Introduction
AMP-activated protein kinase (AMPK), a sensor of
cellular energy status, is one of the major therapeutic targets in
the treatment of metabolic diseases. AMPK is composed of a
catalytic α subunit and regulatory β and γ subunits (1). The AMPK α subunit consists of two
isoforms, α1 and α2; of which α1 is the predominant isoform
expressed in adipocytes (2).
Phosphorylation of Thr172 in the α subunit activates AMPK
>100-fold, and 5′-adenosine monophosphate maintains AMPK in an
activated state by enhancing phosphorylation of the α subunit, as
well as by allosteric activation (3).
Berberine (BBR) is a natural isoquinoline alkaloid,
which has been reported to exert therapeutic effects in a number of
metabolic diseases, such as diabetes and hypercholesterolemia
(4). The beneficial effects of BBR
are attributable to its effect as an AMPK activator. BBR has been
demonstrated to activate AMPK in adipocytes and muscle cells, as
well as in animal models of diabetes and obesity, thus resulting in
reduced fat accumulation and improved insulin sensitivity (5). In addition, BBR has been reported to
inhibit mitochondrial respiratory chain complex I, thereby
activating AMPK (6). However, its
downstream mechanisms have not yet been fully elucidated.
The sterol regulatory element-binding protein
(SREBP) is the major transcription factor involved in regulating
lipogenic gene expression (7).
Previous studies have suggested that BBR reduces fat accumulation
in white adipose tissue and adipocytes via downregulation of SREBPs
(8,9). In the present study, the detailed
molecular mechanisms of the action of BBR were elucidated, whereby
a signaling pathway involving BBR-mediated AMPK activation and
SREBP-1c downregulation in 3T3-L1 adipocytes was identified.
Materials and methods
Chemicals and reagents
Cell culture reagents, including Dulbecco's modified
Eagle's medium (DMEM), fetal bovine serum, calf serum and
antibiotics were obtained from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). AMPKα1 small interfering (si)RNA and control
siRNA were purchased from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Lipofectamine RNAiMAX transfection reagent was purchased
from Invitrogen; Thermo Fisher Scientific, Inc. BBR, insulin,
dexamethasone and 3-isobutyl-1-methylxanthine were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany).
Cell culture and differentiation
3T3-L1 pre-adipocytes were purchased from the
American Type Culture Collection (Manassas, VA, USA) and seeded in
12-well plates at a density of 1.5×105 cells/well with
DMEM containing 10% calf serum at 37°C in 5% CO2
atmosphere with relative humidity of 85–95%. When the cells were
100% confluent following 3 days of culture (taken as day 0), the
cells were induced to differentiate by incubation in a
differentiation induction medium consisting of DMEM, 10% fetal
bovine serum, 1 µg/ml insulin, 0.25 µM dexamethasone and 0.5 mM
3-isobutyl-1-methylxanthine for 2 days. The culture medium was then
replaced with differentiation maintenance medium, consisting of
DMEM, 1 µg/ml insulin and 10% fetal bovine serum on days 2 and 4,
and the cells were harvested on day 7. To examine the effects of
BBR on cell differentiation, BBR was dissolved in dimethylsulfoxide
and diluted >1,000-fold in differentiation maintenance medium.
In the present study, BBR was administered during the late phase of
adipogenic differentiation (days 2–7) in order to analyze the
effects of BBR on fat accumulation and avoid its effects on mitotic
clonal expansion, as BBR was previously reported to inhibit the
mitotic clonal expansion of 3T3-L1 cells (10). In dose-response experiments, cells
were treated with BBR (0, 5, 10, 15 and 20 µM) at day 2 and day 4,
when media were replaced, and harvested at day 7. In time-response
experiments, cells were treated with 0 or 20 µM BBR at day 2 and
day 4, when media were replaced, and harvested at days 3, 5 and 7.
Cells harvested at day 3 were treated with 20 µM BBR at day 2 only.
Control cells were treated with an equal volume of
dimethylsulfoxide diluted in differentiation maintenance medium.
Intracellular fat accumulation in 1.2×106 cells was
measured by 0.3% Oil red O staining solution at room temperature
for 30 min followed by light microscopy with a magnification of
×400. Oil red O dye in intracellular fat droplets was extracted by
adding 700 µl 60% isopropanol per well and shaking at 100 rpm for 2
h at room temperature. The extracted dye was measured by optical
density at 510 nm using the methods described previously (11). Cell viability was determined
according to the manufacturer's protocol using a CellCountEZ™ Cell
Survival assay kit (Rockland Immunochemicals, Inc., Pottstown, PA,
USA), which is based on the ability of viable mammalian cells to
convert hydroxyethyl disulfide into mercaptoethanol (12). Cell number was determined using a
hemocytometer following the harvesting of cells by
trypsinization.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RT-qPCR experiments were performed as described
previously (13). Cells
(1.2×106) were treated with BBR and harvested as
described above. Total RNA was extracted using the RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany), according to the manufacturer's
protocol. RNA (1 µg) was reverse-transcribed at 37°C using the
High-Capacity cDNA Reverse Transcription kit (Applied Biosystems;
Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. qPCR was performed using a StepOne Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.), in
triplicate, in a final volume of 20 µl, which consisted of 10 µl
TaqMan Gene Expression Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) containing Taq polymerase and dNTPs, 1 µl
TaqMan Gene Expression assay (Applied Biosystems; Thermo Fisher
Scientific, Inc.) containing 20X TaqMan probe and primers, cDNA
(1/10th that produced by RT) and distilled water. The reaction
mixtures were preheated at 95°C for 10 min to activate the enzyme,
followed by 40 cycles of melting at 95°C for 15 sec and
annealing/extension at 60°C for 1 min. TaqMan Gene Expression
assays containing TagMan probe and primers were used to evaluate
the mRNA levels of the following genes: peroxisome
proliferator-activated receptor-γ (PPARγ; assay ID, Mm00440945_m1),
CCAAT-enhancer-binding protein α (C/EBPα; assay ID, Mm01265914_s1),
SREBP-1c (assay ID, Mm00550338_m1), lipoprotein lipase (LPL; assay
ID, Mm00434764_m1), fatty acid synthase (FAS; assay ID,
Mm01253292_m1) and acetyl-CoA carboxylase-1 (ACC-1; assay ID,
Mm01304257_m1) and 18S ribosomal (r) RNA (assay ID, Hs99999901_s1).
The 18S rRNA was used as an internal control, as previously
described (11). For each sample,
target gene mRNA levels were normalized against the level of 18S
rRNA, and the ratio of normalized mRNA in each sample to that of
the control sample was determined using the comparative
Cq method (14).
Western blotting
Western blotting was performed using the methods
described previously (13). Cells
(1.2×106) were treated with BBR and harvested as
described above. Cells were lysed for 1 h in ice-cold
radioimmunoprecipitation assay buffer containing 25 mM Tris-HCl (pH
7.6) 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS
and a protease inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The
cell lysates were centrifuged at 19,000 × g for 20 min at 4°C to
remove insoluble materials. The protein concentrations were
determined using a BCA protein assay kit (Pierce; Thermo Fisher
Scientific, Inc.). Protein extracts (50 µg) were resolved by 10%
SDS-PAGE and electro-transferred to nitrocellulose membranes at 150
mA for 1 h. The membranes were then blocked for 2 h at room
temperature with PBS containing 5% skim milk and 0.1% Tween-20 and,
after washing for 10 min three times with PBS containing 0.1% Tween
20, were incubated with primary antibodies (diluted 1:1,000 in the
blocking solution) overnight at 4°C and subsequently with
horseradish peroxidase-conjugated anti-rabbit or anti-mouse
secondary antibodies (diluted 1:1,000 in the blocking solution) for
1 h at room temperature. Protein bands were visualized using Pierce
ECL Western Blotting Substrate, according to the manufacturer's
protocol (Thermo Fisher Scientific, Inc.). An anti-β-Actin antibody
(diluted 1:1,000 in the blocking solution) was used as an
endogenous control to confirm that equal amounts of proteins were
loaded. Anti-phosphorylated (p)-AMPKα (cat. no. 2535), anti-AMPKα
(cat. no. 2603), anti-p-SREBP-1c (cat. no. 9874) and anti-PPARγ
(cat. no. 2430) primary antibodies, as well as anti-mouse IgG (cat.
no. 7076S) and anti-rabbit IgG (cat. no. 7074) secondary antibodies
were purchased from Cell Signaling Technology, Inc. (Danvers, MA,
USA). The anti-SREBP-1c (cat. no. 557036) antibody was purchased
from BD Biosciences (Franklin Lakes, NJ, USA). Anti-C/EBPα (cat.
no. sc-61), anti-TATA box-binding protein (TBP; cat. no. sc-204)
and anti-β-actin (cat. no. sc-47778) primary antibodies were
purchased from Santa Cruz Biotechnology, Inc.
Analysis of nuclear SREBP-1c
levels
Cells (1.2×106) were treated with BBR and
harvested as described above. Cells were harvested using cell
scrapers, and nuclear lysates were prepared using a Nuclear Extract
kit (Active Motif, Inc. Carlsbad, CA, USA) according to the
manufacturer's protocol. The protein concentration of nuclear
lysates was determined using a BCA protein assay kit. To determine
nuclear SREBP-1c protein expression levels, 10 µg nuclear protein
was separated by 10% SDS-PAGE, followed by western blotting using
anti-SREBP-1c antibody followed by secondary antibody. The
procedures and antibodies used for western blotting were identical
to those described above.
Determination of SREBP-1c binding to
target DNA
Binding of nuclear SREBP-1c to the target DNA
sequence, 5′-TCACCTGA-3′, which contains an E-box motif, was
measured using a SREBP-1 Transcription Factor ELISA kit (cat. no.
10010854; Cayman Chemical Company, Ann Arbor, MI, USA). Briefly, 10
µg protein from the nuclear lysates was added to wells of a 96-well
plate that were provided as part of the ELISA kit. The wells
included in the kit were already coated with the 5′-TCACCTGA-3′
oligonucleotide. Following incubation for 1 h at room temperature,
unbound nuclear proteins were removed by washing five times with a
wash buffer provided in the kit. Each well was incubated with 100
µl anti-SREBP-1 (1:100) primary antibody for 1 h at room
temperature followed by 100 µl horseradish peroxidase-conjugated
secondary antibody (1:100) for 1 h at room temperature. Optical
density at 450 nm was measured following the addition of 100 µl
developing solution followed by 100 µl stop solution provided in
the kit.
AMPK activity assay
The kinase activity of AMPK was measured by ELISA
using an AMPK assay kit (cat. no. CY-1182; Cyclex Inc., Columbia,
MD, USA). Cells (1.2×106) were lysed in 0.2 ml lysis
buffer containing 20 mM Tris HCl (pH 7.5) 250 mM NaCl, 10%
glycerol, 0.5% Nonidet P-40, 0.2 mM PMSF, 1 µg/ml pepstatin, 0.5
µg/ml leupeptin, 5 mM NaF, 2 mM Na3VO4, 2 mM
β-glycerophosphate and 1 mM dithiothreitol for 90 min at 4°C,
followed by centrifugation at 22,000 × g for 10 min at 4°C. Cell
lysates (10 µl) and 90 µl kinase reaction buffer with 50 µM ATP
(included in the kit) were added to wells of a 96-well plate coated
with peptides consisting of the sequence surrounding Ser789 of
insulin receptor substrate (IRS)-1, which is efficiently
phosphorylated by AMPK, and incubated at 30°C for 30 min. The
quantity of phosphorylated substrate was measured by incubating
with 100 µl anti-p-IRS-1 antibody at room temperature for 30 min
followed by incubation a with 100 µl horseradish
peroxidase-conjugated secondary antibody at room temperature for 30
min. The primary and secondary antibodies included in the ELISA kit
were prediluted. Subsequently, 100 µl substrate reagent was added
at room temperature for 10 min followed by 100 µl stop solution
included in the kit. Optical density was measured at 450 nm.
Transfection of siRNA
At one day prior to reaching confluence (taken as
day-1), 3T3-L1 cells were incubated in serum-free medium for 1 h
and then transfected with 12.5, 25, 50, or 75 nM AMPKα1 siRNA, or
50 or 75 nM control siRNA using Lipofectamine RNAiMAX transfection
reagent according to the manufacturer's protocol. The following day
(on day 0), the transfected cells were induced to differentiate by
replacing the medium with differentiation-induction medium.
Following 7 days of differentiation into adipocytes, cells were
harvested for Oil red O staining, western blotting, determination
of SREBP-1c binding to target DNA or RT-qPCR analysis using the
aforementioned methods.
Statistical analysis
Data are presented as the mean ± standard deviation
of four replicate experiments. Differences among multiple groups
were determined using one-way analysis of variance followed by post
hoc analysis using Duncan's multiple range test, and comparisons
between two groups were analyzed using an unpaired Student's
t-test. Statistical tests were performed using SPSS software
(version, 14.0; SPSS, Inc., Chicago, IL, USA).
Results
Dose-dependent effect of BBR on AMPK,
SREBP-1c and lipogenesis-associated gene expression
SREBP-1c is synthesized as a 125-kDa precursor,
which is processed by proteolytic cleavage into a mature 68-kDa
form that translocates into the nucleus. Phosphorylation of the
SREBP-1c precursor suppresses its proteolytic maturation (15). In the present study BBR activated
AMPK in a dose-dependent manner (Fig.
1A), which was associated with elevation of p-AMPK and
p-SREBP-1c levels (Fig. 1B). The
increase in p-SREBP-1c levels was accompanied by reduced
proteolytic processing of the 125-kDa SREBP-1c precursor into the
68-kDa mature form (Fig. 1B).
Western blotting of total SREBP-1c revealed gradual proteolytic
degradation of the 125-kDa precursor form into the 68-kDa mature
form (Fig. 1B). The level of the
mature 68-kDa SREBP-1c isoform was decreased in the total cell
lysate and nuclear lysate in a dose-dependent manner following
treatment with BBR, as proteolytic maturation is required for the
nuclear translocation of SREBP-1c (Fig. 1B). Similarly, the binding of
nuclear SREBP-1c to its E-box motif-containing target DNA sequence
was reduced in a dose-dependent manner following BBR treatment
(Fig. 1C).
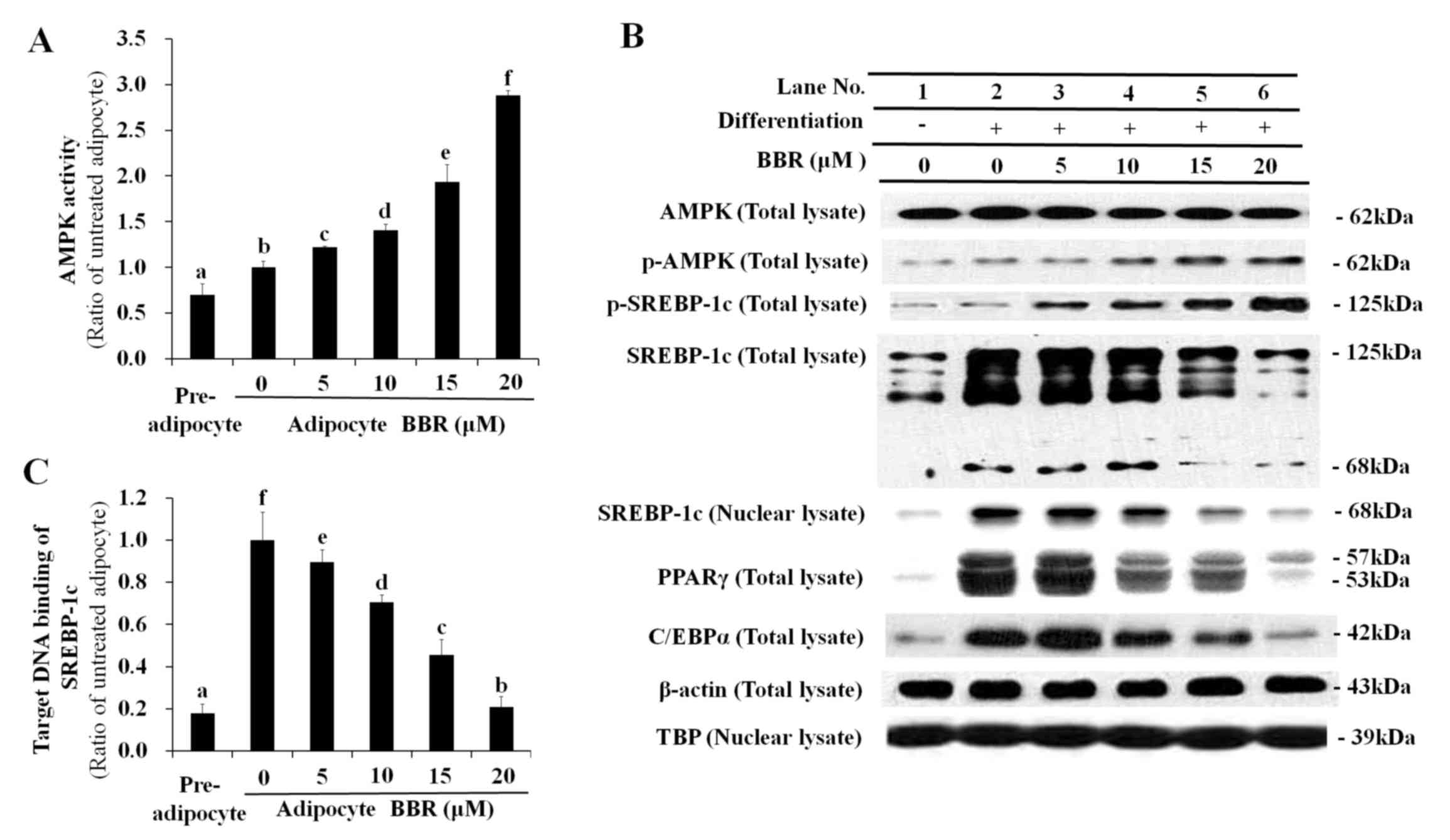 | Figure 1.Dose-dependent effects of BBR on AMPK,
SREBP-1c and lipogenesis-associated protein expression levels.
3T3-L1 adipocytes were treated with 0, 5, 10, 15 or 20 µM BBR from
days 2–7 of differentiation, and cells were harvested on day 7 for
analysis. (A) AMPK activity as determined by its capability to
phosphorylate Ser789 on its substrate, IRS-1. This was measured in
cell lysates by ELISA. (B) The level of p-AMPK and p-SREBP-1c, as
well as the 125-kDa precursor form and mature 68-kDa form, were
measured in total cell lysates by western blotting. Levels of the
68-kDa mature SREBP-1c form were analyzed in nuclear lysates to
determine its nuclear translocation. Levels of the major adipogenic
transcription factors, PPARγ and C/EBPα, were analyzed in total
cell lysates. β-actin and TBP were used as endogenous controls for
protein expression in total cell lysates and nuclear lysates,
respectively. (C) The binding of nuclear SREBP-1c to its E-box
motif-containing target DNA sequence, 5′-TCACCTGA-3′, as determined
by ELISA. Different letters indicate significant differences
between groups (P<0.05). BBR, berberine; AMPK, AMP-activated
protein kinase; SREBP-1c, sterol regulatory element binding
protein-1c; p-AMPK, phosphorylated AMPK; IRS-1, insulin receptor
substrate-1; p-SREBP-1c, phosphorylated SREBP-1c; PPARγ, peroxisome
proliferator-activated receptor-γ; C/EBPα, CCAAT/enhancer-binding
protein α; TBP, TATA box-binding protein. |
SREBP-1c is a transcription factor that binds to the
promoter region of numerous lipogenesis-associated genes (16–21).
As a result of decreased binding of nuclear SREBP-1c to target DNA,
the mRNA expression of lipogenic genes, including FAS, LPL,
SREBP-1c, PPARγ, C/EBPα and ACC-1 were reduced by BBR treatment in
a dose-dependent manner (Fig. 2).
This is consistent with the dose-dependent reduction in PPARγ and
C/EBPα protein expression levels demonstrated in Fig. 1B. Intracellular fat accumulation,
which is mediated by the collective actions of lipogenic genes, was
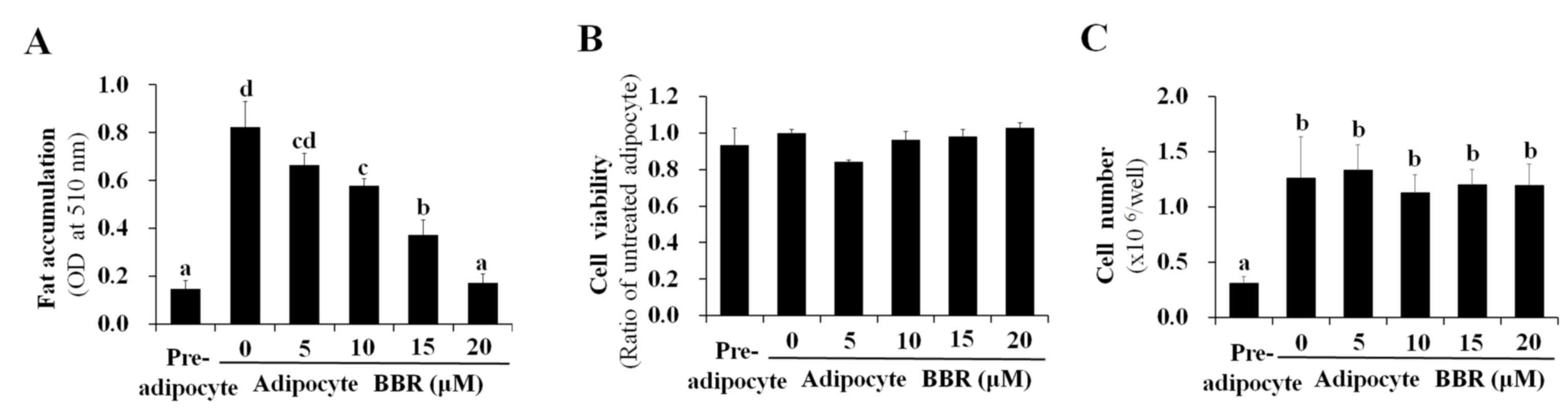
reduced by BBR treatment in a dose-dependent manner (Fig. 3A). BBR exhibited no significant
effect on cell viability, which was maintained at >90% of
untreated adipocytes (Fig. 3B).
This indicates that the BBR-induced suppression of nuclear SREBP-1c
binding to its target DNA, as well as the reduction in lipogenic
gene expression and intracellular fat accumulation, were not caused
by increased cytotoxicity. Previously, BBR was reported to inhibit
the mitotic clonal expansion of 3T3-L1 cells when treated during
the early phase of differentiation (10) and the present study aimed to
elucidate the molecular mechanism of BBR for reducing fat
accumulation without its effects on mitotic clonal expansion. In
the present study, BBR was administered during the late phase of
adipogenic differentiation (from days 2–7) in order to avoid its
effects on mitotic clonal expansion, which occurs during the early
phase of differentiation. As demonstrated in Fig. 3C, increasing concentrations of BBR
demonstrated no significant effects on the number of cells,
demonstrating that mitotic clonal expansion was not affected in
this experimental condition (Fig.
3C).
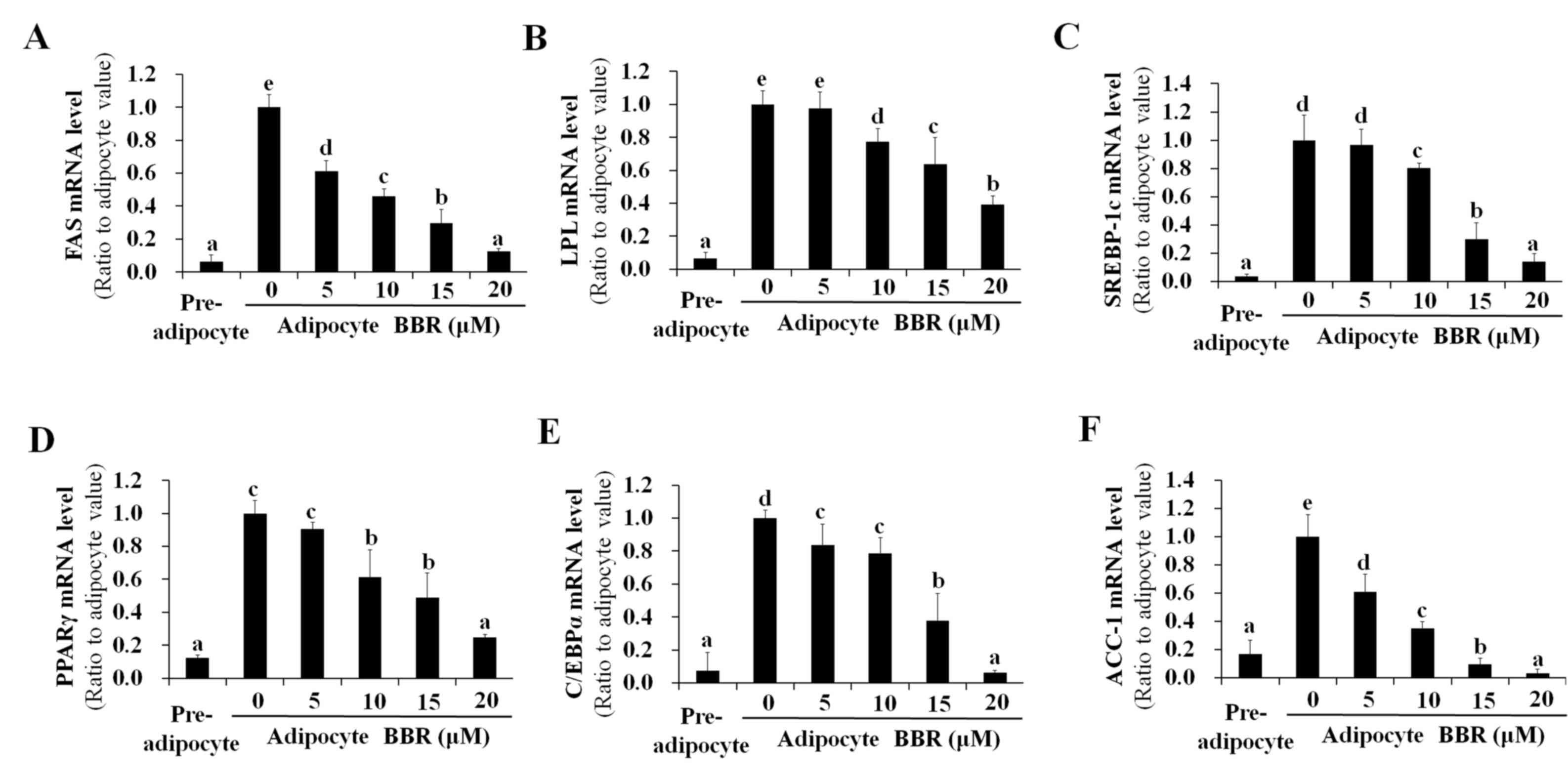 | Figure 2.Dose-dependent effects of BBR on the
expression of lipogenesis-associated genes. 3T3-L1 adipocytes were
treated with 0, 5, 10, 15 or 20 µM BBR from days 2–7 of
differentiation, and cells were harvested on day 7 for analysis.
The mRNA levels of (A) FAS, (B) LPL, (C) SREBP-1c, (D) PPARγ, (E)
C/EBPα and (F) ACC-1 were measured by reverse
transcription-quantitative polymerase chain reaction. Different
letters indicate significant differences between groups
(P<0.05). BBR, berberine; FAS, fatty acid synthase; LPL,
lipoprotein lipase; SREBP1-c, sterol regulatory element binding
protein-1c; PPARγ, peroxisome proliferator-activated receptor-γ;
C/EBPα, CCAAT/enhancer-binding protein α; ACC-1, acetyl-CoA
carboxylase-1. |
Effect of BBR on AMPK, SREBP-1c and
the expression of lipogenesis-associated genes over time
In order to examine the effect of BBR exposure over
time, 3T3-L1 adipocytes were treated with 0 or 20 µM BBR from days
2–7, and were analyzed on days 3, 5 and 7 of adipogenic
differentiation. Under these conditions, the protein expression
levels of p-AMPK and p-SREBP-1c were increased in BBR-treated cells
relative to the untreated cells (Fig.
4A). The protein expression levels of the 125-kDa SREBP-1c
precursor and its processed 68-kDa mature form were reduced in
BBR-treated cells when compared with in untreated cells (Fig. 4A). In addition, the nuclear levels
of mature 68-kDa SREBP-1c were reduced in BBR-treated cells when
compared with untreated cells, as proteolysis is required for
nuclear translocation (Fig. 4A).
The binding of nuclear SREBP-1c to its target DNA sequence was
elevated in a time-dependent manner in untreated cells, which was
significantly suppressed in BBR-treated cells, in a similar manner
to nuclear SREBP-1c levels (Fig.
4B). Intracellular fat accumulation was reduced in BBR-treated
cells when compared with the untreated cells (Fig. 4C). Consistent with these
observations, the mRNA levels of lipogenesis-associated genes,
including SREBP-1c, were significantly suppressed in BBR-treated
cells when compared with the untreated cells (Fig. 5).
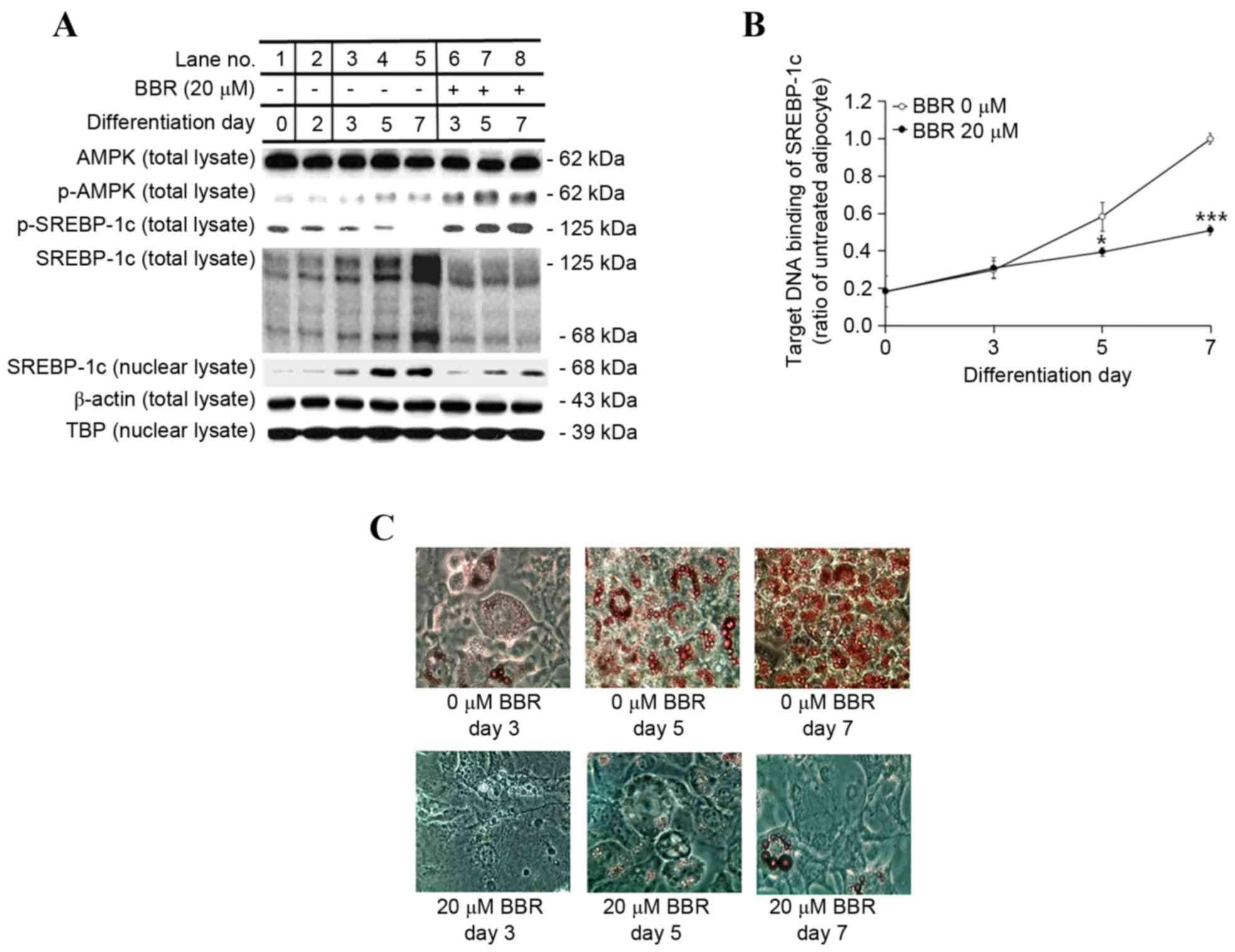 | Figure 4.Effect of BBR on AMPK and SREBP-1c
protein expression and intracellular fat accumulation over time.
3T3-L1 adipocytes were treated with 0 or 20 µM BBR commencing at
day 2, and cells were harvested at days 3, 5 and 7 of
differentiation for time course analyses. (A) The protein levels of
p-AMPK and p-SREBP-1c, as well as the 125-kDa precursor form of
SREBP-1c and the mature 68-kDa form were analyzed in total cell
lysates by western blotting. The level of the mature 68-kDa
SREBP-1c isoform was analyzed in nuclear lysates. β-actin and TBP
were used as endogenous controls for the analysis of protein
expression in total cell lysates and nuclear lysates, respectively.
(B) The binding of nuclear SREBP-1c to its E-box motif-containing
target DNA sequence, 5′-TCACCTGA-3′, was measured by ELISA. (C)
Intracellular fat droplets were stained with Oil red O dye and
examined by light microscopy. Magnification, ×400. *P<0.05 and
***P<0.001 vs. untreated adipocytes at the same day of
differentiation. BBR, berberine; AMPK, AMP-activated protein
kinase; SREBP-1c, sterol regulatory element binding protein-1c;
p-AMPK, phosphorylated AMPK; p-SREBP-1c, phosphorylated-SREBP-1c;
TBP, TATA box-binding protein. |
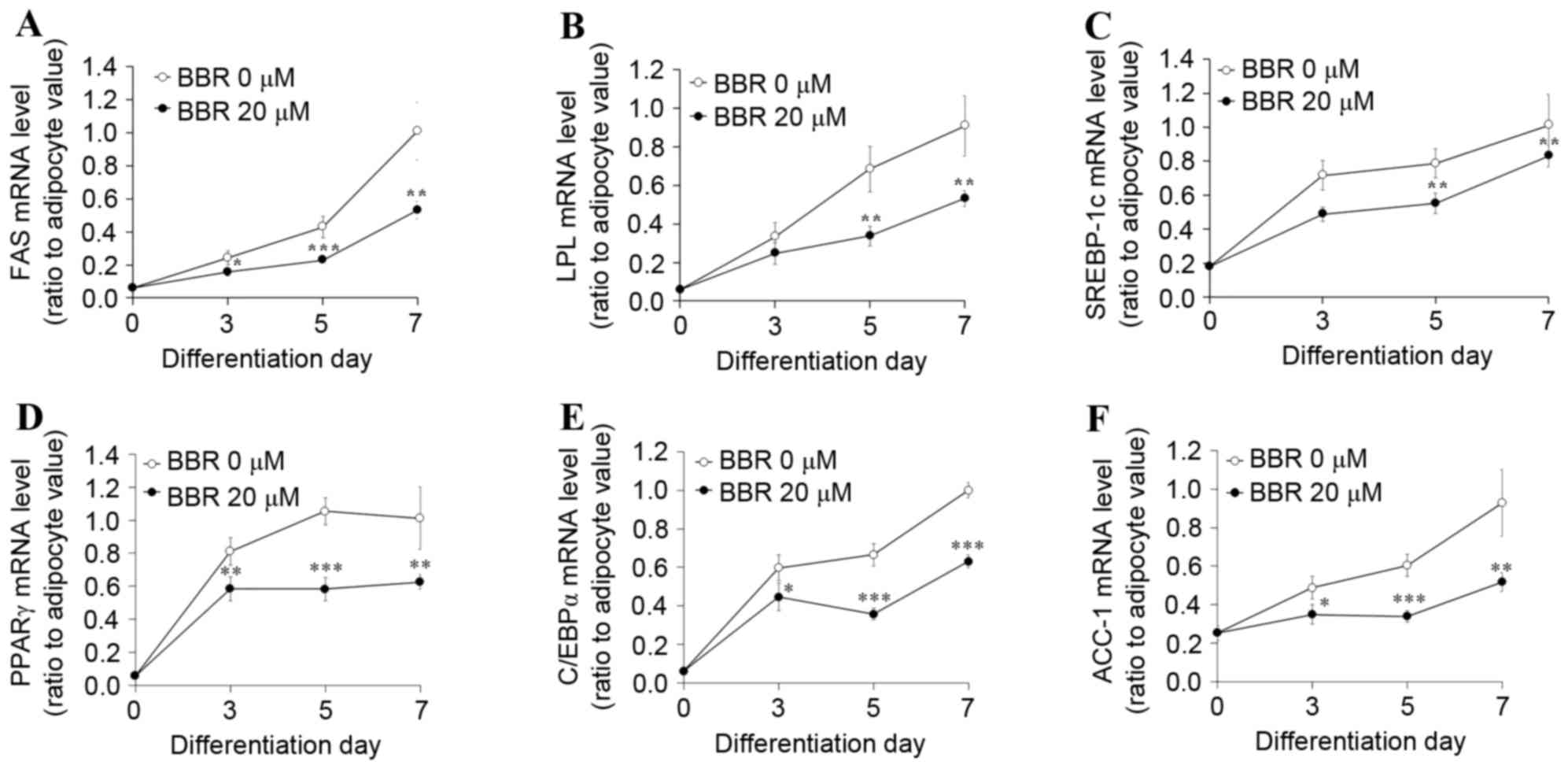 | Figure 5.Effect of BBR on the expression of
lipogenesis-associated genes over time. 3T3-L1 adipocytes were
treated with 0 or 20 µM BBR commencing at day 2 until cells were
harvested at days 3, 5 or 7 of differentiation. The mRNA levels of
(A) FAS, (B) LPL, (C) SREBP-1c, (D) PPARγ, (E) C/EBPα and (F) ACC-1
were measured by reverse transcription-quantitative polymerase
chain reaction. *P<0.05, **P<0.01 and ***P<0.001 vs.
untreated adipocytes at same day of differentiation. BBR,
berberine; FAS, fatty acid synthase; LPL, lipoprotein lipase;
SREBP1-c, sterol regulatory element binding protein-1c; PPARγ,
peroxisome proliferator-activated receptor-γ; C/EBPα,
CCAAT/enhancer-binding protein α; ACC-1, acetyl-CoA
carboxylase-1. |
Effect of AMPK knockdown on SREBP-1c
processing, lipogenesis-associated gene expression and
intracellular fat accumulation
In order to confirm the essential role of AMPK in
BBR-induced SREBP-1c phosphorylation and processing, 3T3-L1 cells
were transfected with different concentrations of control siRNA or
AMPKα1 siRNA. siRNA sequences targeting the α1 isoform of AMPK,
rather than α2, were employed for knockdown experiments in the
present study, as α1 is the predominant isoform expressed in
adipocytes (2). Levels of p-AMPK
and p-SREBP-1c were elevated in adipocytes treated with BBR alone
when compared with untreated adipocytes (Fig. 6A). Levels of p-SREBP-1c were
reduced in a dose-dependent manner following transfection with
increasing concentrations of AMPKα1 siRNA, whereas, this effect was
not observed following transfection with control siRNA (Fig. 6A). This confirmed that SREBP-1c is
a substrate of AMPK. Conversely, the level of total SREBP-1c,
including the 125-kDa precursor form and the mature 68-kDa form,
were reduced in BBR-treated cells when compared with in untreated
cells (Fig. 6A). These levels
increased in a dose-dependent manner in AMPKα1 siRNA-transfected
cells, however, not in control siRNA-transfected cells (Fig. 6A). The nuclear expression levels of
the 68-kDa SREBP-1c form demonstrated a similar pattern to that
observed in the total cell lysate. The binding of nuclear SREBP-1c
to its target DNA sequence, which was suppressed by BBR, was
rescued by AMPKα1 siRNA transfection in a dose-dependent manner
(Fig. 6B). This effect was not
observed following transfection with control siRNA. Intracellular
fat accumulation, which was suppressed by BBR exposure, was rescued
by AMPKα1 siRNA transfection, whereas transfection with control
siRNA demonstrated no effect (Fig.
6C). In addition, the mRNA levels of lipogenesis-associated
genes, including FAS, LPL, SREBP-1c, PPARγ, C/EBPα and ACC-1, which
were suppressed by BBR treatment, were rescued AMPKα1 siRNA
transfection in a dose-dependent manner, whereas the control siRNA
sequences demonstrated no significant effect (Fig. 7). Similar to the protein expression
levels of PPARγ and C/EBPα (Fig.
6A), the mRNA expression levels of these factors were rescued
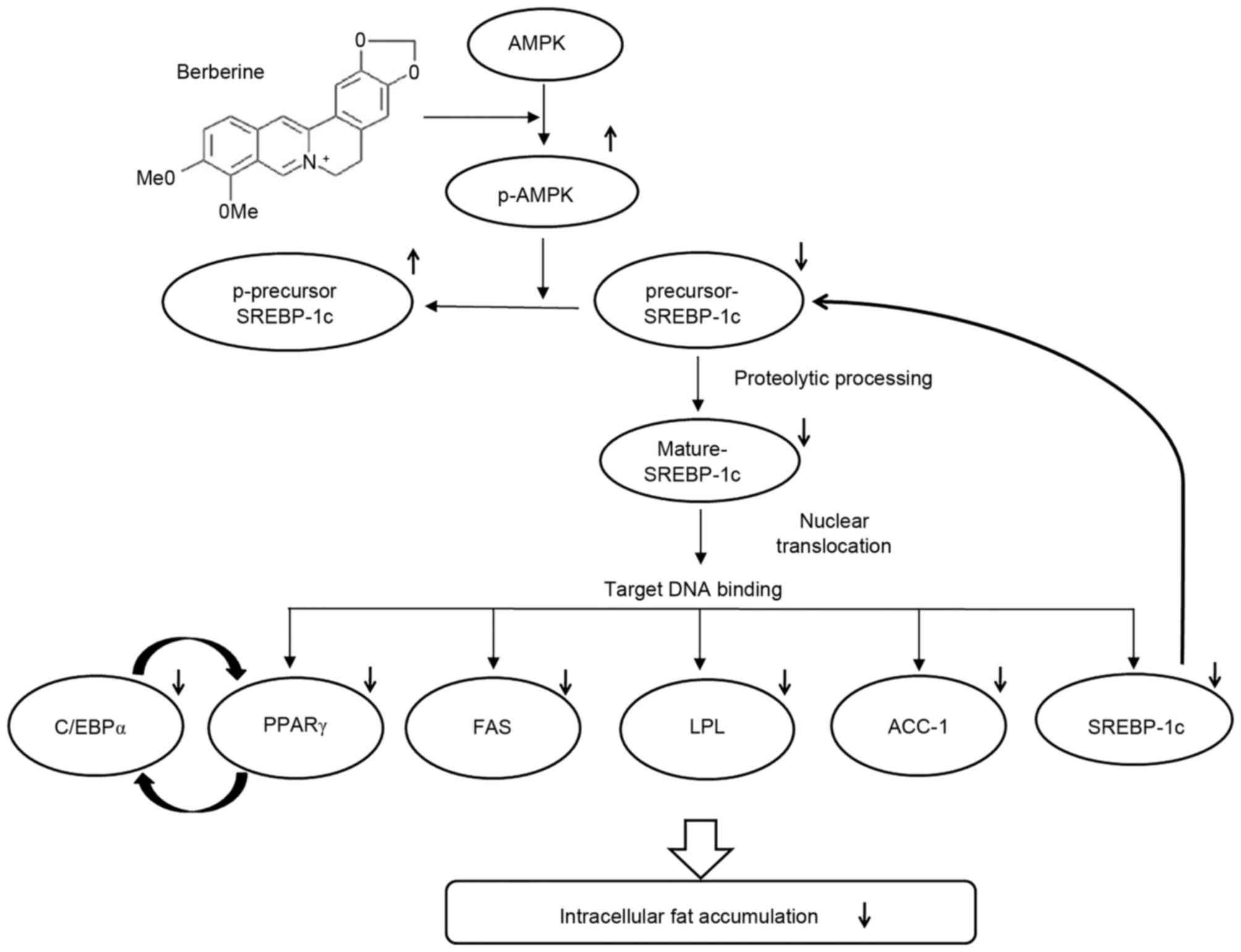
by AMPKα1 siRNA transfection in a dose-dependent manner (Fig. 7). Therefore, the results of the
present study demonstrate that AMPK serves an essential role in the
BBR-induced phosphorylation of SREBP-1c and the suppression of
proteolytic processing, nuclear translocation and target DNA
binding of SREBP-1c, which leads to the downregulation of
lipogenesis-associated gene expression and decreased intracellular
fat accumulation (Fig. 8).
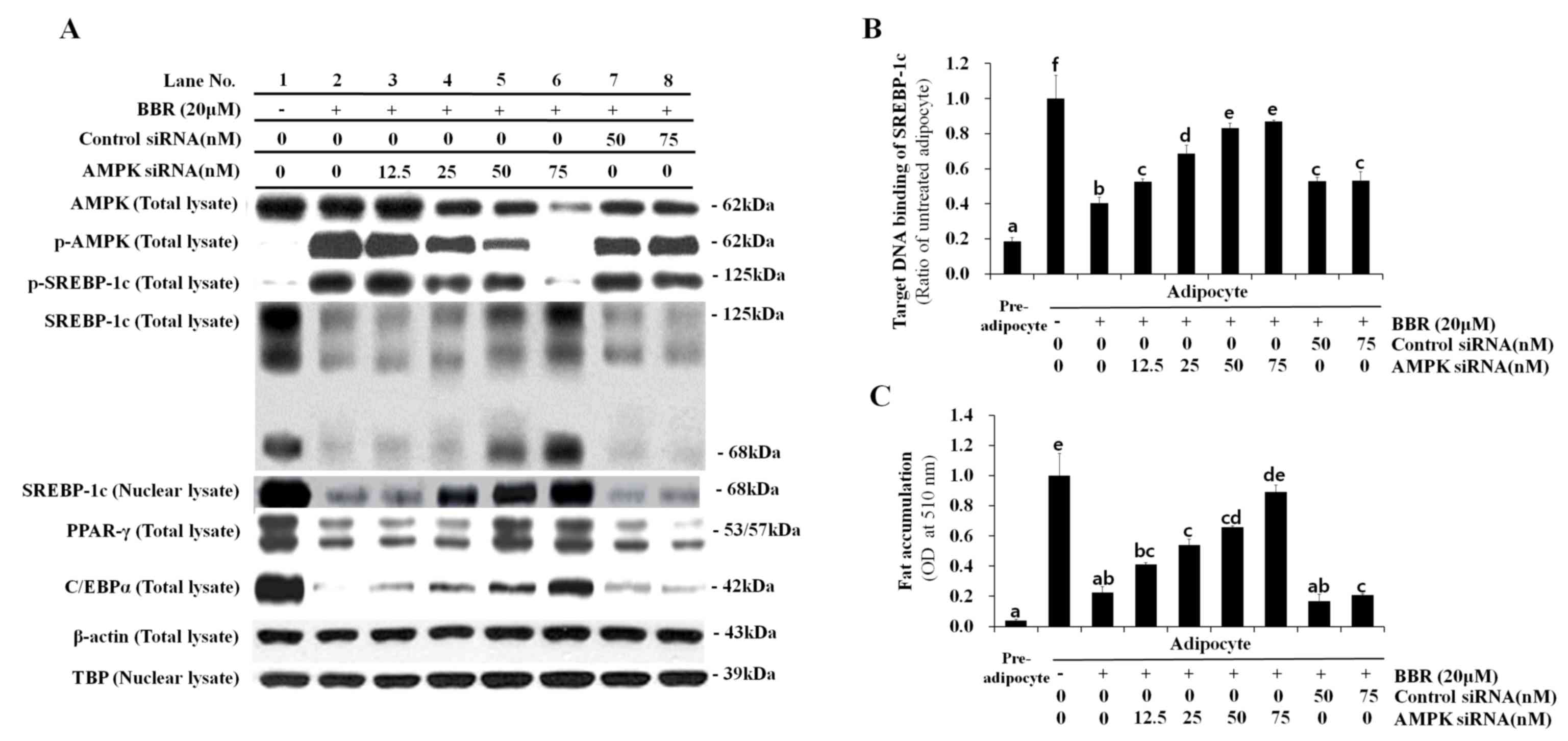 | Figure 6.Effect of AMPK knockdown on SREBP-1c
expression and intracellular fat accumulation following BBR
treatment. 3T3-L1 cells were transfected with 12.5, 25, 50, or 75
nM AMPKα1 siRNA, or 50 or 75 nM control siRNA at one day prior to
induction of differentiation. Cells were treated with 20 µM BBR
from days 2–7 of differentiation, and were harvested on day 7 for
analysis. (A) The protein expression levels of p-AMPK and
p-SREBP-1c, as well as the 125-kDa precursor SREBP-1c isoform and
the mature 68-kDa isoform, were analyzed in total cell lysates by
western blotting. The levels of the mature 68-kDa SREBP-1c isoform
were analyzed in nuclear lysates in order to examine its nuclear
translocation. Levels of the major adipogenic transcription
factors, PPARγ and C/EBPα, were analyzed in total cell lysates.
β-actin and TBP were used as endogenous controls for the analysis
of protein expression in total cell lysates and nuclear lysates,
respectively. (B) The binding of nuclear SREBP-1c to its E-box
motif-containing target DNA sequence, 5′-TCACCTGA-3′, was measured
by ELISA. (C) Oil red O stain in intracellular fat droplets was
extracted using isopropyl alcohol and quantified by measuring the
OD at 510 nm. Different letters indicate significant differences
between groups (P<0.05). AMPK, AMP-activated protein kinase;
SREBP-1c, sterol regulatory element binding protein-1c; BBR,
berberine; siRNA, small-interfering RNA; p-AMPK,
phosphorylated-AMPK; p-SREBP-1c, phosphorylated-SREBP-1c; PPARγ,
peroxisome proliferator-activated receptor-γ; C/EBPα,
CCAAT/enhancer-binding protein α; TBP, TATA box-binding protein;
OD, optical density. |
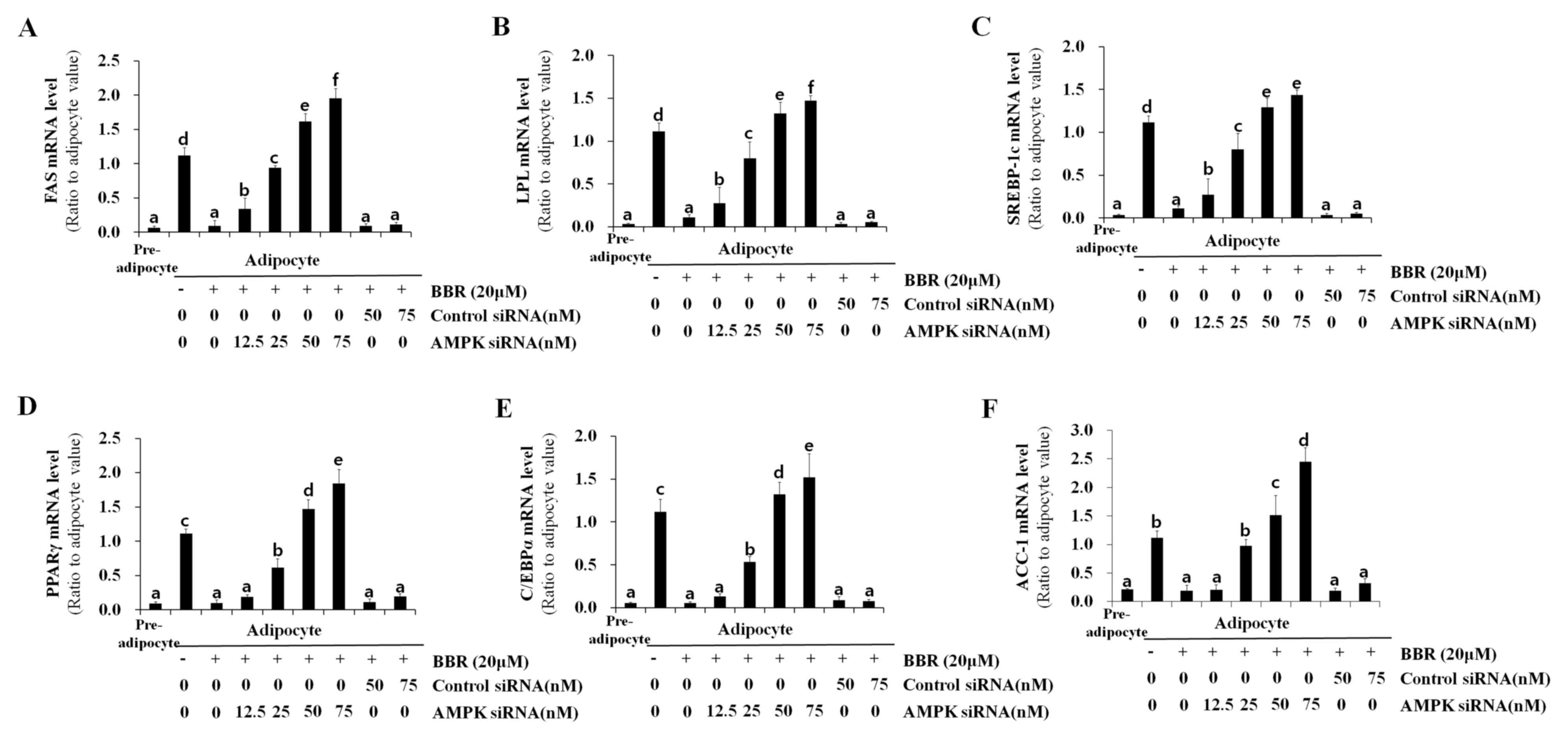 | Figure 7.Effect of AMPK knockdown on the
expression of lipogenesis-associated genes. 3T3-L1 cells were
transfected with 12.5, 25, 50, or 75 nM AMPKα1 siRNA, or 50 or 75
nM control siRNA at one day prior to induction of differentiation.
Cells were treated with 20 µM BBR from days 2–7 of differentiation,
and were harvested on day 7 for analysis. The mRNA level of (A)
FAS, (B) LPL, (C) SREBP-1c, (D) PPARγ, (E) C/EBPα and (F) ACC-1
were measured by reverse transcription-quantitative polymerase
chain reaction. Different letters indicate significant differences
between groups (P<0.05). AMPK, AMP-activated protein kinase;
siRNA, small-interfering RNA; BBR, berberine; FAS, fatty acid
synthase; LPL, lipoprotein lipase; SREBP1-c, sterol regulatory
element binding protein-1c; PPARγ, peroxisome
proliferator-activated receptor-γ; C/EBPα, CCAAT/enhancer-binding
protein α; ACC-1, acetyl-CoA carboxylase-1. |
Discussion
SREBPs are helix-loop-helix-leucine zipper
transcription factors that regulate lipid homeostasis by
controlling the expression of genes required for the synthesis of
fatty acids, triacylglycerol and cholesterol (7). The three SREBP isoforms, SREBP-1a,
SREBP-1c and SREBP-2, serve different roles in lipid metabolism.
SREBP-1c and SREBP-1a arise from the SREBP-1 gene through the use
of alternative first exons, and SREBP-1c is the predominant isoform
in the majority of tissues, including adipose tissue, liver and
skeletal muscle. SREBP-1c is involved in fatty acid synthesis and
lipogenesis, whereas SREBP-2 is primarily involved in cholesterol
synthesis. The transcription of SREBP-1c is induced by insulin, and
the SREBP-1c protein is synthesized as a 125-kDa precursor
containing an N-terminal transcription factor domain and a
C-terminal regulatory domain. Precursor SREBP-1c remains in the
membrane of the endoplasmic reticulum until it is transported to
the Golgi apparatus. In the Golgi apparatus, the membrane-bound
125-kDa precursor SREBP-1c undergoes proteolytic processing to
generate the soluble 68-kDa mature SREBP-1c, which contains a
transcription factor domain and a nuclear localization signal. Once
soluble, the 68-kDa mature SREBP-1c enters the nucleus to bind its
target DNA sequence within the promoters of a number of lipogenic
genes (7,22,23).
In the nucleus, SREBP-1c, also termed adipocyte
determination- and differentiation-dependent factor 1, binds to its
binding sequence, E-box or a sterol regulatory element (SRE) in the
promoter region of many lipogenesis-associated genes, including
that of SREBP-1. In this way, mature SREBP-1c upregulates its own
mRNA expression by binding to the promoter of the SREBP-1 gene
(16). The promoter of the PPARγ
gene contains an E-box motif that mediates its regulation by
SREBP-1 (17). PPARγ immediately
induces C/EBPα to construct a mutual activation loop (24). SREBP-1c has been reported to
transactivate the promoter of the LPL gene in 3T3-L1 cells
(18). The promoter of the ACC
gene contains an E-box motif for SREBP-1c binding, together with
binding sites for additional transcription factors, such as
upstream stimulatory factor (USF) and nuclear factor (NF)-Y
(19). FAS gene transcription is
controlled by USF and SREBP-1c, which bind at the SRE located in
its proximal promoter region (20).
AMPK is a serine/threonine kinase that regulates
metabolic processes by upregulating catabolism and downregulating
anabolism, and its therapeutic importance for metabolic diseases
has led many researchers to search for its downstream target
proteins as well as upstream activators (1). Recently, it was demonstrated that
AMPK phosphorylates precursor SREBP-1c at its Ser372 residue, which
suppresses the proteolytic processing of the precursor 125-kDa
SREBP-1c into the mature 68-kDa SREBP-1c in HepG2 hepatocellular
carcinoma cells and liver tissues of low-density lipoprotein
receptor-deficient diabetic mice (15). However, this AMPK-SREBP-1c
signaling pathway has not been reported as a mechanism of action of
any anti-adipogenic agent in adipocytes.
In the present study, BBR, an AMPK activator of
natural origin, induced the phosphorylation of precursor SREBP-1c
in a time- and dose-dependent manner, suppressing its proteolytic
processing into mature SREBP-1c, its nuclear translocation and
target DNA binding. The ability of nuclear SREBP-1c to bind to its
target DNA sequence was similar to the observed pattern of mRNA
expression of lipogenesis-associated genes in BBR-treated cells.
Knockdown of AMPK was associated with inhibition of BBR-induced
phosphorylation of precursor SREBP-1c, thus confirming that AMPK is
the kinase responsible for phosphorylating SREBP-1c in adipocytes.
AMPKα1 siRNA rescued the proteolytic processing, nuclear
translocation and target DNA binding of SREBP-1c, which was
suppressed by BBR. Accordingly, the expression of
lipogenesis-associated genes, which were suppressed by BBR, was
rescued by AMPKα1 siRNA. SREBP-1c is one of the target genes of
which transcriptions were activated by the binding of mature
SREBP-1c, and levels of SREBP-1c mRNA and SREBP-1c precursor
protein were rescued by AMPKα1 siRNA. The mRNA levels of a number
of lipogenic genes demonstrated similar expression patterns;
however, they did not correspond completely with the ability of
nuclear SREBP-1c to bind to target DNA. The may be due to the
possibility that additional transcription factors, including USF,
NF and C/EBPα, may additionally be involved in these processes in
cooperation with SREBP-1c (19,20).
Acknowledgements
The present study was supported by the Korean Health
Technology Research and Development Project of the Ministry of
Health and Welfare (grant nos. HI15C0075 and HI14C2687).
Glossary
Abbreviations
Abbreviations:
|
ACC
|
acetyl-CoA carboxylase
|
|
AMPK
|
AMP-activated protein kinase
|
|
BBR
|
berberine
|
|
C/EBP
|
CCAAT/enhancer-binding protein
|
|
FAS
|
fatty acid synthase
|
|
IRS
|
insulin receptor substrate
|
|
LPL
|
lipoprotein lipase
|
|
NF
|
nuclear factor
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
SRE
|
sterol regulatory element
|
|
SREBP
|
sterol regulatory element binding
protein
|
|
TBP
|
TATA box-binding protein
|
|
USF
|
upstream stimulatory factor
|
References
|
1
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar
|
|
2
|
Daval M, Diot-Dupuy F, Bazin R, Hainault
I, Viollet B, Vaulont S, Hajduch E, Ferré P and Foufelle F:
Anti-lipolytic action of AMP-activated protein kinase in rodent
adipocytes. J Biol Chem. 280:25250–25257. 2005. View Article : Google Scholar
|
|
3
|
Gowans GJ, Hawley SA, Ross FA and Hardie
DG: AMP is a true physiological regulator of AMP-activated protein
kinase by both allosteric activation and enhancing net
phosphorylation. Cell Metab. 18:556–566. 2013. View Article : Google Scholar :
|
|
4
|
Vuddanda PR, Chakraborty S and Singh S:
Berberine: A potential phytochemical with multispectrum therapeutic
activities. Expert Opin Investig Drugs. 19:1297–1307. 2010.
View Article : Google Scholar
|
|
5
|
Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ,
Shen Y, Ye JM, Lee CH, Oh WK, Kim CT, et al: Berberine, a natural
plant product, activates AMP-activated protein kinase with
beneficial metabolic effects in diabetic and insulin-resistant
states. Diabetes. 55:2256–2264. 2006. View Article : Google Scholar
|
|
6
|
Turner N, Li JY, Gosby A, To SW, Cheng Z,
Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, et al:
Berberine and its more biologically available derivative,
dihydroberberine, inhibit mitochondrial respiratory complex I: A
mechanism for the action of berberine to activate AMP-activated
protein kinase and improve insulin action. Diabetes. 57:1414–1418.
2008. View Article : Google Scholar
|
|
7
|
Jeon TI and Osborne TF: SREBPs: Metabolic
integrators in physiology and metabolism. Trends Endocrinol Metab.
23:65–72. 2012. View Article : Google Scholar
|
|
8
|
Li GS, Liu XH, Zhu H, Huang L, Liu YL, Ma
CM and Qin C: Berberine-improved visceral white adipose tissue
insulin resistance associated with altered sterol regulatory
element-binding proteins, liver × receptors and peroxisome
proliferator-activated receptors transcriptional programs in
diabetic hamsters. Biol Pharm Bull. 34:644–654. 2011. View Article : Google Scholar
|
|
9
|
Hu Y, Kutscher E and Davies GE: Berberine
inhibits SREBP-1-related clozapine and risperidone induced
adipogenesis in 3T3-L1 cells. Phytother Res. 24:1831–1838. 2010.
View Article : Google Scholar
|
|
10
|
Huang C, Zhang Y, Gong Z, Sheng X, Li Z,
Zhang W and Qin Y: Berberine inhibits 3T3-L1 adipocyte
differentiation through the PPARgamma pathway. Biochem Biophys Res
Commun. 348:571–578. 2006. View Article : Google Scholar
|
|
11
|
Gustafson B and Smith U: Cytokines promote
Wnt signaling and inflammation and impair the normal
differentiation and lipid accumulation in 3T3-L1 preadipocytes. J
Biol Chem. 281:9507–9516. 2006. View Article : Google Scholar
|
|
12
|
Li J, Zhang D, Ward KM, Prendergast GC and
Ayene IS: Hydroxyethyl disulfide as an efficient metabolic assay
for cell viability in vitro. Toxicology in Vitro. 26:603–612. 2012.
View Article : Google Scholar :
|
|
13
|
Lee H, Bae S and Yoon Y: The
anti-adipogenic effects of (−)epigallocatechin gallate are
dependent on the WNT/β-catenin pathway. J Nutr Biochem.
24:1232–1240. 2013. View Article : Google Scholar
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X,
Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar :
|
|
16
|
Amemiya-Kudo M, Shimano H, Yoshikawa T,
Yahagi N, Hasty AH, Okazaki H, Tamura Y, Shionoiri F, Iizuka Y,
Ohashi K, et al: Promoter analysis of the mouse sterol regulatory
element-binding protein-1c gene. J Biol Chem. 275:31078–31085.
2000. View Article : Google Scholar
|
|
17
|
Fajas L, Schoonjans K, Gelman L, Kim JB,
Najib J, Martin G, Fruchart JC, Briggs M, Spiegelman BM and Auwerx
J: Regulation of peroxisome proliferator-activated receptor gamma
expression by adipocyte differentiation and determination factor
1/sterol regulatory element binding protein 1: Implications for
adipocyte differentiation and metabolism. Mol Cell Biol.
19:5495–5503. 1999. View Article : Google Scholar :
|
|
18
|
Schoonjans K, Gelman L, Haby C, Briggs M
and Auwerx J: Induction of LPL gene expression by sterols is
mediated by a sterol regulatory element and is independent of the
presence of multiple E boxes. J Mol Biol. 304:323–334. 2000.
View Article : Google Scholar
|
|
19
|
Barber MC, Vallance AJ, Kennedy HT and
Travers MT: Induction of transcripts derived from promoter III of
the acetyl-CoA carboxylase-alpha gene in mammary gland is
associated with recruitment of SREBP-1 to a region of the proximal
promoter defined by a DNase I hypersensitive site. Biochem J.
375:489–501. 2003. View Article : Google Scholar :
|
|
20
|
Griffin MJ and Sul HS: Insulin regulation
of fatty acid synthase gene transcription: Roles of USF and
SREBP-1c. IUBMB Life. 56:595–600. 2004. View Article : Google Scholar
|
|
21
|
Bene H, Lasky D and Ntambi JM: Cloning and
characterization of the human stearoyl-CoA desaturase gene
promoter: Transcriptional activation by sterol regulatory element
binding protein and repression by polyunsaturated fatty acids and
cholesterol. Biochem Biophys Res Commun. 284:1194–1198. 2001.
View Article : Google Scholar
|
|
22
|
Wang X, Sato R, Brown MS, Hua X and
Goldstein JL: SREBP-1, a membrane-bound transcription factor
released by sterol-regulated proteolysis. Cell. 77:53–62. 1994.
View Article : Google Scholar
|
|
23
|
Eberle D, Hegarty B, Bossard P, Ferré P
and Foufelle F: SREBP transcription factors: Master regulators of
lipid homeostasis. Biochimie. 86:839–848. 2004. View Article : Google Scholar
|
|
24
|
Zhao QH, Wang SG, Liu SX, Li JP, Zhang YX,
Sun ZY, Fan QM and Tian JW: PPARγ forms a bridge between DNA
methylation and histone acetylation at the C/EBPα gene promoter to
regulate the balance between osteogenesis and adipogenesis of bone
marrow stromal cells. FEBS J. 280:5801–5814. 2013. View Article : Google Scholar
|