
STAT1 may participate in cell apoptosis through interaction
with YWHAZ in systemic vasculitis. Further experiments and
investigation with larger sample sizes are required to verify these
results. Insights into promising gene targets should lead to novel
and effective strategies, and improved targeted therapy for
systemic vasculitis.
Acknowledgements
This study was supported by the Shanghai Science and
Technology Commission fund (grant no. 15495810202), Medical
Engineering Cross Research fund (Shanghai Jiaotong University;
grant no. YG2013MS08) and Medical Engineering Cross Research fund
(Shanghai Jiaotong Universityy; grant no. YG2014QN09).
References
|
1
|
de Souza AWS: Autoantibodies in systemic
vasculitis. Front Immunol. 6:1842015.PubMed/NCBI
|
|
2
|
Stagnaro C, Cioffi E, Talarico R and Rossa
A Della: Systemic vasculitides: A critical digest of the most
recent literature. Clin Exp Rheumatol. 33(2 Suppl 89): S145–S154.
2015.PubMed/NCBI
|
|
3
|
Jennette JC, Falk RJ, Bacon PA, Basu N,
Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen
EC, et al: 2012 revised international chapel hill consensus
conference nomenclature of vasculitides. Arthritis Rheumatism.
65:1–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang JJ, Preston GA, Alcorta DA, Waga I,
Munger WE, Hogan SL, Sekura SB, Phillips BD, Thomas RP, Jennette JC
and Falk RJ: Expression profile of leukocyte genes activated by
anti-neutrophil cytoplasmic autoantibodies (ANCA). Kidney Int.
62:1638–1649. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wagner EF: Bone development and
inflammatory disease is regulated by AP-1 (Fos/Jun). Annals Rheum
Dis. 69:i86–i88. 2010. View Article : Google Scholar
|
|
6
|
Ordonez L, Bernard I, L'Faqihi-Olive FE,
Tervaert JW, Damoiseaux J and Saoudi A: CD45RC isoform expression
identifies functionally distinct T cell subsets differentially
distributed between healthy individuals and AAV patients. PLoS One.
4:e52872009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kobayashi S, Ito A, Okuzaki D, Onda H,
Yabuta N, Nagamori I, Suzuki K, Hashimoto H and Nojima H:
Expression profiling of PBMC-based diagnostic gene markers isolated
from vasculitis patients. DNA Res. 15:253–265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okuzaki D, Fukushima T, Tougan T, Ishii T,
Kobayashi S, Yoshizaki K, Akita T and Nojima H: Genopal™: A novel
hollow fibre array for focused microarray analysis. DNA Res.
17:369–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Simon R, Lam A, Li MC, Ngan M, Menenzes S
and Zhao Y: Analysis of gene expression data using BRB-array tools.
Cancer Inform. 3:11–17. 2007.PubMed/NCBI
|
|
10
|
Zhao Y and Simon R: BRB-ArrayTools Data
Archive for human cancer gene expression: A unique and efficient
data sharing resource. Cancer Inform. 6:9–15. 2008.PubMed/NCBI
|
|
11
|
Ihaka R and Gentleman R: R: A language for
data analysis and graphics. J Computational and graphical
statistics. 5:299–314. 1996. View Article : Google Scholar
|
|
12
|
Yu G, Wang LG, Han Y and He QY:
ClusterProfiler: An R package for comparing biological themes among
gene clusters. Omics. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reiner-Benaim A: FDR control by the BH
procedure for Two-Sided Correlated Tests with implications to gene
expression Data Analysis. Biom J. 49:107–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Benjamini Y and Hochberg Y: Controlling
the false discovery rate: A practical and powerful approach to
multiple testing. J Royal Statistical Soc. 57:289–300. 1995.
|
|
15
|
Franceschini A, Szklarczyk D, Frankild S,
Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C
and Jensen LJ: STRING v9.1: Protein-protein interaction networks,
with increased coverage and integration. Nucleic Acids Res.
41:D808–D815. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu J and Finley RL: Combining multiple
positive training sets to generate confidence scores for
protein-protein interactions. Bioinformatics. 25:105–111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Csardi G and Nepusz T: The igraph software
package for complex network research. Inter Journal Complex
Systems. 1695:1–9. 2006.
|
|
19
|
Wu G, Dawson E, Duong A, Haw R and Stein
L: ReactomeFIViz: A Cytoscape app for pathway and network-based
data analysis. F1000Res. 3:1462014.PubMed/NCBI
|
|
20
|
Tan PK, Downey TJ, Spitznagel EL Jr, Xu P,
Fu D, Dimitrov DS, Lempicki RA, Raaka BM and Cam MC: Evaluation of
gene expression measurements from commercial microarray platforms.
Nucleic Acids Res. 31:5676–5684. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Van Dongen SM: Graph clustering by flow
simulation. 2001.
|
|
22
|
Huang ZX, Tian HY, Hu ZF, Zhou YB, Zhao J
and Yao KT: GenCLiP: A software program for clustering gene lists
by literature profiling and constructing gene co-occurrence
networks related to custom keywords. BMC bioinformatics. 9:3082008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Matys V, Fricke E, Geffers R, Gössling E,
Haubrock M, Hehl R, Hornischer K, Karas D, Kel AE, Kel-Margoulis
OV, et al: TRANSFAC: Transcriptional regulation, from patterns to
profiles. Nucleic Acids Res. 31:374–378. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Song KH, Kim YH and Kim BY: Sho-saiko-to,
a traditional herbal medicine, regulates gene expression and
biological function by way of microRNAs in primary mouse
hepatocytes. BMC Complement Altern Med. 14:142014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nicolle R, Radvanyi F and Elati M:
CoRegNet: Reconstruction and integrated analysis of co-regulatory
networks. Bioinformatics. 31:3066–3068. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kawai T, Sato S, Ishii KJ, Coban C, Hemmi
H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, et al:
Interferon-alpha induction through Toll-like receptors involves a
direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol.
5:1061–1068. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dillon S, Agrawal A, Van Dyke T, Landreth
G, McCauley L, Koh A, Maliszewski C, Akira S and Pulendran B: A
Toll-like receptor 2 ligand stimulates Th2 responses in vivo, via
induction of extracellular signal-regulated kinase
mitogen-activated protein kinase and c-Fos in dendritic cells. J
Immunol. 172:4733–4743. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wagner EF and Eferl R: Fos/AP-1 proteins
in bone and the immune system. Immunol Rev. 208:126–140. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tadema H, Abdulahad WH, Stegeman CA,
Kallenberg CG and Heeringa P: Increased expression of Toll-like
receptors by monocytes and natural killer cells in ANCA-associated
vasculitis. PLoS One. 6:e243152011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh C, Park S, Lee EK and Yoo YJ:
Downregulation of ubiquitin level via knockdown of polyubiquitin
gene Ubb as potential cancer therapeutic intervention. Sci Rep.
3:26232013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Le Bras S, Loyer N and Le Borgne R: The
multiple facets of ubiquitination in the regulation of notch
signaling pathway. Traffic. 12:149–161. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Piggott K, Deng J, Warrington K, Younge B,
Kubo JT, Desai M, Goronzy JJ and Weyand CM: Blocking the NOTCH
pathway inhibits vascular inflammation in large-vessel
vasculitisclinical perspective. Circulation. 123:309–318. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Verhelst J, Parthoens E, Schepens B, Fiers
W and Saelens X: Interferon-inducible protein Mx1 inhibits
influenza virus by interfering with functional viral
ribonucleoprotein complex assembly. J Virol. 86:13445–13455. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vassilopoulos D and Calabrese LH:
Hepatitis C virus infection and vasculitis: Implications of
antiviral and immunosuppressive therapies. Arthritis Rheum.
46:585–597. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ramana CV, Chatterjee-Kishore M, Nguyen H
and Stark GR: Complex roles of Stat1 in regulating gene expression.
Oncogene. 19:2619–2627. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kawazoe Y, Naka T, Fujimoto M, Kohzaki H,
Morita Y, Narazaki M, Okumura K, Saitoh H, Nakagawa R, Uchiyama Y,
et al: Signal transducer and activator of transcription
(STAT)-induced STAT inhibitor 1 (SSI-1)/suppressor of cytokine
signaling 1 (SOCS1) inhibits insulin signal transduction pathway
through modulating insulin receptor substrate 1 (IRS-1)
phosphorylation. J Exp Med. 193:263–270. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klampfer L: Signal transducers and
activators of transcription (STATs): Novel targets of
chemopreventive and chemotherapeutic drugs. Curr Cancer Drug
Targets. 6:107–121. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin W, Choe WH, Hiasa Y, Kamegaya Y,
Blackard JT, Schmidt EV and Chung RT: Hepatitis C virus expression
suppresses interferon signaling by degrading STAT1.
Gastroenterology. 128:1034–1041. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chan AT, Flossmann O, Mukhtyar C, Jayne D
and Luqmani RA: The role of biologic therapies in the management of
systemic vasculitis. Autoimmu Rev. 5:273–278. 2006. View Article : Google Scholar
|
|
40
|
Aida Y, Sato-Nishiwaki M, Abe S, Kishi H,
Nunomiya K, Yamauchi K, lnoue S and Shibata Y: MafB suppresses
acute inflammatory responses in lipopolysaccharide-stimulated lung
injury in mice. Am J Respir Crit Care Med. 185:A13402012.
|
|
41
|
Nishimura Y, Komatsu S, Ichikawa D, Nagata
H, Hirajima S, Takeshita H, Kawaguchi T, Arita T, Konishi H,
Kashimoto K, et al: Overexpression of YWHAZ relates to tumor cell
proliferation and malignant outcome of gastric carcinoma. Br J
Cancer. 108:1324–1331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kaganoi J, Watanabe G, Okabe M, Nagatani
S, Kawabe A, Shimada Y, Imamura M and Sakai Y: STAT1
activation-induced apoptosis of esophageal squamous cell carcinoma
cells in vivo. Ann Surg Oncol. 14:1405–1415. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jamin C, Duguuguabe G, Okabe M, et al:
STAT1 activation-induced apoptosis of esophageal squamous cell
carcinoma cells in vivo. Annals of Surgical Oncology. 14:1405–1415.
2007. View Article : Google Scholar : PubMed/NCBI
|