Introduction
Acute myeloid leukemia (AML) is a heterogeneous
disease characterized by the malignant clonal proliferation of
immature myeloid cells, which is associated with a poor overall
prognosis (1). To date, only the
acute promyelocytic leukemia subtype of AML has demonstrated
improved therapeutic effects through the use of
differentiation-inducing agents, such as all-trans retinoic acid
and/or arsenic trioxide (2).
However, there are currently no effective treatments for additional
types of AML. Common chemotherapy drug regimens, such as
anthracyclines combined with cytarabine, are not effective for the
treatment of all AML patients, and eventually lead to the
development of drug resistance (3).
In recent years, the demethylation agent,
5-aza-2′-deoxycytidine (5-Aza) has been approved by US Food and
Drug Administration, and applied to treat myelodysplastic syndrome
(MDS) and AML (4). 5-Aza induces
the loss of methylation and reactivates silenced genes via the
irreversible inhibition of DNA methyltransferases (5). A number of previous studies have
reported that 5-Aza induces cell growth inhibition, differentiation
and apoptosis of leukemia cells via distinct mechanisms (6–8). In
particular, Ding et al (8)
demonstrated that 5-Aza increases the sensitivity of
multidrug-resistant HL-60/ADR leukemia cells to aclacinomycin via
the epigenetic modulation of demethylation, which suggests 5-Aza
appears to be safe and effective for the treatment of patients with
high-risk AML. Although 5-Aza monotherapy demonstrates a high
response rate in patients with AML, the complete remission rate
remains low, and eventually all patients experience disease relapse
(9,10). Enhanced tumor invasiveness is
considered to be an important reason underlying the clinical
failure and disease progression that occurs following 5-Aza
treatment (10,11). However, the detailed mechanisms of
5-Aza-induced increases in tumor cell migration and invasion have
not yet been completely elucidated.
In the present study, the effect of 5-Aza on the
growth and differentiation of THP-1 monocytic leukemia cells was
investigated. Of particular note, the molecular mechanism of
5-Aza-induced increases in THP-1 cell migration was proposed. The
results demonstrated the opposing roles of 5-Aza in the treatment
of acute monocytic leukemia, and present a potential strategy to
inhibit the cell migration pathway in the clinical application of
5-Aza.
Materials and methods
Reagents
5-Aza (catalog no. A3656) and the RS102895 (RS) C-C
chemokine receptor type 2 (CCR2) inhibitor (catalog no. R1903) were
purchased from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
Anti-CCR2 (catalog no. 12199S), anti-extracellular signal-regulated
kinase (ERK) (catalog no. 4695S), anti-phosphorylated (p)-ERK
(catalog no. 4376S), anti-AKT (catalog no. 9272S), anti-p-AKT
(catalog no. 4058S), anti-β-actin (catalog no. 4967S) and
horseradish peroxidase-conjugated secondary antibody (catalog no.
7074S) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). The phosphatidylinositol-4,5-bisphosphate
3-kinase (PI3K) inhibitor, LY294002 (catalog no. S1105), the c-Jun
N-terminal kinase (JNK) inhibitor, SP600125 (catalog no. S1460),
the p38 inhibitor, SB203580 (catalog no. S1076), the
mitogen-activated protein kinase (MAPK)/ERK inhibitor, PD98059
(catalog no. S1177) and the NF-κB inhibitor, BMS345541 (catalog no.
8044), were purchased from Selleck Chemicals (Houston, TX, USA).
Transwell chambers were purchased from Corning Incorporated
(Corning, NY, USA). The chemokine (C-C motif) ligand 2 (CCL2)
enzyme-linked immunosorbent assay (ELISA) kit (catalog no. DCP00)
was obtained from R&D Systems, Inc. (Minneapolis, MN, USA). The
bromodeoxyuridine (BrdU) kit (catalog no. 552598) was purchased
from BD Biosciences (Franklin Lakes, NJ, USA). The phycoerythrin
(PE)-conjugated anti-human cluster of differentiation 14 (CD14)
antibody was purchased from eBioscience, Inc. (San Diego, CA, USA).
THP-1 cell line was purchased from the American Type Culture
Collection (Manassas, VA, USA). The bicinchoninic acid protein
assay kit (catalog no. 23225) was purchased from Thermo Fisher
Scientific, Inc., Waltham, MA, USA.
Migration assay
THP-1 cells were cultured in RPMI 1640 (Hyclone; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum (FBS; Biological industries USA, Inc., CT, USA),
100 U/ml ampicillin (Beijing Solarbio Science and Technology Co.,
Ltd., Beijing, China) and 100 U/ml streptomycin (Beijing Solarbio
Science and Technology Co., Ltd.) at 37°C and 5% CO2.
Prior to stimulation, THP-1 cells were divided into different
groups and treated with a certain drug according to experimental
needs. In general, 1×106 pretreated cells were resuspended in 100
µl serum-free RPMI 1640 media and transferred to the upper
Transwell chamber of the 24-well plates in the absence or presence
of a specific drug. Complete media (600 µl) containing 20% FBS was
added to the lower chamber. The plates were placed in the incubator
for 48 h at 37°C. The number of cells that had migrated to the
lower chamber was then determined. An equal volume of 0.1% dimethyl
sulfoxide (DMSO) that was used to reconstitute the drug was used as
a negative control for all experiments.
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
A total of 1×106 THP-1 cells were treated with 0.5
µM 5-Aza or DMSO for 48 h. Total RNA was isolated from 5-Aza or
DMSO treated THP-1 cells using Trizol Reagent (Thermo Fisher
Scientific, Inc.). First-strand cDNA was synthesized using 2 µg of
total RNA in a 20 µl reverse transcriptase reaction mixture using a
cDNA synthesis kit (Beijing Transgen Biotech Co., Ltd., Beijing,
China). The primers used for PCR were as follows: forward, 5′CTC
ATA GCA GCC ACC TTC AT3′ and reverse, 5′TCA CAG CTT CTT TGG GAC
AC3′ for CCL2; forward, 5′TCT CGT TCT CGG TTT ATC AG3′ and reverse,
5′ATT CCC AAA GAC CCA CTC AT3′ for CCR2; forward, 5′TGC ACC ACC AAC
TGC TTA GC3′ and reverse, 5′GGC ATG GAC TGT GGT CAT GA3′ for GAPDH.
Cycling conditions were as follows: An initial predenaturation step
at 94°C for 5 min, followed by 28 cycles of denaturation at 94°C
for 30 sec, annealing at 57°C for 30 sec, extension at 72°C for 30
sec and a final extension step at 72°C for 5 min. The PCR product
was confirmed by electrophoresis on 2% agarose gels, stained with
0.5 µg/ml ethidium bromide solution and detected using a Tanon 1600
ultraviolet detector.
Western blot analysis
A total of 2×106 THP-1 cells were treated with 0.5
µM 5-Aza or 0.1% DMSO for different time points at 4°C. Total
cellular protein (100 µg) was extracted with
radioimmunoprecipitation buffer (Thermo Fisher Scientific, Inc.) at
4°C and quantified using a bicinchoninic acid assay kit (Thermo
Fisher Scientific, Inc.). Proteins were separated on 12% SDS-PAGE
gels and transferred onto polyvinylidene difluoride membranes. The
nonspecific binding of transferred proteins was blocked with PBS
buffer containing 5% non-fat milk powder for 2 h at room
temperature. The membrane was probed with a 1:1,000 dilution of
primary antibodies against p-ERK, ERK, p-AKT, AKT, CCR2 or β-actin
overnight at 4°C, prior to washing three times with Tris-buffered
saline and 0.05% Tween 20 buffer. Membranes were subsequently
probed with a 1:1,000 dilution of horseradish peroxidase-conjugated
secondary antibody. Blotted proteins were detected using an
enhanced chemiluminescence kit (Thermo Fisher Scientific, Inc.) and
bands were quantified using Tanon Image software version 1.10
(Tanon Science and Technology Co., Ltd., Shanghai, China).
ELISA
A total of 1×106 THP-1 cells were treated with 0.5
µM 5-Aza for 96 h. The culture medium was then collected and the
concentration of CCL2 was determined by measuring the absorbance on
a plate reader at a wavelength of 450 nm, according to the
manufacturer's protocol (R&D Systems, Inc.).
Cell differentiation
A total of 2×105 THP-1 cells were cultured in
Dulbecco's modified Eagle's medium containing 10% FBS at 37°C in
the presence of 0.1% DMSO or 0.1 or 0.5 µM 5-Aza for 6 days. During
the incubation period, the cell culture media containing DMSO or
5-Aza were refreshed daily. Cells were stained with a 1:100
dilution of PE-conjugated anti-CD14 antibody for 20 min at room
temperature, and were subsequently analyzed by flow cytometry using
a BD Accuri C6 instrument (BD Biosciences). Cell morphology was
observed by Wright-Giemsa staining. Briefly, 2×104 treated THP-1
cells were centrifuged at 300 × g for 10 min, piptted onto onto
glass slides and then stained with Wright-Giemsa solution (catalog
no. BA4017; Zhuhai Beisuo Biological Technology Co., Ltd., Zhuhai,
China) for 15 min at room temperature. Cells were visualized with a
light microscope (model BA1303IF) and captured using imaging
software version 3.5 (Chongqing Lin Pei Photoelectric Instrument
Co., Ltd., Chongqing, China). Images were captured from three
independent experiments.
Cell counting kit-8 (CCK-8) assay
A total of 1×104 THP-1 cells were seeded in 96-well
plates and treated with 0.1, 0.5, 1.0, 2.0 µM 5-Aza or 0.1% DMSO.
Cell growth was assessed at 0, 24, 48 and 72 h by adding 10 µl
CCK-8 solution (BestBio, Shanghai, China) in the cell medium and
incubated at 37°C for 2.5 h, and the absorbance was measured using
a plate reader at a wavelength of 450 nm.
BrdU staining
A total of 1×106 THP-1 cells were treated with 0.5
µM 5-Aza or DMSO for 48 h. DMSO was used as a negative control.
BrdU was then directly added to the cell culture medium to final
concentration of 10 µM. Cells was gently mixed and returned to the
incubator for 30 min at 37°C. The cells were subsequently
collected, fixed and permeabilized, according to the BrdU kit
protocol (BD Biosciences). Finally, cells were stained with a 1:50
dilution of allophycocyanin-conjugated BrdU antibody for 20 min at
room temperature in the dark, and were analyzed by flow cytometry
using a BD Accuri C6 instrument (BD Biosciences). Inhibition rate
(IR) was calculated as follows: IR=(X -Y)/X, where X and Y are the
percentage of BrdU+ cells in the presence of the DMSO
control and 5-Aza, respectively.
Statistical analysis
Data are presented as the mean ± standard error of
the mean, and were analyzed using GraphPad Prism 5.01 software
(GraphPad Software, Inc., La Jolla, CA, USA). Comparison of the
mean between 2 groups was achieved using the Student's t-test, and
comparisons among multiple groups was achieved by one-way analysis
of variance with a Dunnett's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
5-Aza treatment induced
differentiation and growth inhibition of THP-1 cells
A previous study demonstrated that 5-Aza induces
differentiation and growth inhibition of HL60 human promyelocytic
leukemia cells (12). THP-1 cells,
which are driven by the mixed-lineage leukemia (MLL)-ALL1-fused
gene from chromosome 9 (AF9) fusion gene, are derived from the M5
subtype of AML, and are different to HL60 cells in a number of
respects, including the regulation of gene expression, signaling
pathway involvement and pathogenesis. To investigate whether 5-Aza
inhibits the proliferation of THP-1 cells, cell growth was
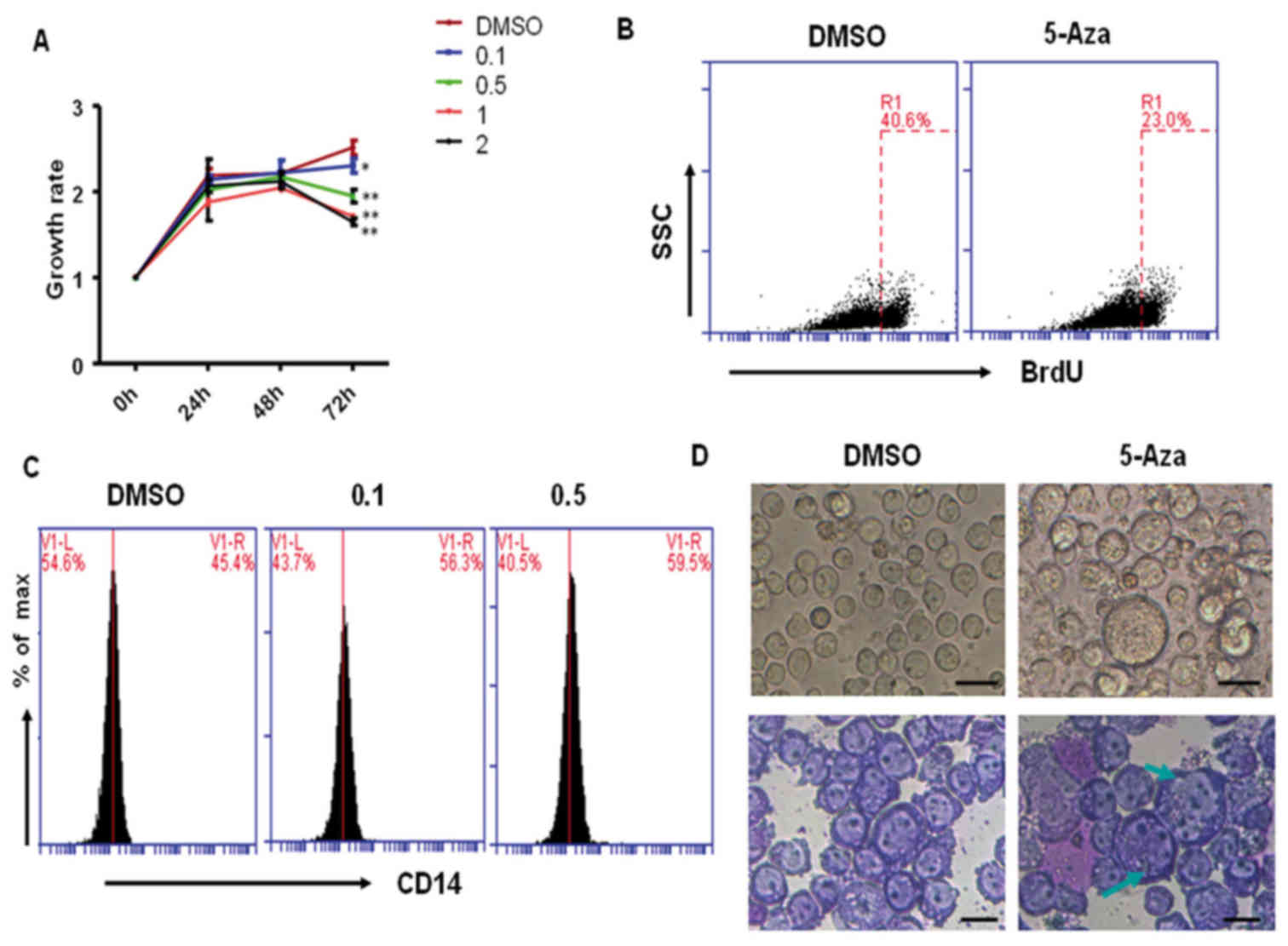
evaluated by CCK-8 staining. As shown in Fig. 1A, 0.1, 0.5, 1 and 2 µM 5-Aza
treatment significantly inhibited THP-1 cell growth at 72 h when
compared with the DMSO-treated control cells. However, at 24 and 48
h, cell growth was not significantly altered among the
5-Aza-treated groups, which suggested that 5-Aza-induced cell
growth inhibition may be dose- and time-dependent. Similarly, the
results from BrdU staining demonstrated that 5-Aza significantly
inhibited THP-1 cell proliferation, and the rate of growth
inhibition reached 43% following treatment with 0.5 µM 5-Aza for 72
h (Fig. 1B).
In order to determine whether 5-Aza treatment
induced differentiation of THP-1 cells, cells that had been
pretreated with 0.5 µM 5-Aza for 6 days were stained with a
PE-conjugated anti-CD14 antibody, and analyzed by flow cytometric
analysis. The results demonstrated that the level of CD14
expression significantly increased in 0.1 or 0.5 µM 5-Aza-treated
cells when compared with that of DMSO-treated cells (Fig. 1C). However, when comparing the 0.5
and 0.1 µM 5-Aza-treatment groups, they demonstrated similar
results, suggesting that low concentrations of 5-Aza may be enough
to induce cell differentiation. In addition, the morphology of
5-Aza-treated cells was observed under an inverted microscope and
following Wright-Giemsa staining. As shown in Fig. 1D, a large proportion of
5-Aza-treated cells exhibited an increase in cell volume (indicated
by green arrows) when compared with the DMSO-treated control group.
These differentiated cells accounted for ~20% of all 5-Aza-treated
cells. These results suggest that 5-Aza treatment weakly induced
the differentiation of THP-1 cells.
5-Aza promotes THP-1 cell
migration
5-Aza may promote cancer cell invasion by
upregulating genes associated with this process. Bernal et
al (10) reported that 5-Aza
upregulated the expression of matrix metalloproteinase (MMP)-9 in
MDS-derived AML cell lines, which may enhance invasiveness in
vitro. In human fibrosarcoma cells, 5-Aza was observed to
enhance tumor cell invasion via transcription-dependent modulation
of MMP-1 expression (11).
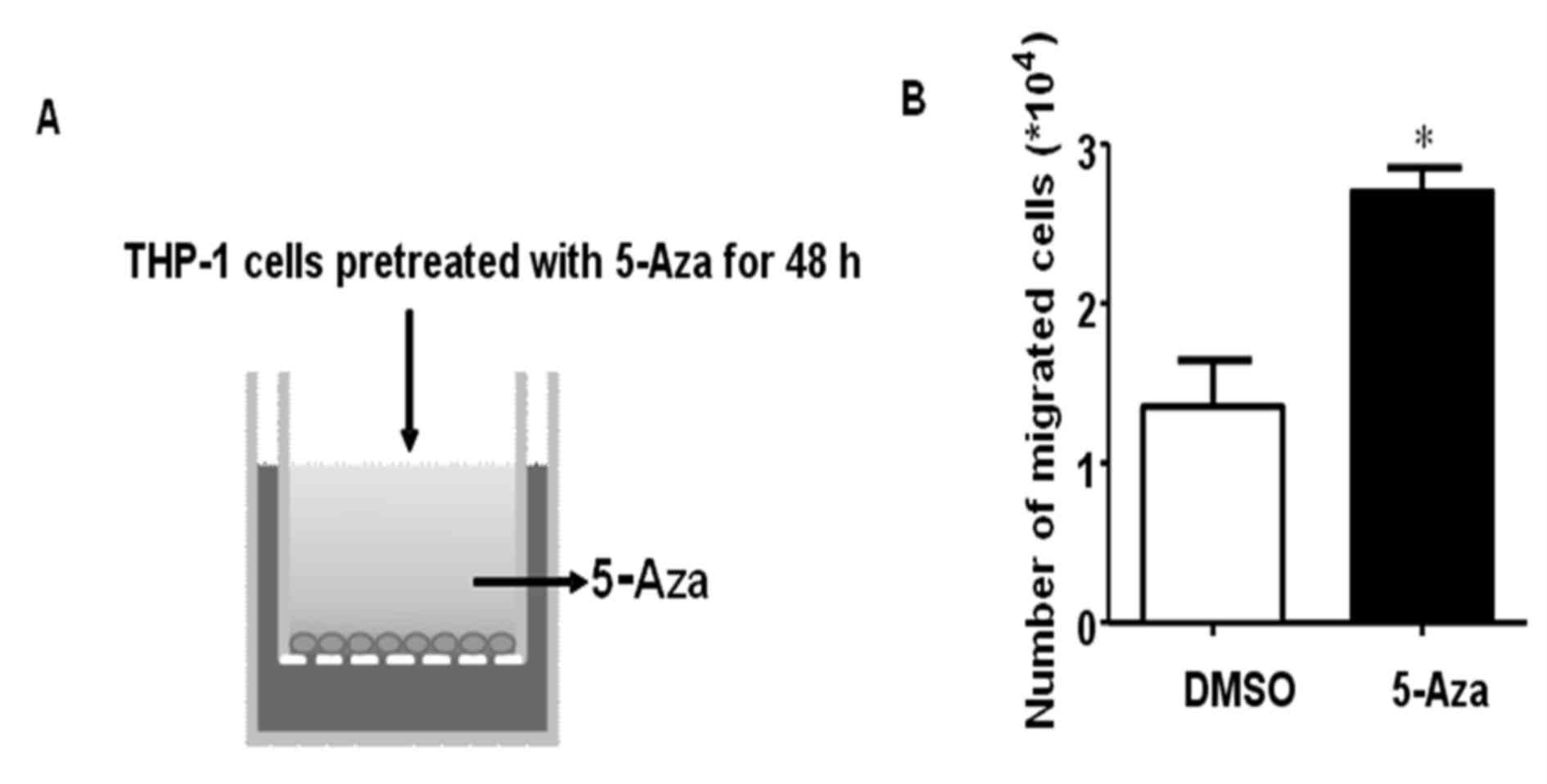
Therefore, the authors of the present study hypothesized that 5-Aza
may promote THP-1 cell migration or invasion. In order to examine
the effects of 5-Aza on THP-1 cell migration, THP-1 cells were
treated with 0.5 µM 5-Aza for 48 h, and then a migration assay was
performed using Transwell chambers (Fig. 2A). The results demonstrated that
5-Aza significantly promoted THP-1 cell migration, and the number
of migrated cells increased ~2-fold when compared with that of the
DMSO control (Fig. 2B).
5-Aza-induced THP-1 cell migration was
mediated by CCL2-CCR2 signaling
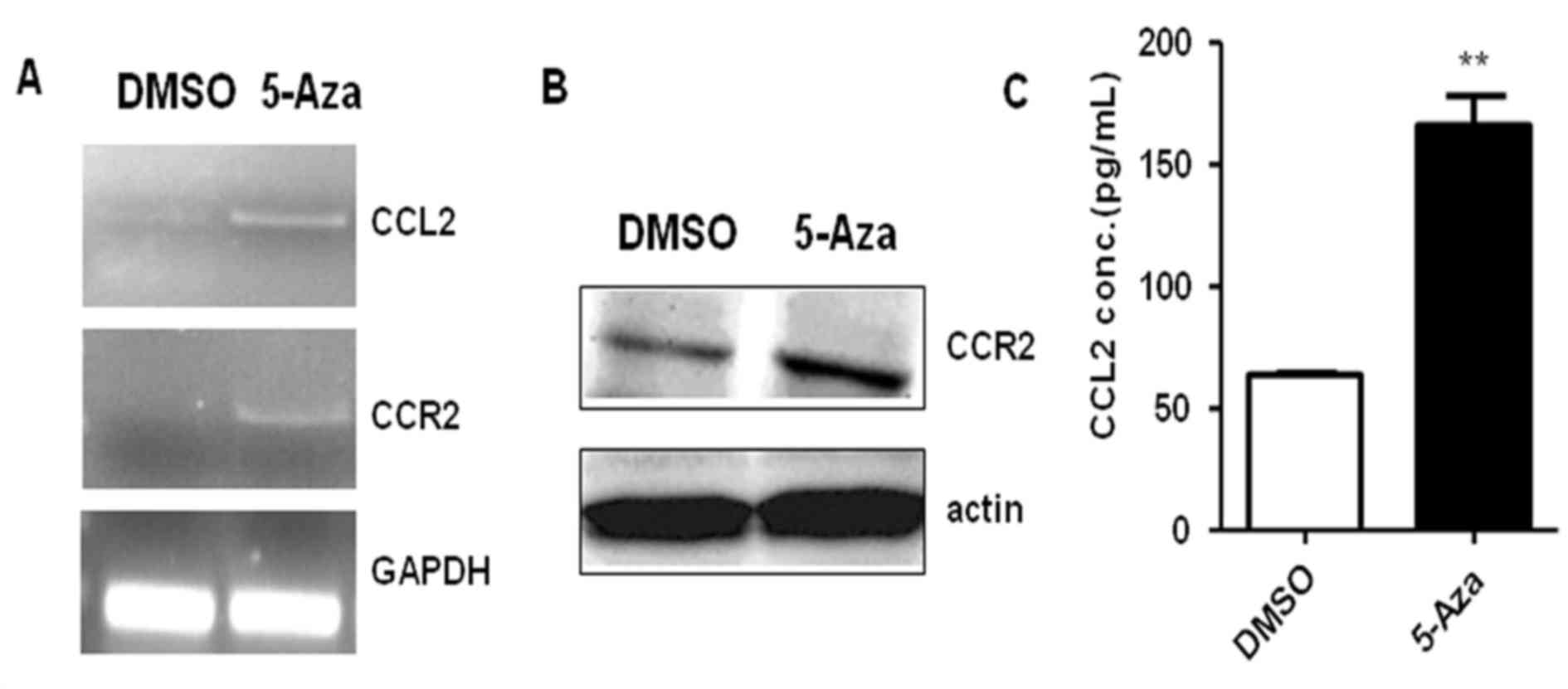
In order to identify the mechanism underlying the
5-Aza-induced increase in THP-1 cell migration, gene expression
profiling of 5-Aza-treated THP-1 cells was performed, using a
Phalanx OneArray® (Phalanx Biotech Group, San Diego, CA, USA).
The preliminary results demonstrated that the
expression levels of numerous genes involved cell migration were
altered, particularly CCR2 and CCL2 (data not shown). CCL2, also
known as monocyte chemoattractant protein-1 (MCP-1), is an
important chemotaxis molecule in monocytic cells that regulates
monocyte migration via CCR2-mediated signaling pathways (13). To investigate whether the CCL2-CCR2
signaling pathway participates in the 5-Aza-induced increase in
THP-1 cell migration, CCR2 and/or CCL2 expression was assayed by
reverse transcription-polymerase chain reaction, western blotting
and ELISA in THP-1 cells treated with 0.5 µM 5-Aza for 72 h. The
results demonstrated that CCL2 and CCR2 mRNA levels were markedly
upregulated in THP-1 cells following 72 h of 5-Aza treatment when
compared with DMSO-treated controls (Fig. 3A). Similarly, an increase in CCR2
protein expression levels were observed in 5-Aza-treated THP-1
cells when compared with DMSO-treated controls (Fig. 3B). The ELISA results indicated that
the concentration of CCL2 in cell culture medium increased 2.6-fold
in the presence of 5-Aza when compared with that of control group
(Fig. 3C). These results suggested
that 5-Aza promoted the expression of CCL2 and CCR2 in THP-1 cells.
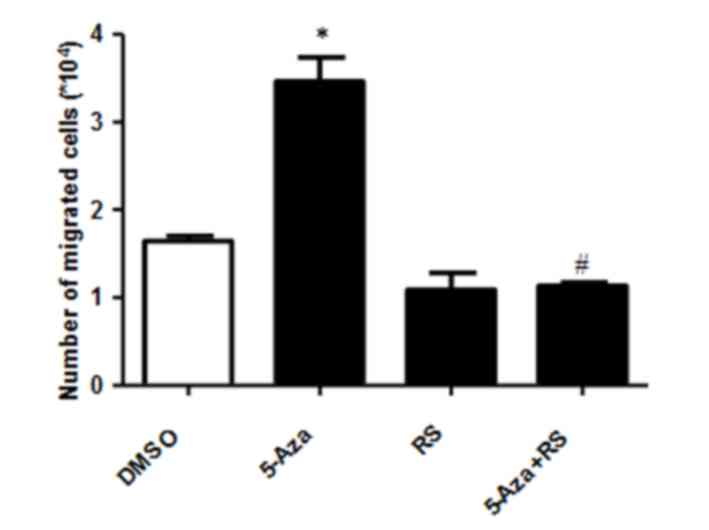
To determine whether this increase in CCL2 and CCR2 expression was
associated with 5-Aza-induced THP-1 cell migration, the migration
capability of cells induced by 5-Aza in the presence of the RS CCR2
inhibitor was examined. As shown in Fig. 4, RS inhibited the 5-Aza-induced
increase in THP-1 cell migration when compared with DMSO or RS-only
treated groups. These results suggest that the 5-Aza-induced
increase in cell migration may be dependent on the CCR2 signaling
pathway.
5-Aza promotes THP-1 cell migration by
CCR2-mediated ERK activation
Our studies demonstrated that the CCL2-CCR2
signaling pathway mediates 5-Aza-induced increases in THP-1 cell
migration (Figs. 3 and 4). In order to investigate the mechanism
underlying CCL2-CCR2-mediated regulation of this process, the role
of CCL2-CCR2 downstream effectors was examined using the following
specific inhibitors: The PI3K inhibitor, LY294002; the JNK
inhibitor, SP600125; the p38 inhibitor, SB203580; the MAPK/ERK
inhibitor PD; and the NF-κB inhibitor, BMS345541. Out of all
inhibitors examine, only PD inhibited the 5-Aza-induced increase in
THP-1 cell migration (data not shown). In addition, the results
demonstrated that 5-Aza promoted the expression of p-ERK following
24 and 36 h of treatment, but not following 18 h (Fig. 5A). This suggested that 5-Aza may
activate ERK indirectly. The protein expression levels of p-AKT
were reduced following treatment with 5-Aza for 18, 24 or 36 h when
compared with DMSO-treated controls (Fig. 5A), suggesting that 5-Aza-induced
growth inhibition may be dependent on the suppression of AKT
signaling pathway To further investigate whether CCL2-CCR2 promotes
cell migration via activation of ERK, the expression levels of
p-ERK were detected by western blot analysis following the
treatment of cells with 5-Aza and/or RS. As shown in Fig. 5B and C, RS inhibited 5-Aza-induced
ERK phosphorylation in THP-1 cells, which suggested that ERK is a
downstream effector of CCL2-CCR2, and ERK activation depends on
CCL2-CCR2 signaling. Notably, treatment of cells with the ERK
inhibitor completely inhibited the 5-Aza-induced increase in THP-1
cell migration (Fig. 5D), which
was similar to that of the CCR2 inhibitor.
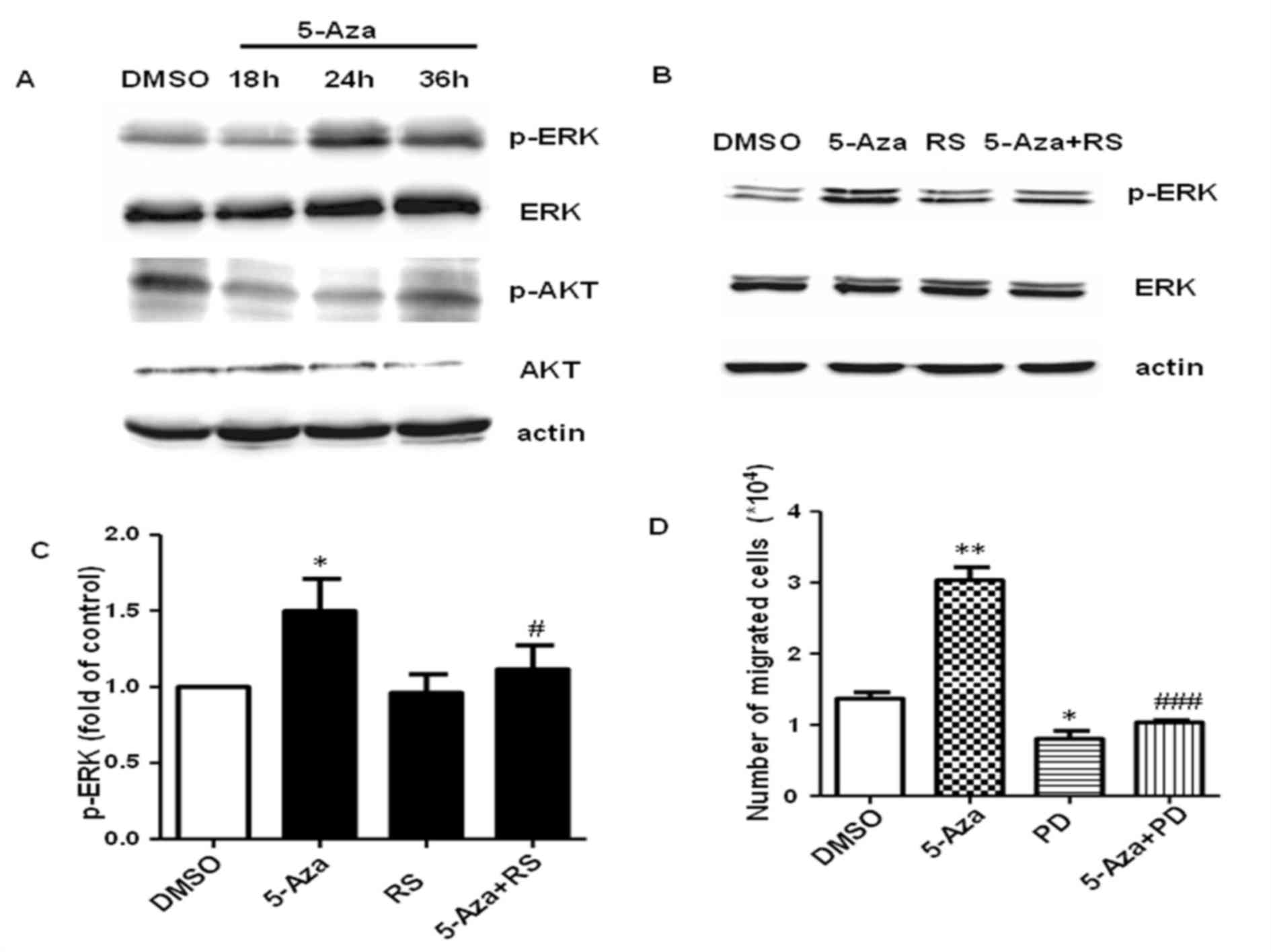 | Figure 5.5-Aza treatment promotes THP-1 cell
migration by CCR2-mediated ERK activation. (A) THP-1 cells were
treated with 0.5 µM 5-Aza for 18, 24 or 36 h and the protein
expression levels of p-ERK, ERK, p-AKT and AKT were analyzed by
western blotting. (B) THP-1 cells were treated with 0.5 µM 5-Aza
with or without 10 µM RS and activation of ERK was assayed by
western blotting. (C) Quantification of p-ERK protein expression
expressed relative to DMSO-treated controls. *P<0.05 vs. DMSO
and #P<0.05 vs. 5-Aza. (D) The migration ability of
THP-1 cells following treatment with 5-Aza with or without 10 µM
PD. Data are presented as the mean ± standard error of the mean
(n=4). *P<0.05 and **P<0.01 vs. DMSO and
###P<0.001 vs. 5-Aza. 5-Aza, 5-aza-2′-deoxycytidine;
CCR2, C-C chemokine receptor type 2; ERK, extracellular
signal-regulated kinase; p-ERK, phosphorylated ERK; DMSO, dimethyl
sulfoxide; RS, RS102895; PD, PD98059. |
Discussion
Several studies have reported that 5-Aza exerts a
number of effects in different leukemia cell lines, including
inhibition of cell proliferation and induction of differentiation
(6,12). THP-1, a monocytic leukemia cell
line driven by the MLL-AF9 fusion gene, was employed for the
purposes of the present study. This THP-1 cell line is different
from additional previously reported cell lines including the gene
expression status, signaling pathway involvement and pathogenesis,
as these cells are driven by the MLL-AF9 fusion gene. The effect of
5-Aza treatment in THP-1 cells was investigated. Using CCK-8 assay
analysis and BrdU staining, the results demonstrated that 5-Aza
treatment inhibited THP-1 cell growth in vitro. The
mechanism underlying the anti-proliferative effects of 5-Aza is
hypothesized to involve the demethylation of genes, including tumor
suppresser genes, cell cycle inhibitors and microRNAs (miRNAs). For
instance, demethylation of p16INK4a by 5-Aza treatment induced cell
growth inhibition of adult T-cell leukemia/lymphoma cells (14), HL60 cells (15), and an MDS cell line (16). In addition, previous studies have
demonstrated that specific miRNAs may be involved in 5-Aza-induced
cell growth arrest, such as miR-125a (17), miR-193a (18). Ufkin et al (17) indicated that 5-Aza-induced
restoration of miR-125a expression led to decreased cell
proliferation, cell cycle progression, and enhanced apoptosis in
NB4 cells via targeting of the the ErbB signaling pathway. In
addition, Gao and colleagues (18)
demonstrated that 5-Aza-induced restoration of miR-193a expression
in AML inhibited c-kit expression, followed by inhibition of cell
growth (18). Nishi et al
(19) reported that the regulatory
region for let-7b expression was hypermethylated in an MLL fusion
gene-driven leukemic cell line, and cell growth was inhibited once
its expression was partially recovered by 5-Aza treatment (19). It is therefore possible that this
mechanism may be responsible for the 5-Aza-induced growth arrest of
MLL-AF9-driven THP-1 cells in the present study. However, the
possibility that additional molecular mechanisms are involved in
the 5-Aza-induced growth arrest of THP-1 cells cannot be
excluded.
In the current study, the level of differentiation
of 5-Aza-induced THP-1 cells was determined by microscopy and by
assessing the expression levels of CD14. The results indicated that
5-Aza produces a similar biological effect on cell differentiation
in THP-1 cells to different types of AML cell lines (12,20).
5-Aza is hypothesized to induce differentiation by inhibiting DNA
methylation and upregulating the expression of transcription
factors associated with differentiation. Koschmieder et al
(20) demonstrated that 5-Aza
induced upregulation of CD14 by enhancing Sp1 transcriptional
activity. In the present study, 5-Aza treatment enhanced CD14
expression in THP-1 cells, which was associated with an increase in
the number of differentiated cells. Therefore, the authors
hypothesized that Sp1 may be critical for this process in THP-1
cells. PU.1 is a member of the Ezb transformation-specific sequence
family of transcription factors and is expressed in granulocytic,
monocytic and B-lymphoid cells (21,22).
5-Aza-induced differentiation of K562 cells is dependent on the
PU.1 expression levels (22).
Alberich-Jordà et al (23)
demonstrated that 5-Aza treatment restored the
CCAAT-enhancer-binding protein (C/EBP)α-C/EBPγ balance and promoted
the differentiation of primary human AML samples, which was
characterized by C/EBPα silencing and C/EBPγ upregulation in
vitro. Therefore the authors of the present study will focus on
investigating the function of these key transcription factors in
5-Aza-induced THP-1 differentiation in future studies.
Besides cell growth arrest and increased
differentiation of THP1 cells by 5-Aza treatment, the results of
the current study demonstrated that 5-Aza significantly promoted
THP-1 migration, and exhibited a 2-fold increase in cell migration
when compared with the controls. To elucidate the underlying
molecular mechanisms, gene expression profiling was performed using
0.5 µM 5-Aza-treated THP-1 cells. Notably, a number of genes
involved in cell migration and invasion were upregulated, including
MMP9, CCR2 and CCL2. A previous study observed upregulated MMP9
expression in a 5-Aza-treated AML cell line and in samples derived
from patients with disease relapse (10). MMP9 serves a critical role in AML
by increasing the invasive properties of malignant myeloblasts,
thus promoting clinical failure of the drug and progression to a
more aggressive disease (10). The
focus of the present study was to investigate the functions of CCL2
and CCR2 in the 5-Aza-induced increase in THP-1 cell migration.
CCR2 is an important marker of monocytic cells, and the binding of
CCL2 to CCR2 results in the chemotaxis of monocytes (24,25).
Therefore, the authors proposed that 5-Aza-induced THP-1 cell
migration may be dependent on the CCL2-CCR2 axis. The results
indicated that 5-Aza promoted CCR2 expression at the
transcriptional and translational levels, and CCL2 is highly
expressed in 5-Aza-treated THP-1 cells and secreted into the
culture medium. The authors initially considered the possibility
that 5-Aza may alter CCL2 and CCR2 expression via
demethylating-dependent or independent mechanisms (26). The level of CCR2 expression is
affected by stress, such as intermittent hypoxia (24). Therefore, the authors proposed that
5-Aza may influence the expression of CCL2 and CCR2 via a
demethylation-independent mechanism.
In the current study, the role of several signaling
pathways involving JNK, ERK, p38 and NF-κB, in the 5-Aza-induced
increase in THP-1 cell migration were investigated using specific
inhibitors. Out of all inhibitors analyzed, only the ERK inhibitor
was observed to prevent the 5-Aza-induced increase in THP-1 cell
migration. In obstructive sleep apnea (OSA), intermittent hypoxia
induces the activation of ERK1/2 and p38 MAPK signaling pathways in
monocytic cells, and increases CCR2 expression and MCP-1-induced
chemotaxis of monocytes (24).
This indicates that the CCL2-CCR2 axis may promote the chemotaxis
and adhesion of monocytes (24).
Yang et al (25)
demonstrated that upregulation of CCL2 and CCR2 in low-metastatic
nasopharyngeal carcinoma cell lines may promote cell migration and
invasion. In addition, dual overexpression of CCL2/CCR2 activated
the ERK1/2 signaling pathway, which sequentially upregulated MMP2
and MMP9. Similarly, Choi et al (27) demonstrated that the MAPK signaling
pathway regulates the invasion and migration of monocytic cells.
These reports are consistent with the results of the current study.
The results further demonstrated that 5-Aza treatment induced cell
migration by activating the CCL2-CCR2-ERK signaling pathway.
In conclusion, the present study demonstrated the
important role of 5-Aza in promoting THP-1 cell differentiation and
growth arrest, which reflects its promising application for the
treatment of patients with MLL-AF9-driven AML in the clinic. In
addition, the results indicated that 5-Aza might promote THP-1 cell
migration by activating the CCL2-CCR2-ERK signaling pathway. These
results will be important for the identification of the molecular
mechanisms underlying the 5-Aza-induced increase in cell migration,
which may lead to the development of strategies that reduce the
likelihood of disease relapse in AML patients treated with 5-Aza.
For instance, inhibition of the ERK or CCR2 signaling pathways in
combination with 5-Aza may be an alternative method for the
clinical treatment of patients with AML.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81570149).
References
|
1
|
Conway O, Brien E, Prideaux S and
Chevassut T: The epigenetic landscape of acute myeloid leukemia.
Adv Hematol. 2014:1031752014.PubMed/NCBI
|
|
2
|
Luesink M, Pennings JL, Wissink WM,
Linssen PC, Muus P, Pfundt R, de Witte TJ, van der Reijden BA and
Jansen JH: Chemokine induction by all-trans retinoic acid and
arsenic trioxide in acute promyelocytic leukemia: Triggering the
differentiation syndrome. Blood. 114:5512–5521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Radujkovic A, Dietrich S, Bochtler T,
Krämer A, Schöning T, Ho AD, Dreger P and Luft T: Azacitidine and
low-dose cytarabine in palliative patients with acute myeloid
leukemia and high bone marrow blast counts-a retrospective
single-center experience. Eur J Haematol. 93:112–117. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Plimack ER, Kantarjian HM and Issa JP:
Decitabine and its role in the treatment of hematopoietic
malignancies. Leuk Lymphoma. 48:1472–1481. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vispé S, Deroide A, Davoine E, Desjobert
C, Lestienne F, Fournier L, Novosad N, Bréand S, Besse J, Busato F,
et al: Consequences of combining siRNA-mediated DNA
methyltransferase 1 depletion with 5-aza-2′-deoxycytidine in human
leukemic KG1 cells. Oncotarget. 6:15265–15282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang F, Dai X and Wang Y:
5-Aza-2′-deoxycytidine induced growth inhibition of leukemia cells
through modulating endogenous cholesterol biosynthesis. Mol Cell
Proteomics. 11:M111.0169152012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shin DY, Park YS, Yang K, Kim GY, Kim WJ,
Han MH, Kang HS and Choi YH: Decitabine, a DNA methyltransferase
inhibitor, induces apoptosis in human leukemia cells through
intracellular reactive oxygen species generation. Int J Oncol.
41:910–918. 2012.PubMed/NCBI
|
|
8
|
Ding B, Wang Z, Jiang X, Li X, Wang C,
Zhong Q, Jiang L, Dai M, Zhang YU, Wei QI and Meng F: Palliative
chemotherapy followed by methylation inhibitor in high-risk acute
myeloid leukemia: An in vitro and clinical study. Mol Clin Oncol.
3:1139–1144. 2015.PubMed/NCBI
|
|
9
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz
G, List A, et al: Efficacy of azacitidine compared with that of
conventional care regimens in the treatment of higher-risk
myelodysplastic syndromes: A randomised, open-label, phase III
study. Lancet Oncol. 10:223–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bernal T, Moncada-Pazos A, Soria-Valles C
and Gutiérrez-Fernández A: Effects of azacitidine on matrix
metalloproteinase-9 in acute myeloid leukemia and myelodysplasia.
Exp Hematol. 41:172–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poplineau M, Schnekenburger M, Dufer J,
Kosciarz A, Brassart-Pasco S, Antonicelli F, Diederich M and
Trussardi-Régnier A: The DNA hypomethylating agent,
5-aza-2′-deoxycytidine, enhances tumor cell invasion through a
transcription-dependent modulation of MMP-1 expression in human
fibrosarcoma cells. Mol Carcinog. 54:24–34. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hassan HT, Veit A and Maurer HR:
Synergistic interactions between differentiation-inducing agents in
inhibiting the proliferation of HL-60 human myeloid leukaemia cells
in clonogenic micro assays. J Cancer Res Clin Oncol. 117:227–231.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Deshmane SL, Kremlev S, Amini S and Sawaya
BE: Monocyte chemoattractant protein-1 (MCP-1): An overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Uenogawa K, Hatta Y, Arima N, Hayakawa S,
Sawada U, Aizawa S, Yamamoto T and Takeuchi J: Azacitidine induces
demethylation of p16INK4a and inhibits growth in adult T-cell
leukemia/lymphoma. Int J Mol Med. 28:835–839. 2011.PubMed/NCBI
|
|
15
|
Su Y, Xu H, Xu Y, Yu J, Xian Y and Luo Q:
Azacytidine inhibits the proliferation of human promyelocytic
leukemia cells (HL60) by demethylation of MGMT, DAPK and p16 genes.
Hematology. 17:41–46. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kimura S, Kuramoto K, Homan J, Naruoka H,
Ego T, Nogawa M, Sugahara S and Naito H: Antiproliferative and
antitumor effects of azacitidine against the human myelodysplastic
syndrome cell line SKM-1. Anticancer Res. 32:795–798.
2012.PubMed/NCBI
|
|
17
|
Ufkin ML, Peterson S, Yang X, Driscoll H,
Duarte C and Sathyanarayana P: miR-125a regulates cell cycle,
proliferation, and apoptosis by targeting the ErbB pathway in acute
myeloid leukemia. Leuk Res. 38:402–410. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao XN, Lin J, Li YH, Gao L, Wang XR, Wang
W, Kang HY, Yan GT, Wang LL and Yu L: MicroRNA-193a represses c-kit
expression and functions as a methylation-silenced tumor suppressor
in acute myeloid leukemia. Oncogene. 30:3416–3428. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishi M, Eguchi-Ishimae M, Wu Z, Gao W,
Iwabuki H, Kawakami S, Tauchi H, Inukai T, Sugita K, Hamasaki Y, et
al: Suppression of the let-7b microRNA pathway by DNA
hypermethylation in infant acute lymphoblastic leukemia with MLL
gene rearrangements. Leukemia. 27:389–397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koschmieder S, Agrawal S, Radomska HS,
Huettner CS, Tenen DG, Ottmann OG, Berdel WE, Serve HL and
Müller-Tidow C: Decitabine and vitamin D3 differentially affect
hematopoietic transcription factors to induce monocytic
differentiation. Int J Oncol. 30:349–355. 2007.PubMed/NCBI
|
|
21
|
Chen HM, Zhang P, Voso MT, Hohaus S,
Gonzalez DA, Glass CK, Zhang DE and Tenen DG: Neutrophils and
monocytes express high levels of PU.1 (Spi-1) but not Spi-B. Blood.
85:2918–2928. 1995.PubMed/NCBI
|
|
22
|
Aoyama S, Nakano H, Danbara M, Higashihara
M, Harigae H and Takahashi S: The differentiating and apoptotic
effects of 2-aza-5′-deoxycytidine are dependent on the PU.1
expression level in PU.1-transgenic K562 cells. Biochem Biophys Res
Commun. 420:775–781. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alberich-Jordà M, Wouters B, Balastik M,
Shapiro-Koss C, Zhang H, Di Ruscio A, Radomska HS, Ebralidze AK,
Amabile G, Ye M, et al: C/EBPγ deregulation results in
differentiation arrest in acute myeloid leukemia. J Clin Invest.
122:4490–4504. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chuang LP, Chen NH, Lin SW, Chang YL, Liao
HR, Lin YS, Chao IJ, Lin Y and Pang JH: Increased C-C chemokine
receptor 2 gene expression in monocytes of severe obstructive sleep
apnea patients and under intermittent hypoxia. PLoS One.
9:e1133042014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Lv X, Chen J, Xie C, Xia W, Jiang
C, Zeng T, Ye Y, Ke L, Yu Y, et al: CCL2-CCR2 axis promotes
metastasis of nasopharyngeal carcinoma by activating ERK1/2-MMP2/9
pathway. Oncotarget. 7:15632–15647. 2016.PubMed/NCBI
|
|
26
|
Nishioka C, Ikezoe T, Yang J, Udaka K and
Yokoyama A: Simultaneous inhibition of DNA methyltransferase and
histone deacetylase induces p53-independent apoptosis via
down-regulation of Mcl-1 in acute myelogenous leukemia cells. Leuk
Res. 35:932–939. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi YJ, Yoon JH, Cha SW and Lee SG:
Ginsenoside Rh1 inhibits the invasion and migration of THP-1 acute
monocytic leukemia cells via inactivation of the MAPK signaling
pathway. Fitoterapia. 82:911–919. 2011. View Article : Google Scholar : PubMed/NCBI
|