Introduction
Tumor necrosis factor (TNF)-α is a 17-kDa oligomeric
glycoprotein, which is primarily stimulated by lipopolysaccharides
(LPS) (1,2). TNF-α exists in two forms,
transmembrane-TNF-α (TM-TNF-α) and soluble (s)-TNF-α (3,4).
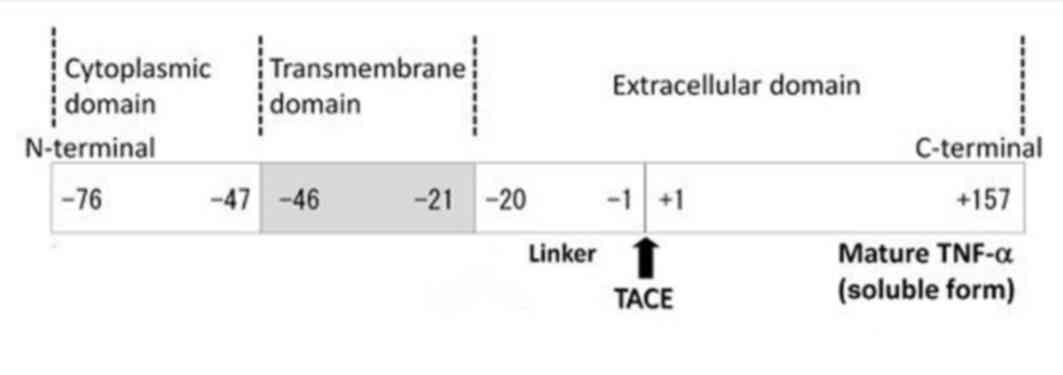
TM-TNF-α is the precursor to s-TNF-α, and its primary structure
consists of 233 amino acid residues, where −76 to −1 amino acids
comprises a leader peptide sequence, −1 to −20 is a linkage segment
and −21 to −46 is a hydrophobic region (5,6).
These sequences comprise the transmembrane segment, whilst the
remainder of the sequence forms the intracellular domain. Under the
effect of the metalloproteinase, tumor necrosis factor-α converting
enzyme, TM-TNF-α extracellular segments 1 to 157 can be hydrolyzed
(Fig. 1). This leads to TM-TNF-α
removal and transformation into s-TNF-α (7–9).
Initially TNF-α is expressed as a membrane protein, and then
following the removal of the extracellular segment by
metalloproteinase, it is transformed into s-TNF-α. This process
requires hydrophobic transmembrane domains in the target protein to
determine transmembrane structure, as well as signaling peptides to
mediate the transport of proteins across the transmembrane. This
process is based on a supported signaling hypothesis (10), whereby the peptide, initially
translated by the mRNA signal sequence in the cytoplasm, is
recognized by the signal recognition particle (SRP), which presents
the signaling peptide recognition site, the translation termination
site and the SRP receptor recognition site. SRP then recognizes the
signaling peptide, which leads to termination of mRNA translation.
SRP subsequently combines with the SRP receptor, also known as the
docking protein (DP), which is located on the endoplasmic
reticulum. SRP then binds guanosine 5′-triphosphate (GTP) at the
GTP catalytic site and transfers the mRNA to a channel protein
known as translocon, which resides in the endoplasmic reticulum in
synergy with DP. mRNA is then transferred to the endoplasmic
reticulum for co-translation. There are two types of signal
generated by this mRNA; the start transfer membrane-anchor sequence
and stop transfer membrane-anchor sequence. When the stop transfer
membrane-anchor sequence signal is generated, mRNA remains on the
surface of the endoplasmic reticulum without entering, and produces
transmembrane proteins during translation (10). The start transfer membrane-anchor
sequence signal induces mRNA to enter the endoplasmic reticulum and
produce multiple transmembrane proteins following translation
(11).
The aim of the present study was to design a
eukaryotic expression vector based on the transformation of
TM-TNF-α into s-TNF-α, which allowed for the hydrolyzation of
membrane fusion proteins in a controlled manner. The sequence of
mRNA encoding the extracellular segment of TM-TNF-α was processed
using enterokinase and FactorXa, and TNF-α mRNA was then inserted
using guiding peptides. The whole recombinant RNA was inserted into
the eukaryotic expression vector pcDNA3.1(+) thus generating a
novel recombinant plasmid. The plasmid was subsequently transfected
into mouse fibroblasts, in order to produce a membrane protein with
an extracellular segment of TNF-α containing a protease cleavage
site in the middle linker region, which was processed with
corresponding enzymes. The target protein was then obtained using
digestive enzyme solutions.
Materials and methods
Materials
Escherichia coli (E. coli) DH5α, E.
coli JM109, pcDNA3.1(+), s-TNF-α cDNA, mouse fibroblast cells
(L929) and mouse embryonic fibroblasts (3T3 cell) were provided by
the laboratory of the Guangdong Medical College Institute
(Guangdong, China), and the human early immature myeloid leukemia
cell line (HL-60) was donated by Professor Yu Lijian (Guangdong
Ocean University, Zhanjiang, Guangdong, China). RPMI-1640,
Dulbecco's modified Eagle's medium (DMEM) and the Lipofectamine™
2000 transfection reagent were purchased from Invitrogen; Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). Fetal bovine serum
(FBS) was purchased from Hangzhou Sijiqing Biological Engineering
Materials Co., Ltd. (Hangzhou, China). Goat anti-mouse
IgG-horseradish peroxidase (HRP) antibody (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA; cat. no. SC-2005; 200 µg/0.5
ml) was purchased online from univ-bio (http://univ.univ-bio.com/; owned by Shanghai You
Ningwei Biotechnology Co., Ltd., Shanghai, China). Mouse anti-human
TNF-α was purchased from BD Biosciences (Franklin Lakes, NJ, USA).
All other chemicals used in this study were national analytical
reagents. The gel imaging analysis system, Molecular Imager
(ChemiDoc™ XRS+), polymerase chain reaction (PCR) instrument
(PTC200 PCR Thermal Cycler), vertical electrophoresis and
electrophoresis apparatus were purchased from Bio-Rad Laboratories,
Inc. (Hercules, CA, USA).
Acquisition of transmembrane TNF-α
cDNA
The expression of TM-TNF-α in human HL-60
promyelocytic leukemia cells was induced by treating cells with
phorbol ester (100 ng/ml) and LPS (100 ng/ml; Beijing LEYBOLD Cable
Technology Co., Ltd., Beijing, China) (12,13).
Total RNA was isolated from HL-60 cells (2×106/ml) using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's recommendations. Using a reverse transcription
(RT)-PCR kit (Takara Bio Inc., Otsu, Japan), total RNA was reverse
transcribed to TM-TNF-α cDNA. The reaction conditions were as
follows: Initial denaturation at 50°C for 30 min, followed by 30
cycles of denaturation at 94°C for 2 min, 94°C for 15 sec and 68°C
for 30 sec, then a final extension at 72°C for 10 min. The TM-TNF
-α gene was amplified using the following primers (reference
sequence, NM_000594.2, https://www.ncbi.nlm.nih.gov/nuccore/25952110): Sense
primer 5′-CGCGGATCCCAGCAAGGACAGCAGAGGACCAG-3′, and anti-sense
primer 5′-CCGGAATTCGAGGCGTTTGGGAAGGTTGGA−3′. The PCR product (904
bp) contained a BamHI restriction site and, downstream of
the amplified DNA fragment, an EcoRI restriction site (New
England Biolabs Ltd., Beijing, China). PCR products were detected
by 2% agarose gel electrophoresis (Gene Tech Biotechnology Co.,
Ltd., Shanghai, China).
Construction of
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
plasmid vectors
NHeI, PmeI and EcoRI
restriction enzymes were purchased from New England Biolabs Ltd.
According to the primer design guidelines (Generay Biotech Co.,
Ltd., Shanghai, China), the enterokinase primer sequences were as
follows: Sense primer (5′ add the NheI restriction site to
cDNA, 162–186 nucleotides), 5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGC-3′,
anti-sense primer, (5′ add the enterokinase and PmeI
restriction site to cDNA, 361–385 nucleotides),
5′-GCGTTTAAACCCTTGTCGTCGTCGTCGCTGATTAGAGAGAGGTCC-3′.
The enterokinase coding region insertion fragment
was 256 bp in length, and the sequence was as follows:
5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGCATGATCCGGGACGTGGAGCTGGCCGAGGAGGCGCTCCCCAAGAAGACAGGGGGGCCCCAGGGCTCCAGGCGGTGCTTGTTCCTCAGCCTCTTCTCCTTCCTGATCGTGGCAGGCGCCACCACGCTCTTCTGCCTGCTGCACTTTGGAGTGATCGGCCCCCAGAGGGAAGAGTTCCCCAGGGACCTCTCTCTAATCAGCGACGACGACGACAAGGGTTTAAACGC-3′.
The primer sequences for FactorXa were as follows: Sense primer (5′
add the NheI restriction site to cDNA, 162–186 nucleotides),
5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGC−3′, anti-sense primer (5′ add
the enterokinase and PmeI restriction site to cDNA, 361–385
nucleotides),
5′-GCGTTTAAACTCGACCCGCGTCCCTCAATGCTGATTAGAGAGAGGTCC−3′. The
FactorXa coding region insertion fragment was 259 bp in length and
the sequence was as follows:
5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGCATGATCCGGGACGTGGAGCTGGCCGAGGAGGCGCTCCCCAAGAAGACAGGGGGGCCCCAGGGCTCCAGGCGGTGCTTGTTCCTCAGCCTCTTCTCCTTCCTGATCGTGGCAGGCGCCACCACGCTCTTCTGCCTGCTGCACTTTGGAGTGATCGGCCCCCAGAGGGAAGAGTTCCCCAGGGACCTCTCTCTAATCAGCATTGAGGGACGCGGGTCGAGTTTAAACGC-3′.
Using TM-TNF-α cDNA as a template and the
aforementioned primers, PCR was performed as follows: Hot start at
98°C for 2 min followed by 30 cycles of denaturation at 98°C for 15
sec, annealing at 68°C for 30 sec, and a final extension step for
at 72°C 10 min. The final holding temperature was 4°C.
The transmembrane region sequence was then annealed
to the enterokinase restriction site and s-TNF-α (700 bp in
length), using the following primer sequences: Fragment 1A primer
(amplicon length, 246 bp), E1 upstream,
5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGC-3′ (5′ add the NheI
restriction site), and E1 downstream,
5′-CCTTGTCGTCGTCGTCGCTGATTAGAGAGAGGTCCCTGGGGGACTCTTCC-3′; fragment
2A primer (amplicon length, 473 bp), TNF -α upstream,
5′-CTAATCAGCGACGACGACGACAAGGGTTTAAACAAGCCTG-3′, and TNF -α
downstream, 5′-CGCGAATTCTCACAGGGCAATGATCCCAAAG-3′ (5′ add the
EcoRI restriction site).
The sequence of transmembrane region was then
annealed to the FactorXa restriction site and s-TNF -α (706 bp in
length) using the following primer sequences: Fragment 1B primer,
(amplicon length, 245 bp), F1 upstream,
5′-CGGCTAGCAAGGACACCATGAGCACTGAAAGC-3′ (5′ add the NheI
restriction site), and F1 downstream,
5′-CCCGCGTCCCTCAATGCTGATTAGAGAGAGGTCCCTGGGGGACTCTTCC-3′; fragment
2B primer (amplicon length, 489 bp), TNF-α upstream,
5′-CTAATCAGCATTGAGGGACGCGGGTCGAGTTTAAACAAGCCTG-3′, and TNF -α
downstream, 5′-CGCGAATTCTCACAGGGCAATGATCCCAAAG-3′ (3′ add the
EcoRI restriction site).
Using the TNF -α transmembrane segment plasmid and
s-TNF-α plasmid as a template together with the associated primers,
the fragment was connected by overlap extension PCR. The overlap
extension PCR was performed by incubating samples at 95°C for 5 min
and 72°C for 10 min.
PCR products were digested with EcoRI and
BamHI before they were cloned into the corresponding
restriction sites in the pcDNA3.1 vector to generate the
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
plasmids for expression. The ligation products were transformed
into chemocompetent E. coli DH5a cells (E. coli DH5a
cells treated by precooled 0.1 mol/l CaCl2) maintained
in LB medium (Shanghai Kehua Bio-Engineering Co., Ltd., Shanghai,
China) and bacterial culture medium containing ampicillin (100
µg/ml; Tiangen Biotech Co., Ltd., Beijing, China), before cultures
were incubated at 37°C for 12 to 16 h in inverted culture. To
select for the recombinant plasmid-positive colonies, 1% agarose
gel electrophoresis was performed, followed by DNA sequencing
(sequenced by Sangon Biotech Co., Ltd., Shanghai, China).
Transfection of 3T3 cells
3T3 cells were seeded into a 60-mm culture dish at a
density of 1×106 cell/dish at 12 h prior to DNA transfection. A
total of 2.5 µg DNA plasmids were diluted in 125 µl opti-MEM medium
(Invitrogen; Thermo Fisher Scientific, Inc.), and Lipofectamine™
2000 transfection reagent was used to transfect 3T3 cells with
plasmid DNA. Following 5 min, the diluted plasmid DNA and
Lipofectamine™ 2000 were mixed together and incubated at room
temperature for 20 min. The mixture was then added to each dish and
incubated at 37°C in a 5% CO2 incubator for 5 h.
Non-transfected 3T3 cells were the control group. Following
incubation, the media was replaced with DMEM (Invitrogen; Thermo
Fisher Scientific, Inc.), supplemented with 10% FBS (Hangzhou
Sijiqing Biological Engineering Materials Co., Ltd.).
Analysis of the expression of TNF-α
RNA
Total cellular RNA was isolated using Trizol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) in 3T3 cells following
48 h of transfection, according to the manufacturer's
recommendations. All processes were conducted in a sterile and
ribonuclease-free environment. Isolated RNA was dissolved in
nuclease-free water and stored at −20°C. RT-PCR was performed using
FastQuant cDNA first strand synthesis kit (Tiangen Biotech Co.,
Ltd., Beijing, China) using total RNA (1 µg) for 5 min at 42°C. The
cDNA samples from RT reactions were amplified using 2X Taq PCR
MasterMix (Tiangen Biotech Co., Ltd.) and the following primers:
Sense, 5′-GGACGCGGGTCGAGTTTAAACAAGCCTG-3′, and anti-sense,
5′-CGAATTCTCACAGGGCAATGATCCC-3′. Thermal cycling conditions were as
follows: Incubation at 50°C for 30 min and 94°C for 2 min, followed
by 30 cycles of 94°C for 15 sec, 68°C for 30 sec and extension at
72°C for 10 min. The PCR products were loaded onto a 1% agarose gel
containing SYBR® Safe DNA Gel Stain (Thermo Fisher Scientific,
Inc.) for electrophoresis at 100 V for 40 min. The gel was then
analyzed and photographed using the BIO-RAD Gel Doc XR + (Bio-Rad
Laboratories, Inc.).
Detection of TNF-α expression in total
protein cell lysates by western blotting
Following incubation of transfected cells for 24 h,
total protein was extracted using a mixture of 1% protease
inhibitors (Merck KGaA, Darmstadt, Germany) and 1%
phenylmethylsulfonyl fluoride (Beyotime Institute of Biotechnology,
Haimen, China) in radioimmunoprecipitation lysis buffer (Beyotime
Biotechnology Co, Shanghai, China). Protein concentration was
determined using a Bradford Protein assay kit (Bi Yuntian
Biological Technology Institution, Shanghai, China). Proteins were
separated on 10 and 5% SDS-PAGE gels and transferred to a
nitrocellulose filter membrane. The membranes were blocked with 5%
non-fat milk in phosphate-buffered saline containing Tween-20
(PBST) for 2 h at room temperature, and were subsequently incubated
with mouse anti-human TNF-α antibody (cat. no. 551220; dilution,
1:1,000 in Primary Antibody Dilution Buffer; Beyotime Institute of
Biotechnology) overnight at 4°C. Following three washes with PBST,
the membrane was then incubated with goat anti-mouse IgG-HRP
antibody (cat. no. sc-2005; dilution, 1:1,000 in Primary Antibody
Dilution Buffer; Beyotime Institute of Biotechnology) for 1 h at
room temperature. In the same manner, the membrane was incubated
with the primary antibody β-Actin (1:1,000; cat. no. 8H10D10; mouse
monoclonal antibody; Cell Signaling Technology, Inc., Danvers, MA,
USA) and then the secondary antibody, goat anti-mouse IgG-HRP, to
determine the β-actin protein expression to be used as an internal
control. Protein bands were visualized using BeyoECL Plus
electro-chemiluminescence reagent (Beyotime Institute of
Biotechnology), following exposure to X-ray film.
Detection of TNF-α in extracellular
digestion mixtures
Following transfection with
pcDNA3.1-TM-enterokinase-TNF-α plasmid, pcDNA3.1-TM-plasmid
FactorXa-TNF-α or control (non-transfected cells) for 24 h, the
media was removed cells were washed 7 or 8 times with PBS to remove
additional proteins. Digestion was performed by applying
enterokinase (2.5 U; GenScript, Piscataway, NJ, USA), 10X
enterokinase buffer [1 M NaCl, 500 mM Tris-HCl, 50 mM
CaCl2], 1X FactorXa/enterokinase Dil/Stor buffer (500 mM
NaCl, 20 mM Tris-HCl, 2 mM CaCl2, 50% glycerol),
FactorXa (3.5 U, Novagen, Merck KGaA); 10X Xa cleavage buffer (1 M
NaCl, 500 mM Tris-HCl) and 1X Xa Dil/Stor buffer to the cells. The
cells were then incubated at 22°C for 6 to 8 h. A dialysis bag was
cut into small sections of appropriate length (10 to 20 cm) and the
pieces were boiled in 2% (w/v) sodium bicarbonate and 1 mmol/l EDTA
(pH 8.0) for 10 min. The sections were thoroughly cleaned using
distilled water and were boiled again in 1 mmol/l EDTA (pH 8.0) for
10 min, and stored at 4°C; EDTA was washed away prior to use. The
cell digestion mixture was then transferred to the dialysis bags
and immersed in dialysis buffer (0.02 mol/l pH8.0 Tris-HCL, 0.05%
Tween-20, 1 mmol/l NaN3) to allow the sample to become
well dialyzed and concentrated. SDS-PAGE analysis was performed on
82×82 mm2 gels, (the percentage of propylene in the
separation gel and concentrate were 12 and 5%, respectively).
Electrophoresis was performed at 140 V for the separation gel and
at 80 V for the concentrate gel. 0.25% Coomassie brilliant blue
staining was applied at room temperature for 15 min and western
blotting was performed as described in the previous section.
Preliminary study of TNF-α activity in
the extracellular digestive mixture
L929 cells were used to determine the activity of
TNF-α in the extracellular digestion mixture, as they are
susceptible to TNF-α. L929 cells were cultured in RMPI-1640
containing 10% BSA and were incubated at 37°C with 5%
CO2. The L929 cells were collected during the
logarithmic growth phase following trypsin digestion (Amresco, LLC,
Solon, OH, USA), and were diluted to a final concentration of 3×105
cells/ml. The cell suspension (100 µl/well) was transferred to a
96-well plate. Following 24 h of incubation, cell adhesion was
complete. A total of 1 µg fresh medium and actinomycin D (Sangon
Biotech Co., Ltd.) were added to the plate. An appropriate dilution
of RPMI-1640 culture containing 1 µg/ml of actinomycin D was added
to the corresponding well with three kinds of TNF-α [hTNF-α
(Guangdong Medical College Institute, Guangdong, China),
pcDNA3.1-TM-enterokinase-TNF-α extracellular digestion mixture (P1)
and pcDNA3.1-TM-FactorXa-TNF-α extracellular digestion mixture
(P2)], to create the concentration range of 5, 25, 125 and 625
pmol/l. Each sample had three parallel wells. A blank group was set
up with only medium. Following incubation at 37°C with 5%
CO2 for 24 h, 10 µl MTT work solution (concentration, 5
mg/ml) was added to each well. The plate was further incubated at
37°C for 4 h. The supernatant was then carefully aspirated and 200
µl/well DMSO was added. The final A570 was measured using an
enzyme-labeled instrument after complete mixing and the death rate
was calculated. The results were expressed as the mean ± standard
deviation of the three-well optical density (OD) values in each
group. The effect of the different concentrations of protein on the
ability of proliferating tumor cells to inhibit the rate of
toxicity is reflected in the killing rate calculated by the
following formula: Cell killing rate (%) =
[(ODcontrol-ODtest)/ODcontrol] × 100%. Specific activity (hTNF-α
activity unit per mg of protein, U/mg) was evaluated by determining
the dilution of the sample at 50% of the killer cell as an activity
unit [U; the median lethal dose (LD50)]. Cells were
cultured for 24 h to allow for the fusion effect on apoptosis and
then cell morphology was observed using a fluorescence
microscope.
Statistical analysis
The results are presented as the mean ± standard
deviation. Data was analyzed using SPSS 17.0 statistical software
(SPSS, Inc., Chicago, IL, USA). A one-way or two-way analysis of
variance was used to compare differences among groups. Two-way
analysis of variance was used to determine the difference between
groups, one-way analysis of variance was used to determine the
difference between each sample. According to the homogeneity test
of variance, when the total variance was the same the least
significant difference (LSD) method was applied. P<0.05 was
considered to indicate a statistically significant difference.
Results
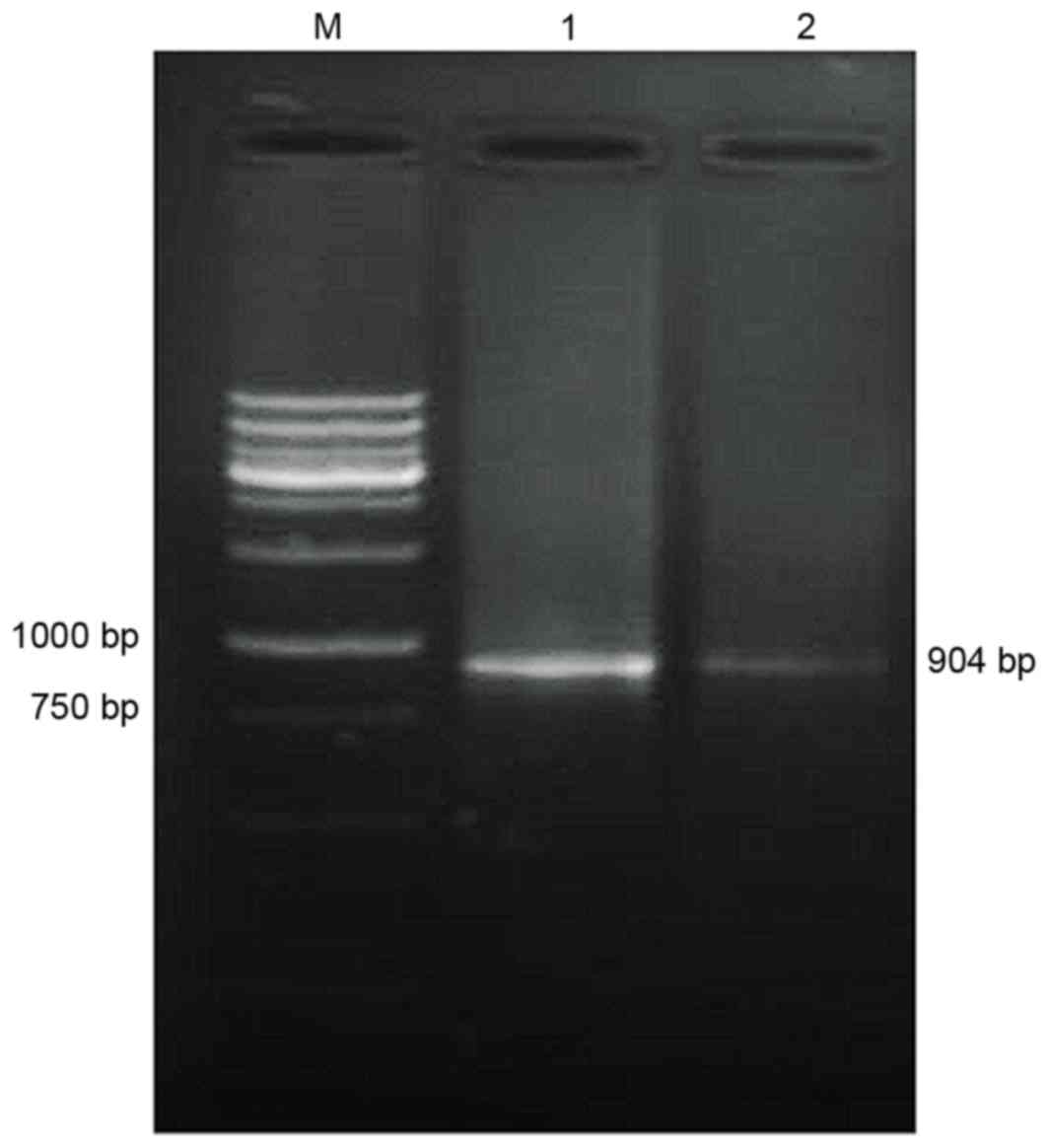
Amplification of TNF-α cDNA
Using the total RNA of the HL-60 cells as a
template, TM-TNF-α cDNA was amplified. Gel electrophoresis
demonstrated that the amplified cDNA matched the size of the target
gene fragments (904 bp; Fig.
2).
Construction and identification of
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
vectors
TM-TNF-α with enterokinase/FactorXa restriction
sites and s-TNF-α cDNA were amplified. Gel electrophoresis
identified four DNA fragments that were 476 bp and 256–259 bp in
length (Fig. 3A). Gel
electrophoresis of TM-TNF-α combined with enterokinase or FactorXa
restriction sites and s-TNF-α cDNA, demonstrated that the DNA bands
were located between 500 bp and 750 bp, with theoretical lengths of
710 bp and 707 bp (Fig. 3B). Gel
electrophoresis of the pcDNA3.1 vector combined with the
TM-enterokinase or FactorXa-TNF-α cDNA fragments, demonstrated that
there were three DNA fragments displayed as selected positive
recombinants following double-digestion with
EcoRI/NheI. The molecular mass of the fragments
matched the pcDNA3.1 vector (5,428 bp in length) and the
TM-enterokinase and FactorXa-TNF-α cDNA sequences (between 707 and
710 bp in length; Fig. 3C). The
DNA sequencing results revealed that the recombinant plasmid vector
was successfully constructed (Fig. 3D
and E).
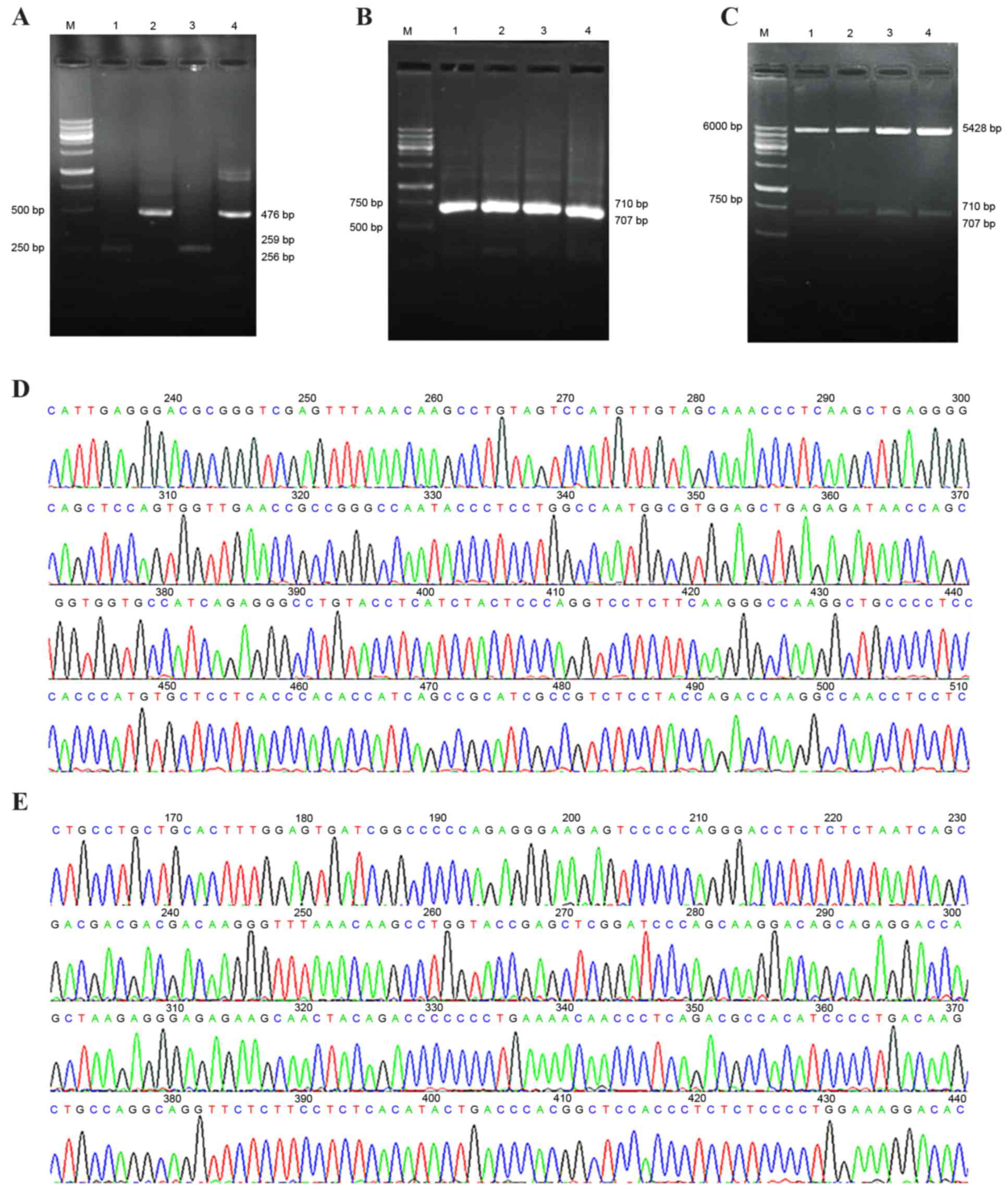 | Figure 3.Construction and identification of
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
vectors. (A) Overlapping PCR fragments analyzed by 2% agarose gel
electrophoresis. M, wide-range DNA marker (100–6,000 bp); lane 1,
the transmembrane domain fragment with an enterokinase restriction
site; lanes 2 and 4, soluble-TNF-α fragment; lane 3, the
transmembrane domain fragment containing a FactorXa restriction
site. (B) Overlapping PCR products analyzed by 2% agarose gel
electrophoresis. M, wide range DNA marker (100–6,000 bp); lanes 1
and 2, fragment with an enterokinase restriction site; lanes 3 and
4, fragment with a FactorXa restriction site. (C) Restriction
enzyme (EcoRI and NheI) digestion of positive cloning
vector analyzed by 2% agarose gel electrophoresis. M, wide-range
DNA marker (100–6,000 bp); lanes 1 and 2, the fragment with an
enterokinase restriction site; lanes 3 and 4, the fragment with a
FactorXa restriction site. (D) DNA sequencing analysis of the
positively cloned recombinant pcDNA3.1-TM-enterokinase-TNF-α
plasmid. (E) DNA sequencing analysis of the positively cloned
pcDNA3.1-TM-FactorXa-TNF-α recombinant plasmid. TM, transmembrane;
TNF-α, tumor necrosis factor-α; PCR, polymerase chain reaction. |
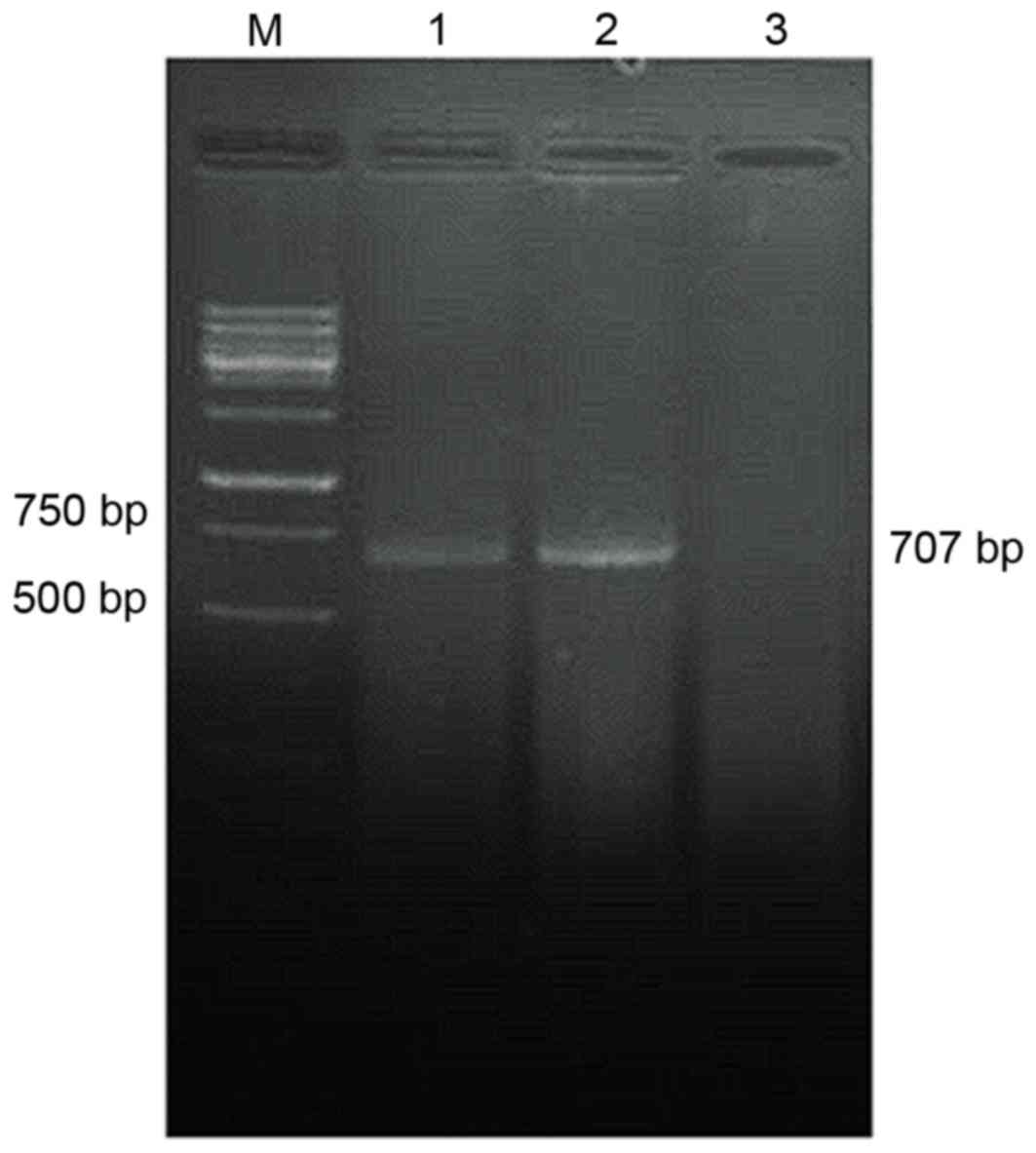
Detection of
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
plasmid expression by RT-PCR
The RT-PCR results demonstrated that the amplified
pcDNA3.1-TM-enterokinase and FactorXa-TNF-α cDNA gene fragments
were ~707 bp in length, whereas no TNF-α cDNA was detected in the
non-transfected control group (Fig.
4).
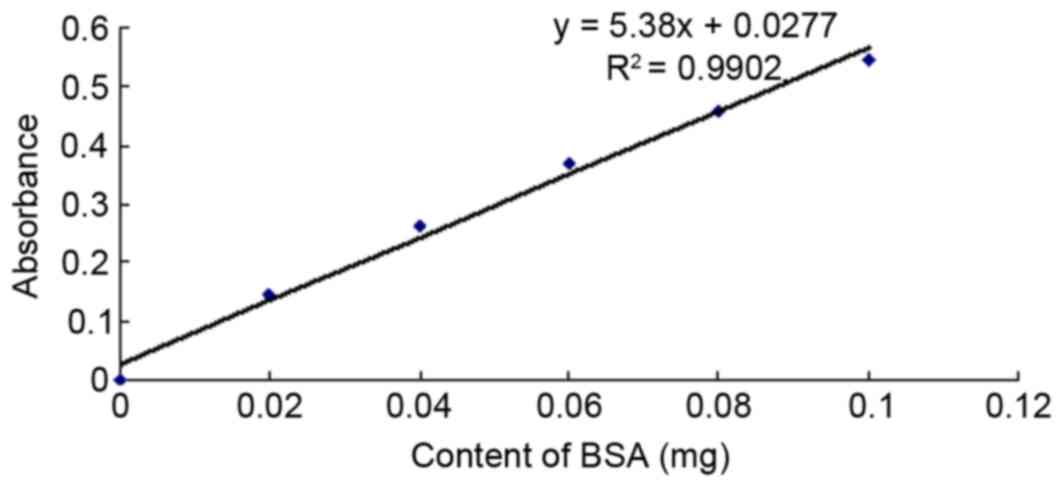
Determination of protein content
According to protein concentration analysis using a
Bradford assay, the BSA standard protein concentration was 1 mg/ml.
Fig. 5 displays the standard
curve, regression equation and the trend line of the standard
protein of the BSA. The standard protein curve and regression
equations indicated that the pcDNA3.1-TM-enterokinase-TNF-α group,
pcDNA3.1-TM-FactorXa-TNF-α group and non-transfected control group
exhibited total protein values of 14.1, 12.9 and 12.6 mg/ml,
respectively.
TNF-α expression in total protein
extracts detected by western blotting
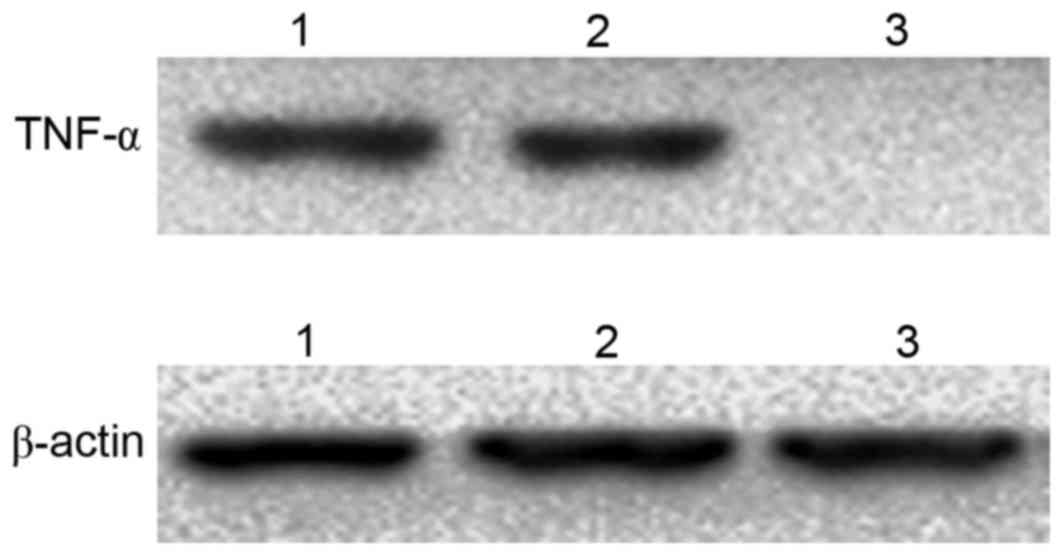
The western blotting results revealed that TNF-α was
detected in 3T3 cells transfected with
pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
plasmids following digestion with the corresponding enzymes,
enterokinase and FactorXa, respectively (Fig. 6). By contrast, no TNF-α expression
was observed in the control group (Fig. 6).
Detection of TNF-α in the
extracellular digestive enzymatic solution by SDS-PAGE and western
blotting
SDS-PAGE was performed on the protein extracts from
digested cells and the results demonstrated that there was a marked
difference between the transfection group and the control group.
The difference was more marked between 14.3 and 20 kDa. s-TNF-α is
17 kDa in size and when compared with control group, the
transfected with pcDNA3.1-TM-enterokinase-TNF-α plasmid and the
transfected with pcDNA3.1-TM-FactorXa-TNF-α plasmid had a deeper
color, the cell medium digest with FactorXa had a lighter color and
the cell medium digest with enterokinase's was the closest in color
to the control, suggesting that s-TNF-α may have been present in
the digestive solution (Fig. 7A).
The results of Western blotting showed after that two kinds of
plasmid transformed 3T3 cells were digestion with the corresponding
enzyme (enterokinase and FactorXa). TNF-α was detected in
pcDNA3.1-TM-FactorXa-TNF-α plasmid-transfected, while the
pcDNA3.1-TM-enterokinase-TNF-α plasmid-transfected or control cells
were not detected. The western blotting results revealed that the
level of protein in the cells transfected with
pcDNA3.1-TM-FactorXa-TNF-α plasmid-transfected was significantly
higher when compared with blank cells. However, the level of
protein in the cells transfected with
pcDNA3.1-TM-enterokinase-TNF-α plasmid-transfected was not
significantly different when compared with the blank group. TNF-α
was detected in pcDNA3.1-TM-FactorXa-TNF-α plasmid-transfected
cells; whereas it was not detected in
pcDNA3.1-TM-enterokinase-TNF-α plasmid-transfected or control cells
(Fig. 7B). These results indicated
that the pcDNA3.1-TM-FactorXa-TNF-α plasmid achieved the expected
effect, as the initial expression of the fusion protein was a
transmembrane protein and the extracellular segments were
hydrolyzed by FactorXa to form s-TNF-α.
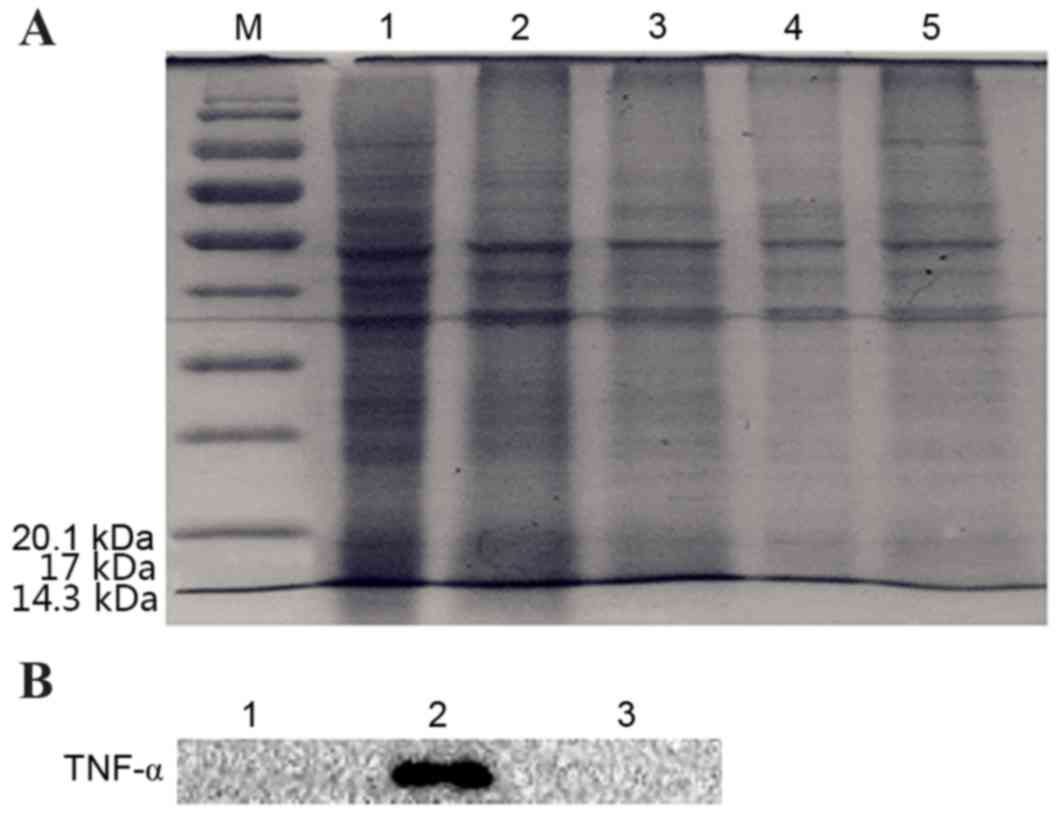 | Figure 7.Detection of TNF-α in extracellular
digestive enzymatic solutions from 3T3 cells that were transfected
with pcDNA3.1-TM-enterokinase-TNF-α and pcDNA3.1-TM-FactorXa-TNF-α
by SDS-PAGE and western blotting. (A) Digestion solution analyzed
by SDS-PAGE. M, protein marker; lane 1, transfection with
pcDNA3.1-TM-FactorXa-TNF-α plasmid; lane 2, transfection with
pcDNA3.1-TM-enterokinase-TNF-α plasmid; lane 3, cell medium
digested by FactorXa; lane 4, cell medium digested by enterokinase;
lane 5, control. (B) Detection of TNF-α following digestion by
western blotting. Lane 1, transfection with
pcDNA3.1-TM-enterokinase-TNF-α plasmid; lane 2, transfection with
pcDNA3.1-TM-FactorXa-TNF-α plasmid; lane 3, control. TM,
transmembrane; TNF-α, tumor necrosis factor-α. |
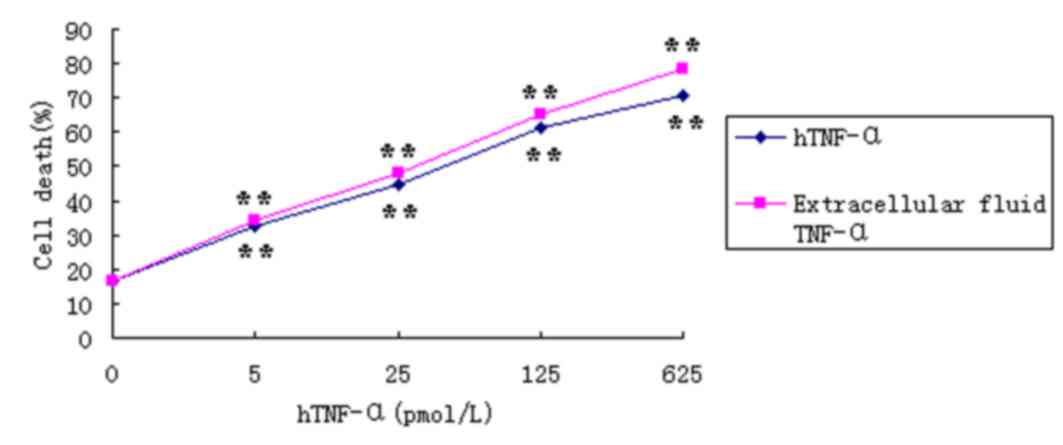
TNF-α bioactivity in the extracellular
digestive liquid
The activity of TNF-α was measured by its toxic
effects on L929 cells (14).
Following treatment of L929 cells with varying concentrations of
hTNF-α or TNF-α from the extracellular digestion solutions for 24
h, the proliferation rate of the L929 cells decreased in a
dose-dependent manner (Fig. 8).
The activity results of hTNF and pcDNA3.1-TM-FactorXa-TNF-α are
presented and compared in Table
II. The results indicate that the two were similar in activity.
Based on the results shown in Tables
I and II, the level of
cytotoxicity of TNF-α extracted from extracellular fluid was
marginally higher when compared with the hTNF-α positive control.
Incubation of L929 cells with 625 pmol/l TNF-α derived from
extracellular fluid for 24 h, demonstrated an inhibition rate of
78.24% (Fig. 8 and Table I). In addition, the specific
activity of TNF-α from the extracellular fluid was 30% higher when
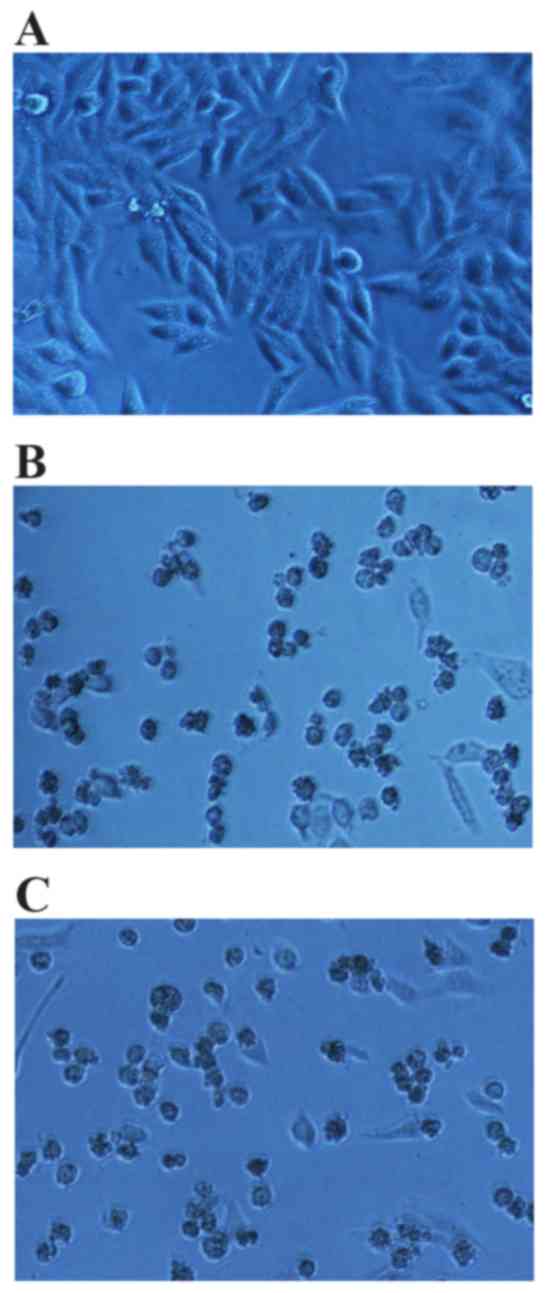
compared with the hTNF-α positive control. Following 24 h of
drug-processing, inverted microscopy revealed that the cells in the
negative control group were fusiform or irregular fusiform, with
sharp outlines, high levels of light refractivity and cell
attachment, and were enlarged (Fig.
9A). In addition, very few dead cells were observed in the
untreated control group. In the hTNF-α-treated group and the
extracellular fluid TNF-α-treated group, the majority of cells were
enlarged, displayed irregular cell borders, poor light refractivity
and a small proportion of cells exhibited dissolved nuclear
membranes or were floating in the medium (Fig. 9B and C).
 | Table II.Comparison of the specific activity
of different proteins. |
Table II.
Comparison of the specific activity
of different proteins.
| Sample | LD50
(pmol/l) | Specific activity
(U/mg) |
|---|
| hTNF-α | 42.09 |
1.78×107 |
|
pcDNA3.1-TM-FactorXa-TNF-α | 28.17 |
2.32×107 |
| Table I.MTT assay results demonstrating the
effect of TNF-α on L929 cell viability. |
Table I.
MTT assay results demonstrating the
effect of TNF-α on L929 cell viability.
| Group | OD570
(mean ± standard deviation) | Inhibition rate
(%) |
|---|
| Negative control,
pmol/l |
| 0 | 0.537±0.05 | – |
| hTNF-α, pmol/l |
| 5 | 0.363±0.017 | 32.64 |
| 25 | 0.297±0.01 | 44.81 |
| 125 | 0.209±0.018 | 61.10 |
| 625 | 0.158±0.021 | 70.64 |
| Extracellular
TNF-α, pmol/l |
| 5 | 0.355±0.022 | 34.06 |
| 25 | 0.280±0.016 | 48.02 |
| 125 | 0.188±0.014 | 65.18 |
| 625 | 0.117±0.011 | 78.24 |
Discussion
Expression vectors, the most significant elements in
the expression system, require high expression levels, high
stability and extensive application (9). In addition, expression vectors must
contain a replication origin, selection markers and a strong
promoter and transcription terminator. Fusion mRNA (plasmids
containing two genes fused together) facilitates the initiation of
translation as well as the purification of the corresponding
protein, and a fusion protein with a low molecular weight may
increase the stability of mRNA (15,16).
In a number of expression systems, the fusion protein is commonly
produced in the cell and is extracted from the cell inclusion as
the target protein. Prokaryotic cells, which lack eukaryotic
protein processing and modification systems, produce cell
inclusions when there is a high level of protein translation, or
when the cell undergoes hydrophobic interactions, ionic reactions,
degradation, protein misfolding or there is a reducible environment
in the host cell (17).
Conformation-dependent metaproteins in the cell inclusion require
purification and renaturation before they are transformed into
active proteins. Due to the diversity in structure, function and
molecular weight, different types of protein require distinct and
specific optimum renaturation conditions, which greatly limit the
application of prokaryotic expression systems (18,19).
In eukaryotes, proteins are commonly modified and processed so that
the majority are biologically active, and their overexpression will
impact the host cells. Additional types of expression systems
secrete proteins outside of the cells, and the target protein is
subsequently collected from the culture media. For instance, insect
cells secrete heterologous proteins via signal peptides prior to
productive modification and processing (e.g. glucosylation and
phosphorylation), which produce proteins with biological
characteristics of antigenicity and enzyme activity similar to that
of the natural protein (20,21).
Therefore, this expression system has been researched in-depth over
the last few years. Makadiya et al (22) investigated the most suitable
production system for spikes of protein and constructed a
eukaryotic secretion expression system that was transfected into
yeast, insect cells and mammalian cells. The group demonstrated
that mammalian cells were the optimal cells to use as they obtained
the highest yield and their secreted protein activity was not
affected. In addition, Stock et al (23) established a novel expression
platform in the well characterized eukaryotic microorganism,
Ustilago maydis. I, for the production of challenging
proteins.
The glutathione-S-transferase (GST) system, maltose
binding protein system, protein kinase A system, protein
purification tag fusion system and galactosidase system, are all
examples of successful fusion expression vectors (24). Proteins with purification tags,
including the widely applied GST, histidine and FLAG tags,
facilitate purification (25–29).
Secreted expression vectors contain signal peptides that enable
transfer of the protein to the periplasm, outer membrane and the
medium, thus avoiding degradation of target proteins and excessive
metabolic load of host bacteria (30). Accordingly, distinct signal
peptides are required for different proteins, and signal peptides
including the human lysosome, alkaline phosphatase of E.
coli, the N-terminal leader sequence of pectatelyase B of
Erwinia carotovora CE, PelB and the outer membrane protein A
precursor are commonly used.
The primary structure of TM-TNF-α consists of 233
amino acid residues, of which −21 to −46 contains the transmembrane
region that forms the hydrophobic α-helix; this region is highly
conserved, as mutations will not allow for the formation of
transmembrane domains (9). The aim
of the present study was to anchor the target protein on the outer
cell membrane using this domain structure. Linkage peptides are
present between the TM-TNF-α transmembrane structure and s-TNF-α,
which is less conserved and functions as a linkage, which is
replaceable. Therefore, these linkage peptides were substituted
with the enterokinase and FactorXa enzyme loci in the present
study. As long as the transmembrane structure of the fusion protein
is formed, its extracellular segment should contain the
controllable enzyme digestion site. Two expression vectors,
pcDNA3.1-TM-FactorXa-TNF-α and pcDNA3.1-TM-enterokinase-TNF-α
plasmids, were successfully constructed by replacing the
extracellular segment with FactorXa (cutting site,
Ile-Glu-Gly-Arg-↓-Gly-Ser) separately, thus inducing controllable
hydrolysis of s-TNF-α. Therefore, the fusion protein is initially
produced in the transmembrane protein form. In the presence of
enterokinase or FactorXa the extracellular segment detaches, which
generates the target protein. Thus, target proteins are produced
when enterokinase or FactorXa is added to the culture medium of
transfected cells. In addition, the production of unwanted proteins
is effectively reduced. Jia et al (31) used the small ubiquitin-like
modifier-mmTNF-α fusion protein in the cytoplasm of E. coli
to generate high TNF-α expression. It produced soluble and
insoluble proteins with excellent activities; however, the
purification and separation process was complicated. Alizadeh et
al (32) performed experiments
based on the optimization of the fusion system with GST-labeled
TNF-α in E. coli. However, GST was required to cut off the
coagulation protease, which was then purified. In the present
study, a PmeI enzyme digestion site (GTTT↓AAAC) was inserted
into the linkage between the transmembrane segment and the
extracellular target protein, thus generating blunt ends. This
facilitated the insertion of additional target proteins by
combining the target protein sequence and the vector. It is
important that the vectors have the forward inserted sequence
(33). Generating a novel vector
in this way may influence the host cell as well as productivity,
due to overexpression of specific active proteins. However, the
vectors generated in the present study successfully overcame this
issue by anchoring the active region onto the outer cytomembrane.
In addition, the relative content of target proteins was improved
by the level of control of restriction enzyme digestion, where ~24
h was required to reach the maximum (data not shown), and the
operation was simple. The vector constructed in this experiment
produced no loss of biological activity, and was generated using a
simple, rapid and effective method.
The present study s-TNF-α was not detectable in
cells transfected with pcDNA3.1-TM-enterokinase-TNF-α plasmids.
There are a number of reasonable explanations for this. Firstly,
the fusion protein was not produced in the form of a transmembrane
protein, due to the insertion of the enterokinase digestion site
affecting the formation of the α-helix. In addition, the
hydrophobic structure in this region may induce the formation of
cell inclusions, thus impeding detection of the protein in the cell
dissociation solution. Secondly, the peptide conformation may have
inhibited the enterokinase digestion site, which results in a lack
of enterokinase accessibility and no production of the target
protein. Finally, it is possible that the enterokinase was
inactivated and thus did not possess normal a hydrolysis function.
Therefore, the correct design of this plasmid construct requires
further investigation in order to achieve the desired results.
Acknowledgements
The present study was supported by the First Science
and Technology Program of Guangdong (grant no. 2008B030301023) and
the Science and Technology Program of Higher Learning Institutions
in Dongguan (grant nos. 200910815264 and 2012108102016).
References
|
1
|
Itai T, Tanaka M and Nagata S: Processing
of tumor necrosis factor by the membrane-bound TNF-alpha-converting
enzyme, but not its truncated soluble form. Eur J Biochem.
268:2074–2082. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang AM, Creasey AA, Ladner MB, Lin LS,
Strickler J, van Arsdell JN, Yamamoto R and Mark DF: Molecular
cloning of the complementary DNA for human tumor necrosis factor.
Science. 228:149–154. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kriegler M, Perez C, DeFay K, Albert I and
Lu SD: A novel form of TNF/cachectin is a cell surface cytotoxic
transmembrane protein: Ramifications for the complex physiology of
TNF. Cell. 53:45–53. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Black RA, Rauch CT, Kozlosky CJ, Peschon
JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P,
Srinivasan S, et al: A metalloproteinase disintegrin that releases
tumour-necrosis factor-alpha from cells. Nature. 385:729–733. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abu-Amer Y, Erdmann J, Alexopoulou L,
Kollias G, Ross FP and Teitelbaum SL: Tumor necrosis factor
receptors types 1 and 2 differentially regulate osteoclastogenesis.
J Biol Chem. 275:27307–27310. 2000.PubMed/NCBI
|
|
6
|
Nagano K, Alles N, Mian AH, Shimoda A,
Morimoto N, Tamura Y, Shimokawa H, Akiyoshi K, Ohya K and Aoki K:
The tumor necrosis factor type 2 receptor plays a protective role
in tumor necrosis factor-alpha-induced bone resorption lacunae on
mouse calvariae. J Bone Miner Metab. 29:671–681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Horiuchi T, Mitoma H, Harashima S,
Tsukamoto H and Shimoda T: Transmembrane TNF-alpha: Structure,
function and interaction with anti-TNF agents. Rheumatology
(Oxford). 49:1215–1228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bjornberg F, Lantz M, Olsson I and
Gullberg U: Mechanisms involved in the processing of the p55 and
the p75 tumor necrosis factor (TNF) receptors to soluble receptor
forms. Lymphokine Cytokine Res. 13:203–211. 1994.PubMed/NCBI
|
|
9
|
Jonasson P, Liljeqvist S, Nygren PA and
Ståhl S: Genetic design for facilitated production and recovery of
recombinant proteins in Escherichia coli. Biotechnol Appl Biochem.
35:91–105. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blobel G and Dobberstein B: Transfer of
proteins across membranes. I. Presence of proteolytically processed
and unprocessed nascent immunoglobulin light chains on
membrane-bound ribosomes of murine myeloma. J Cell Biol.
67:835–851. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Pennica D, Nedwin GE, Hayflick JS, Seeburg
PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB and Goeddel DV:
Human tumour necrosis factor: Precursor structure, expression and
homology to lymphotoxin. Nature. 312:724–729. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hehlgans T and Pfeffer K: The intriguing
biology of the tumour necrosis factor/tumour necrosis factor
receptor superfamily: Players, rules and the games. Immunology.
115:1–20. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qin YW, Cheng C, Wang HB, Gao YJ, Shao XY
and Shen AG: Role of P38 in LPS induced TNF-α expression in Schwann
cells. Chin J Biochemistry Mol Biol. 05:382–387. 2007.(In
Chinese).
|
|
14
|
Li YP, Pei YY, Ding J, Shen ZM, Zhang XY,
Gu ZH and Zhou JJ: PEGylated recombinant human tumor necrosis
factor alpha: Preparation and anti-tumor potency. Acta Pharmacol
Sin. 22:549–555. 2001.PubMed/NCBI
|
|
15
|
Mao H: A self-cleavable sortase fusion for
one-step purification of free recombinant proteins. Protein Expr
Purif. 37:253–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SO and Lee YI: High-level expression
and simple purification of recombinant human insulin-like growth
factor I. J Biotechnol. 48:97–105. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singh SM and Panda AK: Solubilization and
refolding of bacterial inclusion body proteins. J Biosci Bioeng.
99:303–310. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Daly R and Hearn MT: Expression of
heterologous proteins in Pichia pastoris: A useful experimental
tool in protein engineering and production. J Mol Recognit.
18:119–138. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chaudhuri TK, Horii K, Yoda T, Arai M,
Nagata S, Terada TP, Uchiyama H, Ikura T, Tsumoto K, Kataoka H, et
al: Effect of the extra n-terminal methionine residue on the
stability and folding of recombinant alpha-lactalbumin expressed in
Escherichia coli. J Mol Biol. 285:1179–1194. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H, Shaffer PL, Huang X and Rose PE:
Rapid screening of membrane protein expression in transiently
transfected insect cells. Protein Expr Purif. 88:134–142. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng J, Ren F, Yu X, Lai BC and Wang Y:
Expression of HPV16E6 protein with insect-baculovirus expression
system. J Northwest Univ (Natural Sci Ed). 05:775–778. 2008.(In
Chinese).
|
|
22
|
Makadiya N, Brownlie R, van den Hurk J,
Berube N, Allan B, Gerdts V and Zakhartchouk A: S1 domain of the
procine epidemic diarrhea virus spike protein as a vaccine antigen.
Virol J. 13:572016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stock J, Sarkari P, Kreibich S, Brefort T,
Feldbrügge M and Schipper K: Applying unconventional secretion of
the endochitinase Cts1 to export heterologous proteins in Ustilago
maydis. J Biotechnol. 161:80–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arnau J, Lauritzen C, Petersen GE and
Pedersen J: Current strategies for the use of affinity tags and tag
removal for the purification of recombinant proteins. Protein Expr
Purif. 48:1–13. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hu J, Qin H, Gao FP and Cross TA: A
systematic assessment of mature MBP in membrane protein production:
Overexpression, membrane targeting and purification. Protein Expr
Purif. 80:34–40. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klein S, Geiger T, Linchevski I,
Lebendiker M, Itkin A, Assayag K and Levitzki A: Expression and
purification of active PKB kinase from Escherichia coli. Protein
Expr Purif. 41:162–169. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leviatan S, Sawada K, Moriyama Y and
Nelson N: Combinatorial method for overexpression of membrane
proteins in Escherichia coli. J Biol Chem. 285:23548–23556. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim DJ, Jang HJ, Pyun YR and Kim YS:
Cloning, expression, and characterization of thermostable DNA
polymerase from Thermoanaerobacter yonseiensis. J Biochem Mol Biol.
35:320–329. 2002.PubMed/NCBI
|
|
29
|
Magliery TJ, Wilson CG, Pan W, Mishler D,
Ghosh I, Hamilton AD and Regan L: Detecting protein-protein
interactions with a green fluorescent protein fragment reassembly
trap: Scope and mechanism. J Am Chem Soc. 127:146–157. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han S, Li H, Jin Z, Huang D, Ren C and Lin
Y: Yeast cell surface display and its application of enzymatic
synthesis in non-aqueous phase. Sheng Wu Gong Cheng Xue Bao.
25:1784–1788. 2009.(In Chinese). PubMed/NCBI
|
|
31
|
Jia D, Yang H, Wan L, Cheng J and Lu X:
Production of bioactive, SUMO-modified, and native-like TNF-α of
the rhesus monkey, Macaca mulatta, in Escherichia coli. Appl
Microbiol Biotechnol. 93:2345–2355. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alizadeh AA, Hamzeh-Mivehroud M,
Farajzadeh M, Moosavi-Movahedi AA and Dastmalchi S: A simple and
rapid method for expression and purification of functional TNF-α
using GST fusion system. Curr Pharm Biotechnol. 16:707–715. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Evans DH, Willer DO and Yao XD: DNA
joining method US Patent 7575860 B2. March 7–2001, issued August
18, 2009.
|