Introduction
Chronic obstructive pulmonary disease (COPD) is a
group of diseases associated with cigarette smoke, and it is
becoming a primary cause of mortality and morbidity in humans. COPD
is the fourth leading cause of mortality worldwide and the World
Health Organization predicts it will become the third leading cause
by 2030 (1,2). The disease is characterized by
chronic airway inflammation, airflow obstruction, alveolar wall
destruction and oxidant/antioxidant imbalance (3). There is a strong correlation between
cigarette smoking and COPD; ~15% of heavy smokers develop COPD.
Compared with non-smoker COPD patients, active smokers exhibit an
accelerated decline in lung function and a higher mortality rate
(3). Cigarette smoke is a
worldwide risk factor in the pathogenesis of COPD, and it is a
mixture that contains >1015–17 oxidant and free
radical molecules and >5,000 chemicals per inhalation. These
cause oxidative damage and alveolar epithelial cell death, which
have been implicated in COPD pathogenesis. Exposure to cigarette
smoke-induced oxidative stress becomes a burden to the lungs of
smokers (4). Therefore, it is
important to investigate potential antioxidant defenses against
cigarette smoke-induced cell damage.
Nuclear factor erythroid 2 like 2 (NFE2L2; Nrf2)
regulates the antioxidant defense system and, as a central basic
leucine zipper transcription factor, modifies oxidative responses
and inflammation. Under physiological conditions, Nrf2 is
sequestered in the cytoplasm via an interaction with Kelch-like ECH
associated protein 1 (Keap1). However, upon exposure to oxidative
stimuli, Nrf2 dissociates from Keap1 and translocates into the
nucleus, where it dimerizes with Maf proteins and specifically
binds to the antioxidant responsive element (ARE) to initiate
transcription of target genes. In the absence of oxidative stress,
Keap1 suppresses Nrf2 signaling constitutively by preventing Nrf2
translocation to the nucleus (5).
Numerous protective antioxidant responses are associated with Nrf2
function. Nrf2 regulates ‘direct’ and ‘indirect’ antioxidant
enzymes, including superoxide dismutase, γ-glutamylcysteine
synthetase, heme oxygenase-1 and NADPH:quinone oxidoreductase-1,
among others. These enzymes exert cytoprotective, antioxidant and
anti-inflammatory effects in the respiratory system (6). Upregulation and activation of Nrf2
enhances expression of antioxidant genes and protects cells from
oxidative injury (7). Thus, Nrf2
is an important link between the regulation of antioxidant gene
expression and cell survival (8).
Deficiency of Nrf2 in a murine model confirmed the direct
association between alveolar wall destruction, antioxidant gene
regulation and enhanced susceptibility to cigarette smoke-induced
emphysema (9). Previous studies
have reported that Nrf2 downregulation is linked to increased
oxidative stress and pathogenesis in the lungs of patients with
COPD (10,11).
Sulforaphane [1-isothiocyanate-(4R)-(methylsulfinyl)
butane; SFN] is a natural isothiocyanate contained in cruciferous
vegetables, including broccoli, kale, cole crops and cabbage.
Experimental studies have demonstrated that it exhibits
cytoprotective effects, particularly against oxidative stress, by
inducing the expression of antioxidant enzymes (12,13).
SFN induces ARE expression through disruption of the Keap1-Nrf2
complex to release Nrf2, by interacting directly with sulfhydryl
residues on Keap1 (14). In
addition, SFN activates the MAPK pathway, resulting in
phosphorylation of Keap1 and subsequent release of Nrf2 (15). Another study indicated that SFN
inhibits Keap1-dependent proteasomal degradation to stabilize the
Nrf2 protein (16). Previous
studies reported that mice lacking the Nrf2 gene do not develop
cancer, if they are treated with broccoli, SFN or other
Nrf2-activating drugs (17,18).
These reports indicated that the protective effect of broccoli or
SFN requires a functional Nrf2 response, and that activation of
Nrf2 is involved in protection from free radical-induced diseases,
including COPD.
However, the potential protective effect and
mechanism of SFN against cigarette smoke extract (CSE)-induced
alveolar epithelial cell type II damage has not been fully
elucidated to date. The present study aimed to investigate the
potential mechanism underlying the protective effects of SFN on
RLE-6TN lung epithelial cells. The results demonstrated that SFN
attenuated CSE-induced cell injury, and Nrf2 may have an important
role in the process.
Materials and methods
Culture of RLE-6TN cells
The RLE-6TN rat lung epithelial type II cell line
was obtained from the Advanced Research Center and Modern Analysis
and Testing Center of Central South University (Changsha, China).
Cells were grown in Dulbecco's modified Eagle's medium (DMEM;
HyClone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10–12% fetal bovine serum (HyClone; GE Healthcare Life
Sciences) and 100 IU/ml penicillin/streptomycin, in an atmosphere
of 5% CO2 at 37°C. Cells were subcultured using 0.25%
trypsin and 0.02% EDTA every 4 to 5 days. Medium was replaced with
fresh every 2 days. Cells in the logarithmic phase were used for
subsequent experiments.
CSE preparation
CSE was freshly combusted and obtained through an
experimental apparatus with an airflow driven continually by
vacuum. The smoke from three cigarettes was bubbled into 20 ml DMEM
at the rate of one cigarette/2 min. The resulting suspension was
regarded as 100% CSE and diluted to the indicated concentrations
with DMEM. The obtained CSE was filtered through a 0.22-µm sterile
filter prior to addition to cell cultures and was used within 30
min of preparation. Control group medium was prepared by bubbling
air into 20 ml DMEM and filtering.
Cytotoxicity assay
Cell toxicity was determined by an MTT assay.
RLE-6TN cells were seeded into 96-well plates at a density of 1×104
cells/well. Following cell adherence, complete medium was replaced
with serum-free DMEM for 16 h. The cells were subsequently
incubated with 1, 5, 10, 15 or 20% CSE in serum-free DMEM. After 24
h, MTT reagent (5 mg/ml) was added into each well and incubated for
a further 4 h at 37°C, following which crystals were solubilized
with 150 µl dimethyl sulfoxide (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany). The absorbance was measured at a wavelength of
560 nm with a BioTek Elx800 plate reader (BioTek Instruments Inc.,
Winooski, VT, USA).
To observe the effect of SFN (Sigma-Aldrich; Merck
KGaA) on cell viability, RLE-6TN cells were incubated with 0.5, 1,
2, 4 or 8 µM SFN in serum-free DMEM. The MTT assay was performed
after 24 h incubation.
Cell cycle analysis
RLE-6TN cells were seeded into 6-well plates at a
density of 2×106 cells/well. The CSE group was treated with 5% CSE
for 24 h and the CSE + SFN group was pretreated with 0.5 µM SFN for
12 h prior to 5% CSE treatment for 24 h. The control group was
treated with air. Cells were subsequently harvested using 0.25%
trypsin and 0.02% EDTA, washed three times with PBS by centrifuging
at 800 × g/min for 5 min at room temperature and fixed with 70%
pre-chilled ethanol for 12 h at 4°C. Cells were washed with PBS
again and stained with 50 µg/ml propidium iodide (PI;
Sigma-Aldrich; Merck KGaA) for 30 min in the dark. Finally, cell
cycle distribution was detected by flow cytometry and analyzed with
Multicycle for Windows (Epics XL; Beckman Coulter, Inc., Brea, CA,
USA).
Apoptosis analysis
Apoptosis was determined by a Fluorescein
Isothiocyanate (FITC)-conjugated Annexin V/PI Double Staining kit
(Sigma-Aldrich; Merck KGaA). RLE-6TN cells were plated, treated and
harvested as in the cell cycle analysis section. Cells were
subsequently resuspended in 500 µl annexin V binding buffer
according to the manufacturer's protocol. Aliquots of the cells
were incubated with 5 µl annexin V-FITC and 10 µl PI at room
temperature for 10 min in the dark. Apoptotic cells were detected
by flow cytometry and analyzed with Expo 32 software (Epics XL;
Beckman Coulter, Inc.).
Measurement of reactive oxygen species
(ROS) levels
RLE-6TN cells were treated with CSE for 24 h as
described above in the cell cycle analysis section, and
intracellular ROS levels were detected by staining with
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA;
Sigma-Aldrich; Merck KGaA). H2DCFDA (10 µM) was added to
the cell pellet and incubated at 37°C for 30 min. Fluorescence was
measured by flow cytometry with excitation at a wavelength of 485
nm and emission at a wavelength of 545 nm, and analyzed with System
II software version 3.0 (Epics XL; Beckman Coulter, Inc.).
Morphology
Following CSE treatment, RLE-6TN cell morphological
alterations were observed by inverted phase-contrast microscopy and
fluorescence microscopy. Cells were washed twice with PBS and
stained with 0.5 µg/ml Hoechst 33258 for 10 min in the dark.
Following staining, cells were washed three times with PBS. The
nuclear morphology was observed using a Nikon ECLIPSE Ti-U system
(Nikon Corporation, Tokyo, Japan). The excitation wavelength was
350 nm.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RLE-6TN cells were seeded into 6-well plates at a
density of 1×105 cells/well. Following serum starvation, SFN
pretreatment and CSE treatment as aforementioned, total RNA was
isolated using TRIzol® reagent (Takara Biotechnology Co., Ltd.,
Dalian, China), according to the manufacturer's protocol. RNA
samples were reverse-transcribed using the PrimeScript™ RT Reagent
kit (Takara Biotechnology Co., Ltd.), according to the
manufacturer's protocol. The cDNA products were used immediately
for qPCR, using primers specific for Nrf2 (forward,
5′-CTGCCATTAGTCAGTCGCTCTC-3′; and reverse,
5′-TCGGCTGGGACTTGTGTTC-3′) and β-actin (forward,
5′-GGAGATTACTGCCCTGGCTCCTA-3′; and reverse,
5′-GACTCATCGTACTCCTGCTTGCTG-3′), and the SYBR® Premix Ex Taq TM II
kit (Takara Biotechnology Co., Ltd.), according to the
manufacturer's protocol. Gene expression was monitored using the
Rotor-Gene 3000 system (Qiagen GmbH, Hilden, Germany), following a
standardized protocol. In brief, reactions were incubated at 95°C
for 10 sec, and subsequently amplified for 45 cycles at 95°C for 5
sec followed by 60°C for 20 sec in each cycle. Relative
quantification was performed using the following formula:
ΔCq=Cq (target gene)-Cq (β-actin)
and fold=2−(ΔCq2-ΔCq1), in which ΔCq1
represents the value of control and ΔCq2 represents the
value of sample (19). Results
were presented as ratios between Nrf2 and the housekeeping
reference gene, β-actin.
Western blot analysis
RLE-6TN cells were plated and treated as in the
RT-qPCR section. Total protein was isolated using
radioimmunoprecipitation assay lysis buffer (Applygen Technologies,
Inc., Beijing, China), according to the manufacturer's protocol.
After centrifugation at with 10,000 × g/min for 20 min at 4°C, the
protein concentration of the supernatant was determined using a
Bicinchoninic Acid Protein assay kit (Applygen Technologies, Inc.).
Protein samples (40 µg) were separated on 12% polyacylamide gels at
120 V for 2.5 h and transferred onto polyvinylidene difluoride
membranes at 250 mA for 1.5 h. Membranes were blocked with 5%
non-fat milk powder in TBS containing 0.1% Tween-20 (TBST) for 1 h
at room temperature and subsequently probed with a polyclonal
rabbit anti-Nrf2 antibody (cat. no. ab31163; 1:1,000; Abcam,
Cambridge, UK) or a mouse anti-β-actin antibody (cat. no. ab8226;
1:1,000; Abcam) overnight at 4°C. Following three washes with TBST,
DyLight 800-labeled goat anti-rabbit/mouse secondary antibodies
(cat. nos. 042-07-15-06 and 042-07-18-06, respectively; 1:10,000;
SeraCare Life Sciences, Inc., Milford, MA, USA) were incubated with
membranes for 1 h at room temperature. The bands were detected
using an Odyssey Infrared Imaging System (LI-COR Biosciences,
Lincoln, NE, USA) and analyzed with ImageJ software version 1.50i
(National Institutes of Health, Bethesda, Maryland, USA).
Statistical analysis
Experimental data are presented as the mean ±
standard error. SPSS software version 21.0 (IBM SPSS, Armonk, NY,
USA) was used to analyze statistical significance between groups by
paired Student's t-test or one-way analysis of variance followed by
Fisher's least significant difference post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
SFN treatment protects against
CSE-induced cell injury
Following adherence, RLE-6TN lung epithelial cells
were exposed to different concentrations of CSE (1, 5, 10, 15 or
20%) for 24 h. Cell viability was subsequently measured by MTT
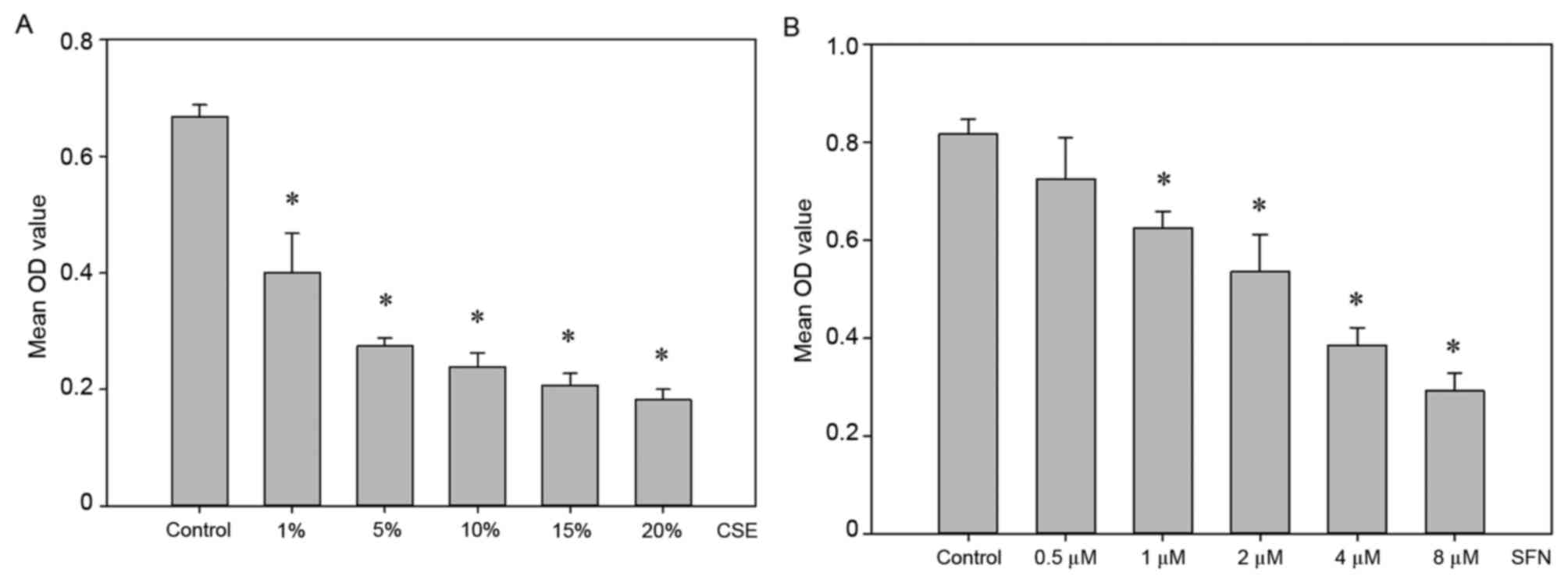
assay. As demonstrated in Fig. 1A,
CSE treatment resulted in a significant decrease in cell viability
compared with control cells, in a dose-dependent manner
(P<0.05). A concentration of 5% CSE was selected for further
experiments as it reduced cell viability by ~50% and cells also
remained attached to the bottom of the flask.
To examine the cytotoxic effect of SFN on RLE-6TN
cells and to select the appropriate concentration to pretreat cells
for further experiments, another MTT assay was performed. Cells
were exposed to different concentrations of SFN (0.5, 1, 2, 4 or 8
µM) for 24 h. As demonstrated in Fig.
1B, no significant difference was observed in the 0.5 µM SFN
treatment group compared with the control untreated group
(P>0.05). Therefore, this concentration was selected for the
pretreatment of cells in subsequent experiments.
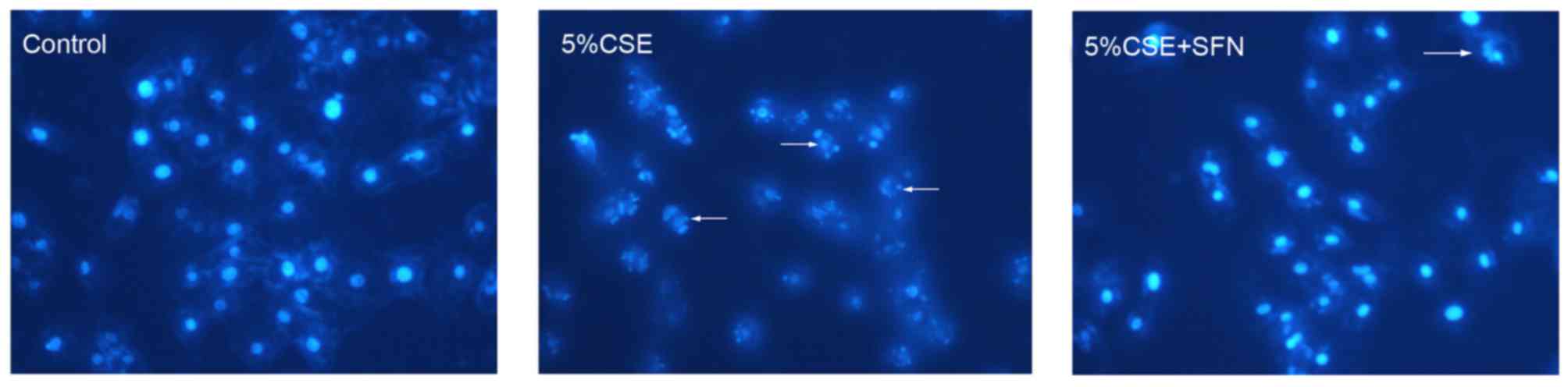
By Hoechst 33258 staining, cells treated with CSE
exhibited a shrunken morphology and nuclei fragmentation compared
with control air-treated cells (Fig.
2). These morphological characteristics are features of
apoptotic cells. Following pretreatment with SFN for 12 h, the
cells exposed to CSE exhibited no clear alterations in morphology
compared with control cells (Fig.
2), suggesting that SFN had a protective effect against
CSE.
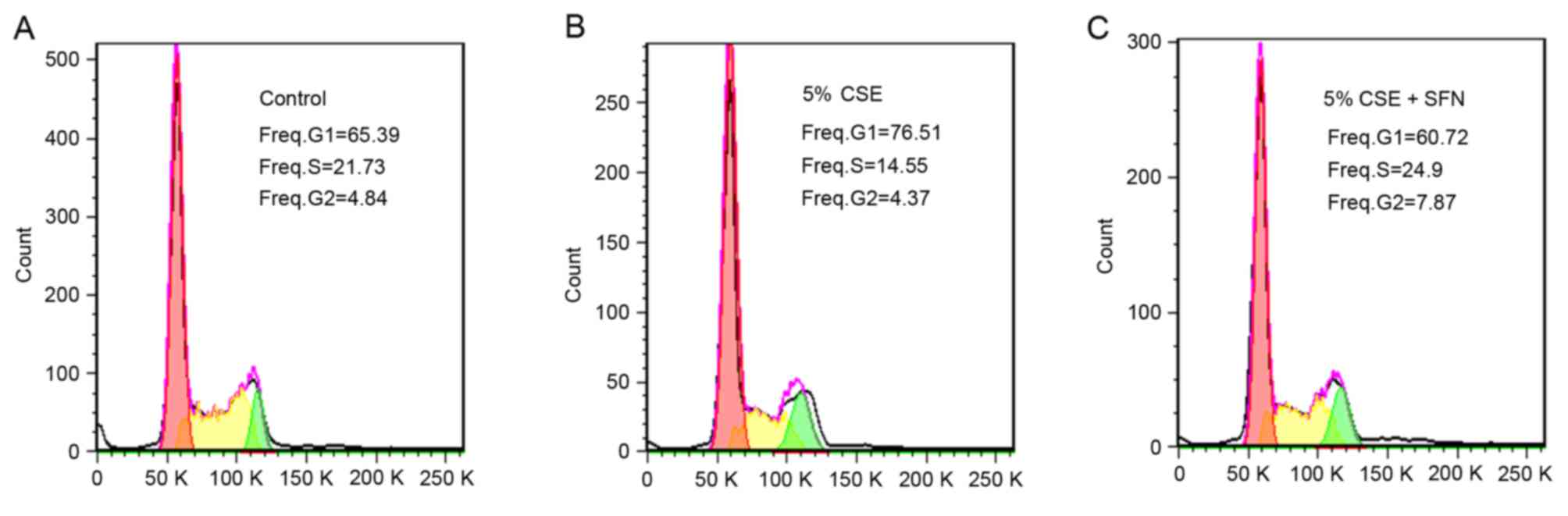
To further confirm the protective effect of SFN
against CSE on lung epithelial cells, cell cycle analysis was
performed. The results demonstrated that, in the control group, the
percentage of cells in the G1 phase was 60.56±3.68% and in the S
phase was 22.26±0.24% (Fig. 3A;
Table I). Following treatment with
5% CSE, the proportion of cells in the G1 phase increased to
65.88±2.78%, whereas the proportion in the S phase decreased to
12.84±0.52% (Fig. 3B; Table I). However, pretreatment with SFN
reversed this effect, with the percentage of cells in the G1 and S
phases recovering to 58.69±3.94 and 19.98±0.52%, respectively
(Fig. 3C; Table I). Although the difference in G1
phase values between the groups was not statistically significant,
similar changes in the mean values between control, CSE and CSE +
SFN groups were observed in several independent experiments, these
results indicate that CSE may inhibit cell growth by inducing cell
cycle arrest in the G1 phase, and that SFN may protect cells
against CSE.
 | Table I.Cell cycle progression, apoptosis and
ROS levels were assessed by flow cytometry. |
Table I.
Cell cycle progression, apoptosis and
ROS levels were assessed by flow cytometry.
|
| Group |
|---|
|
|
|
|---|
| Parameter | Control | CSE | SFN + CSE |
|---|
| Proportion in G1
phase (%) | 60.56±3.68 | 65.88±2.78 | 58.69±3.94 |
| Proportion in S phase
(%) | 22.26±0.24 |
12.84±0.52a |
19.98±0.52b |
| Apoptotic proportion
(%) | 0.16±0.21 |
78.08±0.97a |
6.12±0.33a,b |
| ROS levels (%) | 20.87±0.94 |
86.63±1.21a |
28.03±0.26a,b |
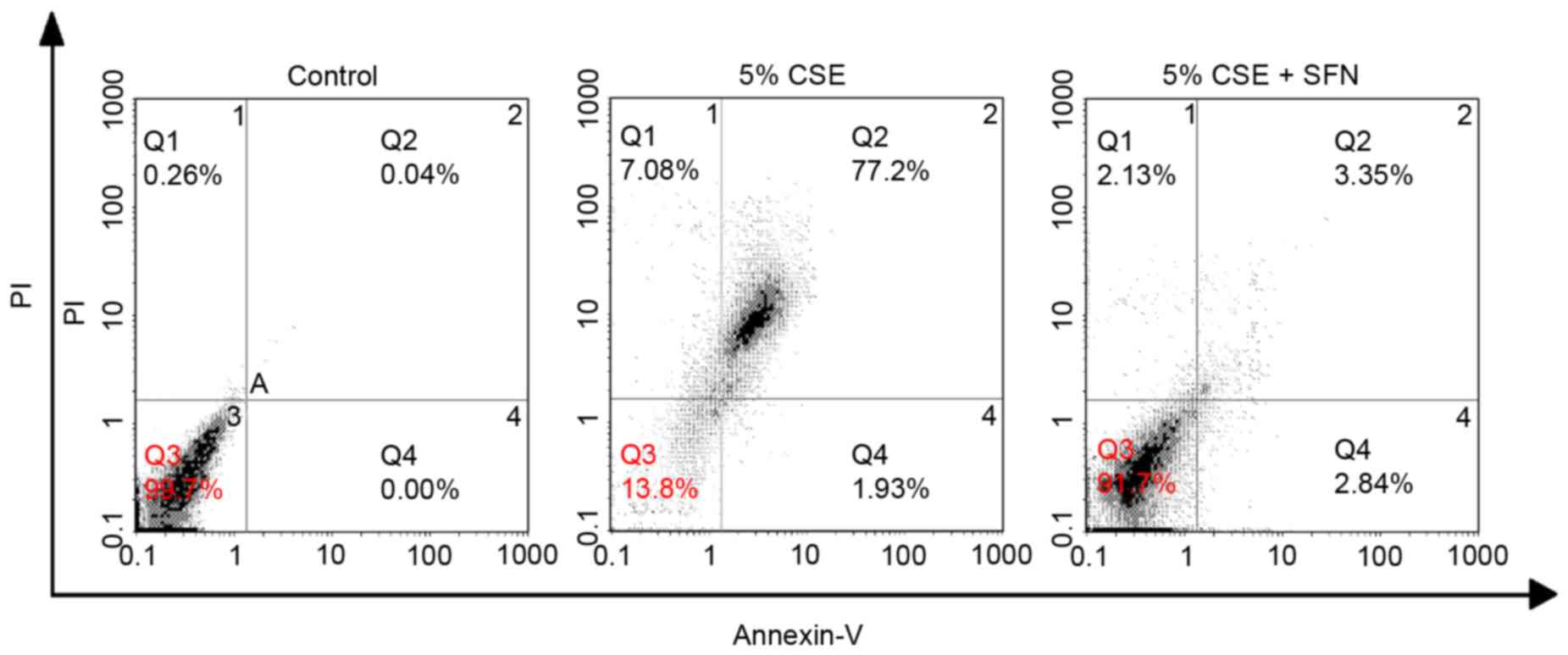
Apoptotic cell death induced by CSE in the presence
or absence of SFN pretreatment was additionally quantified using
annexin V-FITC/PI double staining. As demonstrated in Table I and Fig. 4, the percentage of apoptotic cells
(Q2+Q4 quadrants) in the control group was 0.16±0.21%, whereas this
was increased to 78.08±0.97% in the CSE treatment group. SFN
pretreatment again exhibited a protective effect against
CSE-induced apoptosis, with the percentage of apoptotic cells
decreasing to 6.12±0.33% in the SFN group.
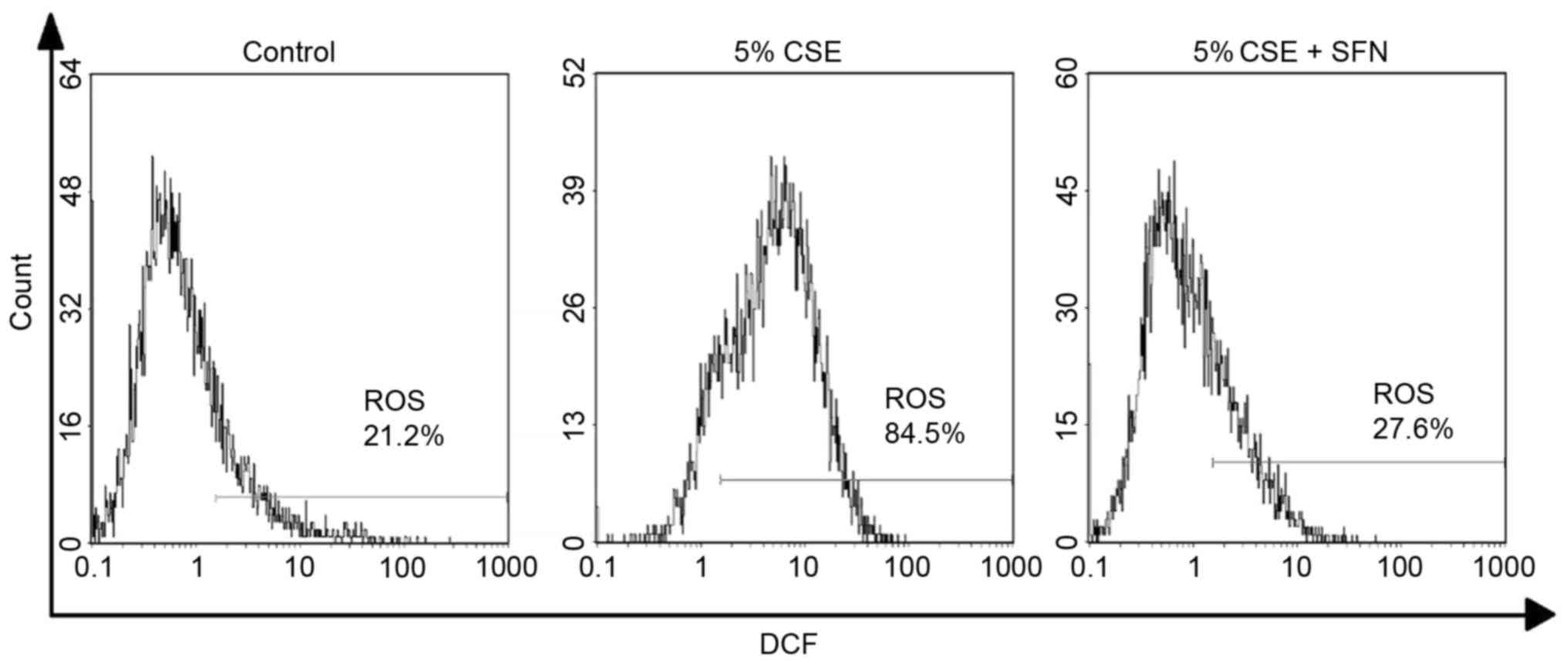
Finally, the H2DCFDA probe was used to
examine intracellular ROS levels. The results demonstrated that CSE
increased intracellular ROS levels, with 86.63±1.21% positive
cells, compared with 20.87±0.94% in the control cells; this
percentage was decreased to 28.03±0.26% by pretreatment with SFN
(Fig. 5; Table I).
SFN increases Nrf2 expression levels
following CSE exposure
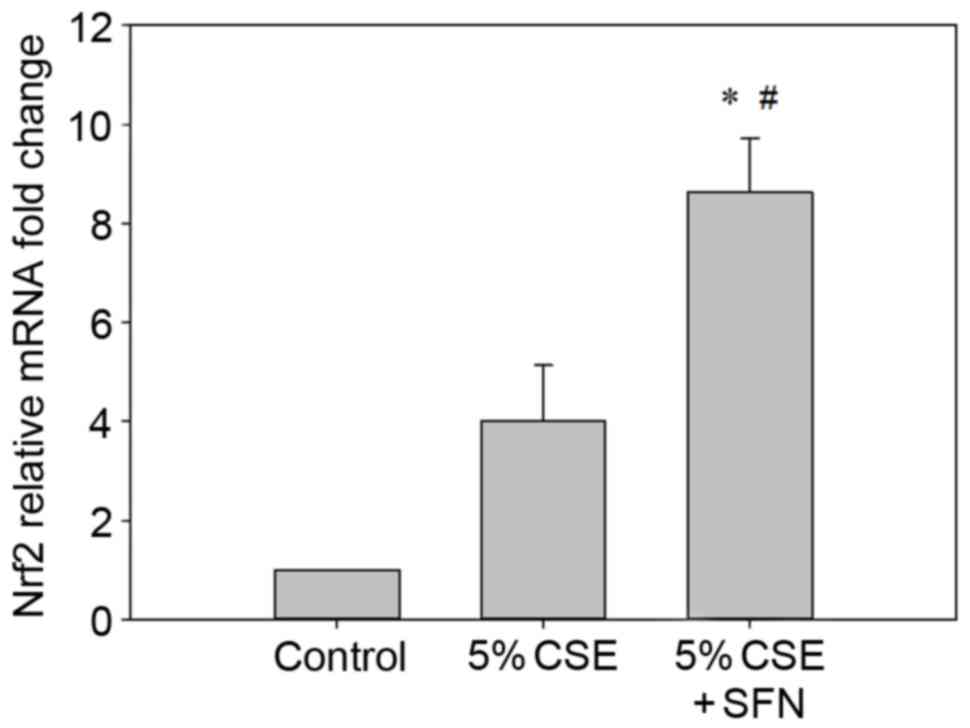
The mRNA expression levels of Nrf2 were quantified
by RT-qPCR. Following pretreatment with SFN for 12 h and exposure
to CSE for 24 h, Nrf2 mRNA expression levels were significantly
elevated (>8-fold or >2-fold) compared with the control group
or CSE-only group (P<0.05; n=3; Fig. 6).
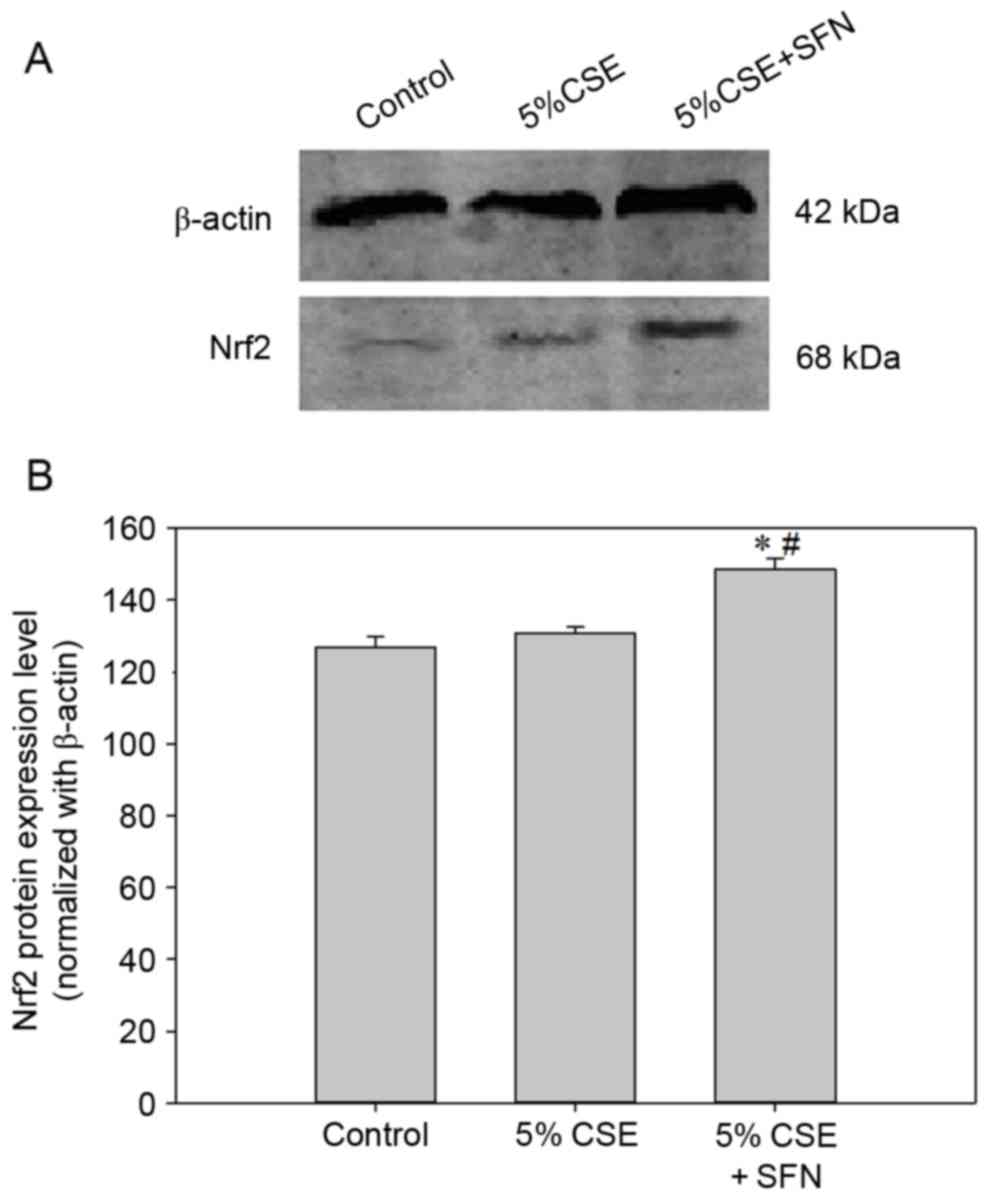
The protein levels of Nrf2 were detected by western
blotting, and were consistent with the RT-qPCR data. The SFN group
had significantly increased protein levels of Nrf2 compared with
control and CSE groups (Fig.
7).
Discussion
Alveolar epithelial cells are comprised of two
primary types, I and II. Although the surface area occupied by type
II cells is small, it may serve an important role in lung function.
A primary culture, despite representing the best experimental
system, is not suitable for long-term experiments as type II cells
will transdifferentiate into type I-like cells. Thus, the RLE-6TN
cell line is a useful and appropriate immortalized cell line for
the study of alveolar cell function due to its similarity to
alveolar type II epithelial cells (20).
Cigarette smoke is well known to induce oxidative
stress and serve a pathological role in CSE-induced lung diseases
including COPD. Alveolar epithelial cells represent the initial
target of CSE. It is therefore essential to protect alveolar
epithelial cells from oxidative stress injury induced by CSE
(21,22).
SFN is chemically defined as an isothiocyanate, and
is abundant in cruciferous vegetables including broccoli and
Brussels sprouts. For a long time, SFN has been considered a
cytoprotective agent based on its ability to induce and activate
antioxidant enzymes. A concentration of 0.5 µM SFN did not
significantly reduce cell viability in the present study, however,
SFN may be toxic at higher concentrations as observed in the in
vitro MTT assay of the present study, which may limit the use
of high doses of SFN. The Nrf2-ARE pathway has been investigated as
a potential underlying mechanism. Nrf2 is known to be activated by
phosphorylation. Various protein kinase pathways lead to Keap1-Nrf2
dissociation, and Nrf2 translocation from the cytoplasm to the
nucleus to bind to the ARE. Nrf2 is largely responsible for the
basal and inducible expression of proteins involved in drug
metabolism, the oxidative stress response and cytoprotection
(23).
The present study investigated the effects of SFN on
CSE-induced toxicity in RLE-6TN cells.
Using 5% CSE to stimulate RLE-6TN cells following
pretreatment with 0.5 µM SFN for 12 h, the results of the present
study suggested that SFN-induced Nrf2 may serve an important role
in protecting RLE-6TN cells from oxidative injury caused by CSE.
Compared with the CSE and control groups, the SFN group exhibited
significantly increased levels of Nrf2 and attenuated CSE-induced
cell injury. The possible mechanism underlying Nrf2 protection is
that the activation of Nrf2 may promote the synthesis and release
of antioxidant enzymes through the Nrf2-ARE pathway. In addition,
the results of the present study demonstrated a reduction in the
percentage of cells in the S phase following CSE treatment; and SFN
pretreatment alleviated this reduction, which indicated that SFN
may have its protective role by modulating the cell cycle; however,
the underlying mechanisms remain unclear. Apoptosis was induced by
CSE and the effects of SFN pretreatment were in accordance with the
morphological observations reported by Kosmider et al
(24). Furthermore, ROS serves a
critical role in lung epithelial cell damage, particularly in
CSE-induced injury. The results of the present study suggested that
the Nrf2-ARE pathway may promote antioxidant enzymes and reduce ROS
levels in the cell. These findings support the hypothesis that SFN
may protect alveolar epithelial cells from CSE injury by activating
the Nrf2-ARE pathway.
In conclusion, the results of the present study
indicate that CSE inhibits RLE-6TN cell proliferation and induces
apoptosis. Pretreatment with SFN may alleviate cell injury and
reduce ROS damage, potentially via increased Nrf2 expression. These
results suggest that Nrf2 may be a potential therapeutic target for
the treatment of COPD.
Acknowledgements
Zongxian Jiao was supported by the Fundamental
Research Funds for the Central Universities (grant no.
lzujbky-2013-223) and the Project sponsored by The Scientific
Research Foundation for Returned Overseas Chinese Scholars (State
Education Ministry, Beijing, China).
References
|
1
|
Jansson SA, Backman H, Stenling A,
Lindberg A, Rönmark E and Lundbäck B: Health economic costs of COPD
in Sweden by disease severity-has it changed during a ten years
period? Respir Med. 107:1931–1938. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Decramer M, Janssens W and Miravitlles M:
Chronic obstructive pulmonary disease. Lancet. 379:1341–1351. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tamimi A, Serdarevic D and Hanania NA: The
effects of cigarette smoke on airway inflammation in asthma and
COPD: Therapeutic implications. Respir Med. 106:319–328. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang H, Shih A, Rinna A and Forman HJ:
Exacerbation of tobacco smoke mediated apoptosis by resveratrol: An
unexpected consequence of its antioxidant action. Int J Biochem
Cell Biol. 43:1059–1064. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Goven D, Boutten A, Leçon-Malas V,
Boczkowski J and Bonay M: Prolonged cigarette smoke exposure
decreases heme oxygenase-1 and alters Nrf2 and Bach1 expression in
human macrophages: Roles of the MAP kinases ERK(1/2) and JNK. FEBS
Lett. 583:3508–3518. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Regoli F and Giuliani ME: Oxidative
pathways of chemical toxicity and oxidative stress biomarkers in
marine organisms. Mar Environ Res. 93:106–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tasaki M, Kuroiwa Y, Inoue T, Hibi D,
Matsushita K, Kijima A, Maruyama S, Nishikawa A and Umemura T: Lack
of nrf2 results in progression of proliferative lesions to
neoplasms induced by long-term exposure to non-genotoxic
hepatocarcinogens involving oxidative stress. Exp Toxicol Pathol.
66:19–26. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cano M, Thimmalappula R, Fujihara M, Nagai
N, Sporn M, Wang AL, Neufeld AH, Biswal S and Handa JT: Cigarette
smoking, oxidative stress, the anti-oxidant response through Nrf2
signaling and Age-related macular degeneration. Vision Res.
50:652–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rangasamy T, Cho CY, Thimmulappa RK, Zhen
L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM and
Biswal S: Genetic ablation of Nrf2 enhances susceptibility to
cigarette smoke-induced emphysema in mice. J Clin Invest.
114:1248–1259. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki M, Betsuyaku T, Ito Y, Nagai K,
Nasuhara Y, Kaga K, Kondo S and Nishimura M: Down-regulated
NF-E2-related factor 2 in pulmonary macrophages of aged smokers and
patients with chronic obstructive pulmonary disease. Am J Respir
Cell Mol Biol. 39:673–682. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malhotra D, Thimmulappa R, Navas-Acien A,
Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P,
et al: Expression of concern: Decline in NRF2-regulated
antioxidants in chronic obstructive pulmonary disease lungs due to
loss of its positive regulator, DJ-1. Am J Respir Crit Care Med.
178:592–604. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ping Z, Liu W, Kang Z, Cai J, Wang Q,
Cheng N, Wang S, Wang S, Zhang JH and Sun X: Sulforaphane protects
brains against hypoxic-ischemic injury through induction of
Nrf2-dependent phase 2 enzyme. Brain Res. 1343:178–185. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guerrero-Beltrán CE, Calderón-Oliver M,
Pedraza-Chaverri J and Chirino YI: Protective effect of
sulforaphane against oxidative stress: Recent advances. Exp Toxicol
Pathol. 64:503–508. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bergström P, Andersson HC, Gao Y, Karlsson
JO, Nodin C, Anderson MF, Nilsson M and Hammarsten O: Repeated
transient sulforaphane stimulation in astrocytes leads to prolonged
Nrf2-mediated gene expression and protection from
superoxide-induced damage. Neuropharmacology. 60:343–353. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu R, Hebbar V, Kim BR, Chen C, Winnik B,
Buckley B, Soteropoulos P, Tolias P, Hart RP and Kong AN: In vivo
pharmacokinetics and regulation of gene expression profiles by
isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther.
310:263–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Iida K, Itoh K, Kumagai Y, Oyasu R,
Hattori K, Kawai K, Shimazui T, Akaza H and Yamamoto M: Nrf2 is
essential for the chemopreventive efficacy of oltipraz against
urinary bladder carcinogenesis. Cancer Res. 64:6424–6431. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xu C, Huang MT, Shen G, Yuan X, Lin W,
Khor TO, Conney AH and Kong AN: Inhibition of
7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in
C57BL/6 mice by sulforaphane is mediated by nuclear factor
E2-related factor 2. Cancer Res. 66:8293–8296. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burkhardt BR, Lyle R, Qian K, Arnold AS,
Cheng H, Atkinson MA and Zhang YC: Efficient delivery of siRNA into
cytokine-stimulated insulinoma cells silences Fas expression and
inhibits Fas-mediated apoptosis. FEBS Lett. 580:553–560. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Oda K, Yumoto R, Nagai J, Katayama H and
Takano M: Mechanism underlying insulin uptake in alveolar
epithelial cell line RLE-6TN. Eur J Pharmacol. 672:62–69. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nadigel J, Audusseau S, Baglole CJ,
Eidelman DH and Hamid Q: IL-8 production in response to cigarette
smoke is decreased in epithelial cells from COPD patients. Pulm
Pharmacol Ther. 26:596–602. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thorne D and Adamson J: A review of in
vitro cigarette smoke exposure systems. Exp Toxicol Pathol.
65:1183–1193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bryan HK, Olayanju A, Goldring CE and Park
BK: The Nrf2 cell defence pathway: Keap1-dependent and -independent
mechanisms of regulation. Biochem Pharmacol. 85:705–717. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kosmider B, Messier EM, Chu HW and Mason
RJ: Human alveolar epithelial cell injury induced by cigarette
smoke. PLoS One. 6:e260592011. View Article : Google Scholar : PubMed/NCBI
|