Introduction
The most prevalent inflammatory bowel diseases
(IBDs) include ulcerative colitis and Crohn's disease, which are
induced by several factors, including genetic defects,
environmental stimuli, immune abnormalities and microbiota
(1). Susceptible hosts may be
associated with defective bacterial clearance, leading to
exaggerated immune responses and subsequent tissue damage;
substantial evidence has shown that abnormal innate immunity,
specifically in microbial recognition and its subsequent
elimination, are involved in the development of IBD (2,3). IBD
exhibits an exaggerated T cell response (Th1/Th17) towards luminal
microbiota, which results in the breakdown of mucosal tolerance to
enteric bacteria (4).
Toll-like receptors (TLRs) are involved in the
development of innate immunity and subsequent development of
adaptive immunity for the clearance of bacterial infection. Thus,
TLRs are key components involved in the innate defense against
pathogens. Lipopolysaccharide (LPS) is an integral component of the
G− bacterial cell wall, which can be recognized by TLR4,
whereas CpG-dsDNA can be recognized by TLR-9 (5). TLR4 and TLR-9 have been confirmed to
be overexpressed in IBD and induce Th1/Th17 differentiation
(6,7). Once TLRs recognize a particular
component, they recruit adaptor proteins and initiate downstream
signaling cascades. According to the protein interaction
information obtained from the online updated Search Tool for the
Retrieval of Interacting Gene 10 database (http://string-db.org/), which has a confidence score
for every protein interaction, TLR4 and TLR-9 have interactions
with cluster of differentiation 40 (CD40).
The CD40/CD40L system is crucial in its involvment
in the differentiation of Th1 and Th17 cells (8,9).
CD40 and CD40L are overexpressed in both forms of IBD (10,11),
indicating that the CD40/CD40L pathway has a key pathogenic role in
intestinal inflammation and is a rational target for therapeutic
intervention. Previous reports have shown that shutting down the
CD40/CD40L system is effective in reducing inflammation in in
vitro cellular systems and in in vivo animal models of
experimental colitis (12,13). Several signaling pathways have been
coupled to CD40, for example, P38 mitogen-activated protein kinase
(MAPK)/tumor necrosis factor (TNF) α has been shown to enlarge
systemic inflammation and promote the differentiation of Th1 and
Th17 cells (14), whereas signal
transducer and activator of transcription (STAT)-3/interleukin
(IL)-6 has been shown to control systemic inflammation and inhibit
the differentiation of Th1 and Th17 cells (15). However, whether LPS or CpG-dsDNA
affect the expression of CD40 in colonic epithelial cells, which
downstream signaling is involved and what effects are induced by
different signaling pathways remain to be elucidated.
In the present study, dextran sodium sulfate, which
is a common agent used for the establishment of IBD in animal
models, was used to injure colonic epithelial cells, and the
protective effects of pretreatment with different concentrations of
LPS and of CpG-dsDNA were observed. In addition, the present study
examined whether pretreatment with LPS or CpG-dsDNA induces
protective effects against IBD by affecting the expression CD40 and
its downstream signaling.
Materials and methods
Cell culture
The human colonic epithelial cell line (HT-29) was
purchased from Cell Biologics, Inc. (Chicago, IL, USA). The cell
line was cultured in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) supplemented
with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), 100 U/ml penicillin and 100 µg/ml streptomycin
at 37°C in 5% CO2. At 90% confluence, the HT-29 cells,
with an initial density of 1×105 cells/well, were challenged with
various concentrations of microbial cell components, including LPS
from Escherichia coli (E.coli O111:B4; Beyotime Institute of
Biotechnology, Haimen, China) at concentrations of 50, 20 and 10
µg/ml, or CpG-dsDNA from double-stranded genomic DNA of E.coli K12
(Invivogen, San Diego, CA, USA) at concentrations of 50, 20 and 10
µg/ml at 37°C for 24 h. The cells were then stimulated with 20
µg/ml DSS (MP Biomedicals, Santa Ana, CA, USA) at 37°C for a
further 24 h.
Wound repair assay
A wound repair assay was performed, as previously
described (16). Briefly, the
HT-29 cells were cultured in 12-well plates to 90% confluence and a
wound was made in the confluent monolayer by mechanical scraping.
The wound was observed and recorded every 4 h for a total of 24 h
using video microscopy (Olympus Corporation, Tokyo, Japan). The
repair index was calculated according to a linear regression
equation of the remaining wound area over time.
Flow cytometric analysis
The cells from a six-well plate were harvested and
washed twice in phosphate-buffered saline (PBS), counted, and
re-suspended in 1% BSA (RayBiotech, Norcross, GA, USA) containing
0.01% NaN3. For flow cytometric analysis, 106 cells were
incubated with mouse anti-human CD40-PE antibody (dilution, 1:200;
cat. no. 9821–09; SouthernBiotech, Birmingham, AL, USA) at 37°C for
1 h. The cells were then washed with washing buffer three times,
re-suspended in 0.5 ml PBS and analyzed using a flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA). All incubations were
performed on ice. Anti-CD11b (dilution, 1:200; cat. no.
FHP011b-100; 4A Biotech Co., Ltd., Beijing, China) was used for
isotype controls. The expression levels of antigen were calculated
as a percentage of positive cells in the total cells using
CytExpert software version 1.1.10.0 (Beckman Coulter, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA was extracted from the cell samples using TRIzol
reagent (Takara Bio, Inc., Otsu, Japan). The RT-qPCR analysis was
performed using cDNA generated from 3 µg of total RNA using the
SuperScript II Reverse Transcriptase kit (Takara Bio, Inc.). The
following PCR primer sequences were used: STAT-3, forward
5′-CATACCCTTGTGGATCGTGCACG-3′ and reverse
5′-GGCAAAGGCTTACTGATAAACTTGA-3′; P38 MAPK, forward
5′-CGCATCTGAACTGTTGTAGGGTG-3′ and reverse
5′-TGTCTTTGTGGGAGGGTAAGACA-3′; 18S, forward
5′-GGTTCCTTTGGTCGCTCGC-3′ and reverse 5′-CTGCTGCCTTCCTTGGATGTG-3′.
The reactions were performed using SYBR Green Master mix (Takara
Bio, Inc.) and quantitatively measured using an ABI Prism 7900HT
Sequence Detection system (Thermo Fisher Scientific, Inc.). The
following thermal cycler parameters were used: A single cycle of
95°C for 30 sec and 40 cycles of denaturation (95°C for 15 sec) and
combined annealing/extension (60°C for 45 sec). The relative mRNA
expression of target genes was normalized with that of the
endogenous 18S control gene (Applied Biosystems; Thermo Fisher
Scientific, Inc.) (17).
Western blot analysis
The cells were disrupted by homogenization on ice
and centrifuged at 12,000 g for 30 min at 4°C, followed by
collection of supernatants. Equal quantities of protein (80 µg)
were separated by 10% SDS-PAGE and transferred onto a
nitrocellulose membrane. The membranes were blocked in 5% (w/v)
skim milk and incubated with antibodies against human p-STAT3
(dilution, 1:1,000; cat. no. ab30647; Abcam, Cambridge, UK), STAT-3
(dilution, 1:1,000; cat. no. 9939; Cell Signaling Technology, Inc.,
Danvers, MA, USA), phosphorylated (p)-P38 MAPK (dilution, 1:1,000;
cat. no. sc-101759; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), p38MAPK (dilution, 1:1,000; cat. no. sc-7149; Santa Cruz
Biotechnology, Inc.) and β-actin (dilution, 1:2,000; cat. no.
sc-7210; Santa Cruz Biotechnology, Inc.), respectively. The blots
were then incubated with horseradish peroxidase-conjugated
secondary antibodies (dilution, 1:2,000; cat. no. sc-2054; Santa
Cruz Biotechnology, Inc.) and detected using enhanced
chemiluminescence (Thermo Fisher Scientific, Inc.). The
quantification of protein expression was performed using AlphaEase
software version 2200 (Alpha Innotech Corporation; ProteinSimple,
San Jose, CA, USA). The ratios of band intensity were calculated
using the value of the band intensity of the experimental protein
divided by that of β-actin. Data are expressed as the mean ±
standard deviation of the mean of three replicate experiments.
Determination of the levels of IL-6
and TNFα
The supernatants were assayed for IL-6 and TNFα
using ELISA kits according to the manufacturer's protocol (R&D
Systems, Inc., Minneapolis, MN, USA). The results are expressed as
pg/ml of protein in each sample.
Effects of MAPK and STAT-3 inhibitors
on bacterial component-induced mechanisms
The HT-29 cells (5×105 cells/well) in 6-well plates
(90% confluence) were pretreated with LPS or CpG-dsDNA for 24 h and
then treated with DSS for 30 min, followed by treatment with MAPK
inhibitor (SB 203580; Beyotine Institute of Biotechnology) to a
final concentration of 500 nM or STAT-3 inhibitor (S31-201; Santa
Cruz Biotechnology Inc.) to a final concentration of 100 µM at 37°C
for 30 min. The MAPK inhibitor, SB 203580, is a pyridinyl imidazole
inhibitor widely used to inhibit the phosphorylation and activation
of p38 MAPK. The STAT-3 inhibitor, S31-201, inhibits the STAT-3
transcription factor by inhibiting the phosphorylation and
dimerization events necessary for activation. The cells were then
subjected to protein analysis for p38 or STAT-3. Wound repair was
assayed following treatment with DSS for 24 h.
Statistical analysis
All data are expressed as the mean ± standard
deviation. The differences among groups were analyzed using one-way
analysis of variance and the Bonferroni method was used for post
hoc tests. Statistical analysis was performed by using the SPSS
15.0 software package (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
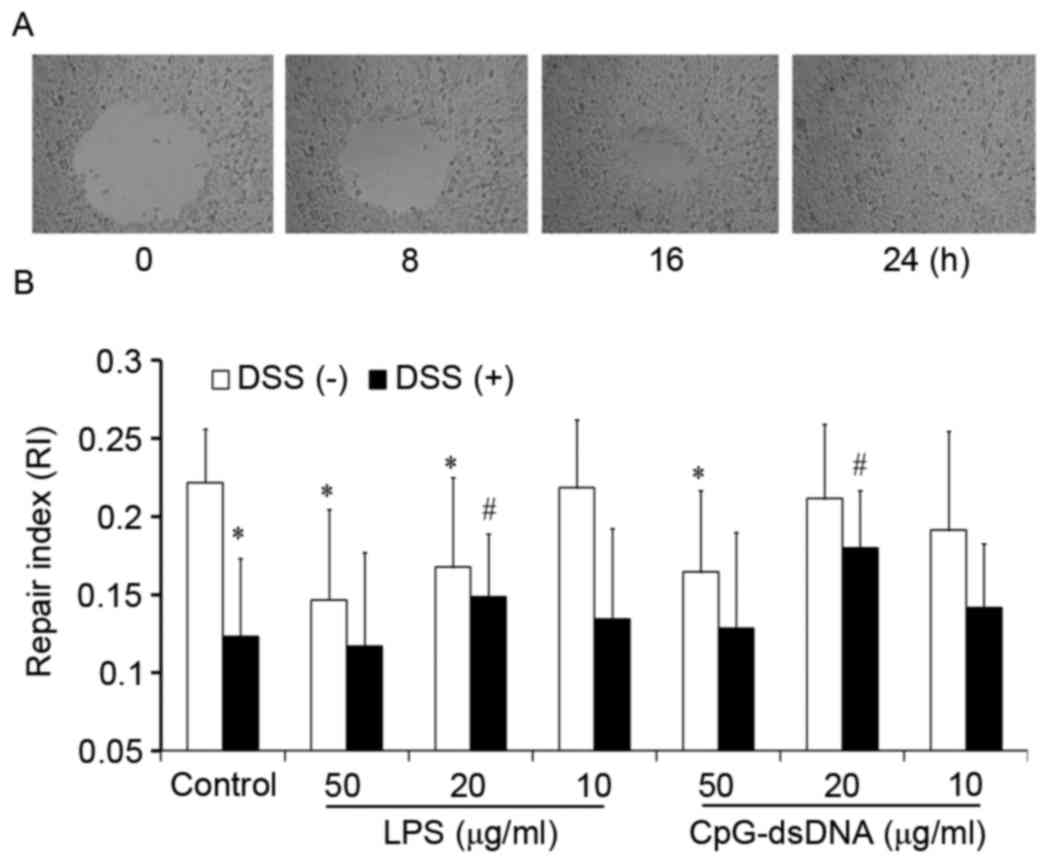
Effects of LPS and CpG-dsDNA on wound
repair of HT-29 cells
A wound was introduced in every well with a closure
area of ~2.98±0.124 mm2. Following 24 h, the remaining
wound area in the control group was decreased to zero (Fig. 1A). The repair of HT-29 cells was
inhibited by LPS at concentrations of 50 and 20 µg/ml, and
inhibited by CpG-dsDNA at the concentration of 50 µg/ml. The repair
was inhibited by DSS, and either 20 µg/ml of LPS or 20 µg/ml of
CpG-dsDNA were found to abrogate the inhibitory effects induced by
DSS (Fig. 1B).
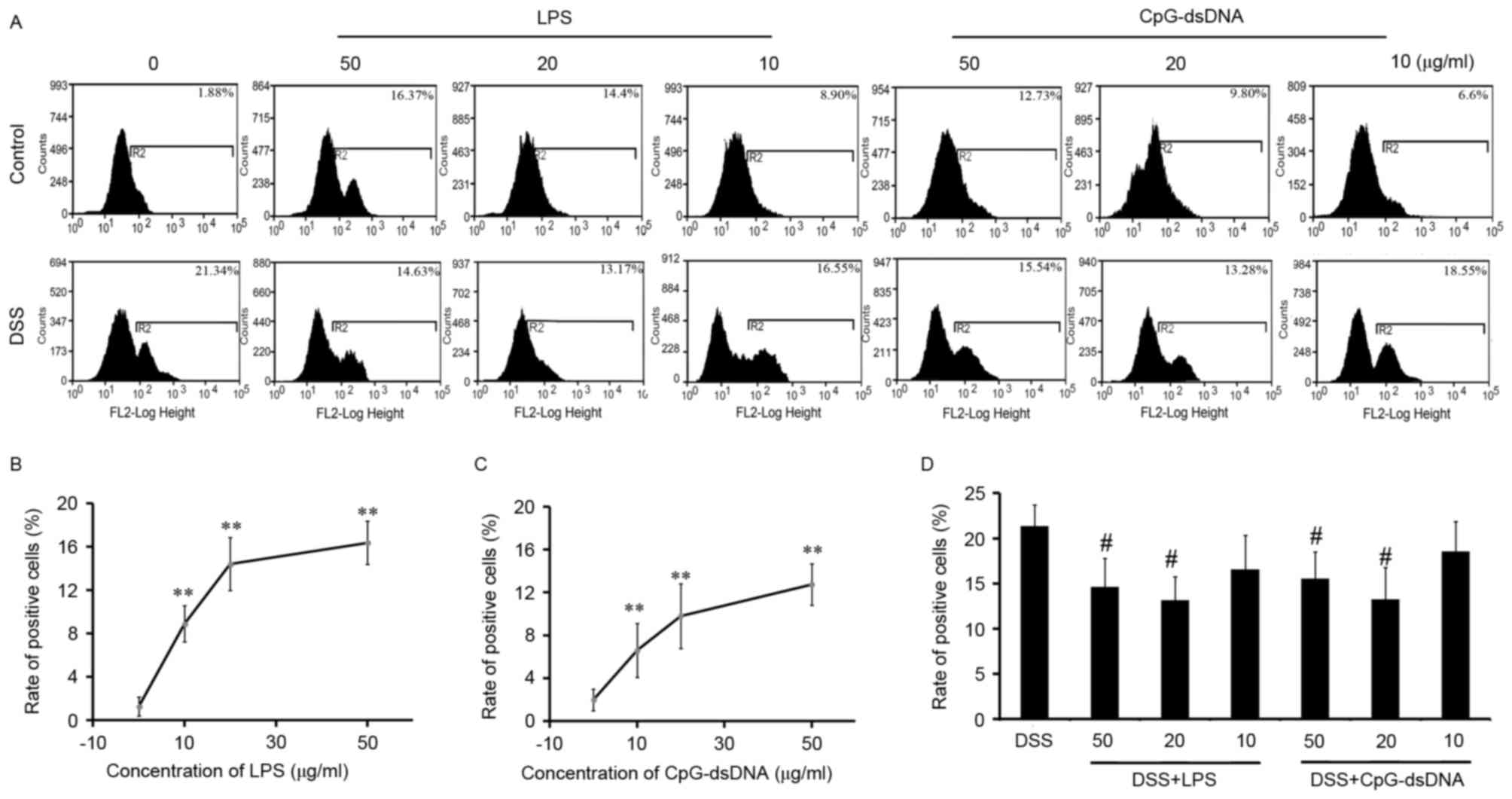
Pretreatment with LPS or CpG-dsDNA on
expression of CD40 in HT-29 cells
Treatment of the normal HT-29 cells with LPS (50, 20
and 10 µg/ml) or CpG-dsDNA (50, 20 and 10 µg/ml) produced a
significant increase in the expression of CD40 in a
concentration-dependent manner (Fig.
2A to C; P<0.01), with no statistically significant
differences between LPS and CpG-dsDNA (Fig. 2B and C). Pretreatment with LPS at
50, 20 and 10 µg/ml decreased the DSS-induced expression of CD40 by
31.44, 38.28 and 27.18%, respectively (Fig. 2A and D). Pretreatment with
CpG-dsDNA at 50, 20 and 10 µg/ml concentrations decreased the
DSS-induced expression of CD40 by 22.45, 38.77 and 13.07%,
respectively, compared with the DSS group (Fig. 2A and D). As 20 µg/ml of LPS or
CpG-dsDNA had the highest protective effects on DSS injury and the
most potent inhibition of CD40 molecules, this concentration was
selected for the further experiments.
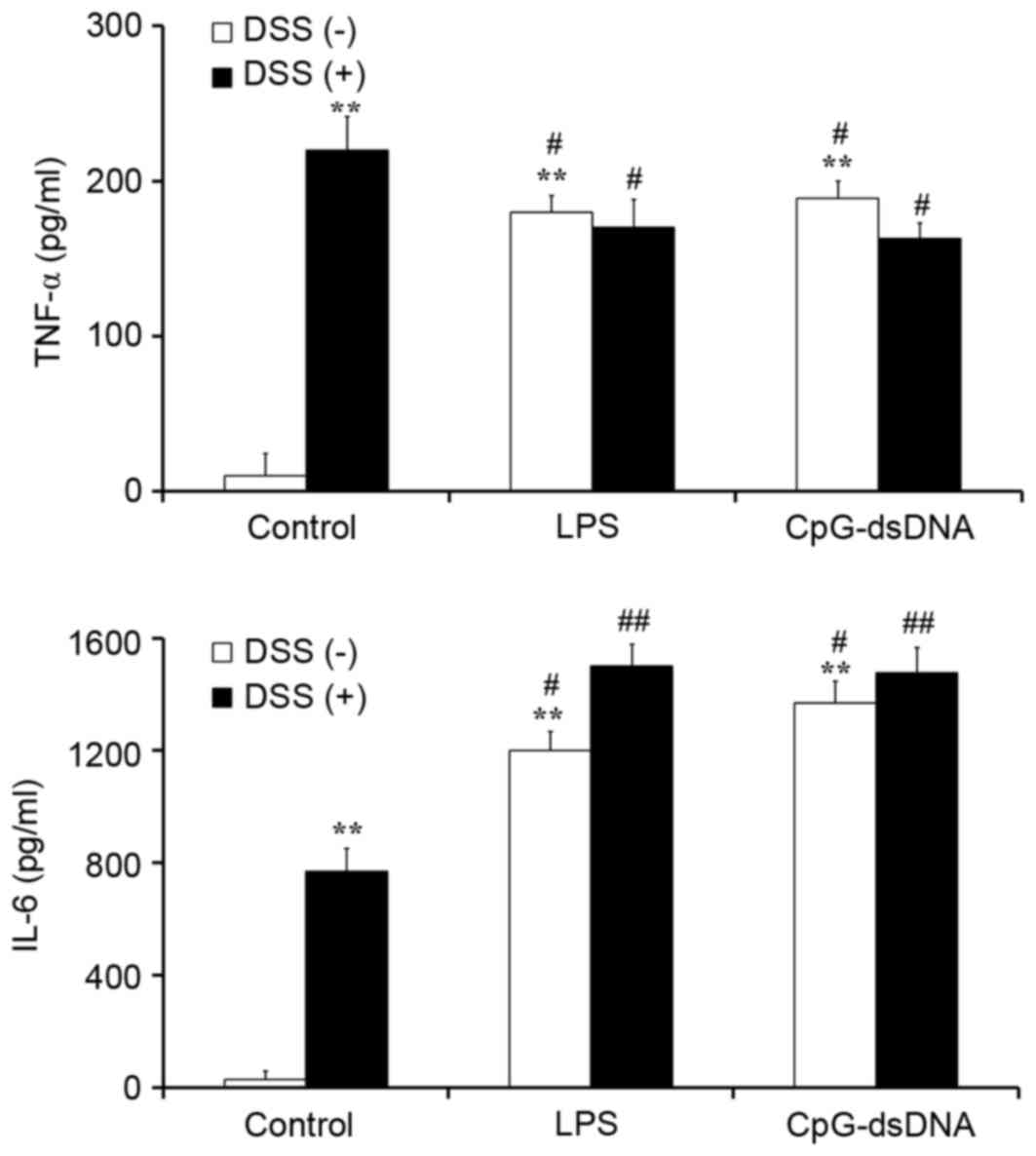
Pretreatment with CpG-dsDNA or LPS
promotes the production of IL-6 and inhibits the production of TNF
α in DSS-treated HT-29 cells
To elucidate the protective mechanism of LPS and of
CpG-dsDNA on DSS-treated HT-29 cells, ELISA was performed in a
collected culture supernatant to estimate the secretory levels of
TNF α and IL-6. The results showed that LPS (20 µg/ml) and
CpG-dsDNA (20 µg/ml) promoted the secretion of TNF α and IL-6 in
normal HT-29 cells, with no statistically significant differences
between them (Fig. 3). The
DSS-treated HT-29 cells expressed a relatively higher level of TNFα
and relatively lower level of IL-6, compared with the cells
pretreated with LPS or CpG-dsDNA (P<0.05). Pretreatment with LPS
(20 µg/ml) or CpG-dsDNA (20 µg/ml) decreased expression of the
pro-inflammatory cytokine TNFα and increased the secretion of IL-6,
compared with the DSS group (Fig.
3).
Pretreatment with LPS or CpG-dsDNA
inhibits p38 MAPK and promotes STAT-3 in DSS-treated HT-29
cells
MAPK signaling is important during inflammatory
responses. To analyze the role of MAPK in the cells, HT-29 cells
were cultured with or without DSS following pretreatment with LPS
(20 µg/ml) or CpG-dsDNA (20 µg/ml). The cells were the subjected to
mRNA analysis and protein analysis for activated p38. The data
showed that LPS and CpG-dsDNA promoted the mRNA expression of MAPK
in normal HT-29 cells (P<0.01). An increased mRNA expression of
p38 was found in DSS-treated HT-29, which was significantly
inhibited by LPS or CpG-dsDNA pretreatment (Fig. 4A; P<0.05). The protein
expression and activation of P38 exhibited the same tendancy as the
mRNA expression.
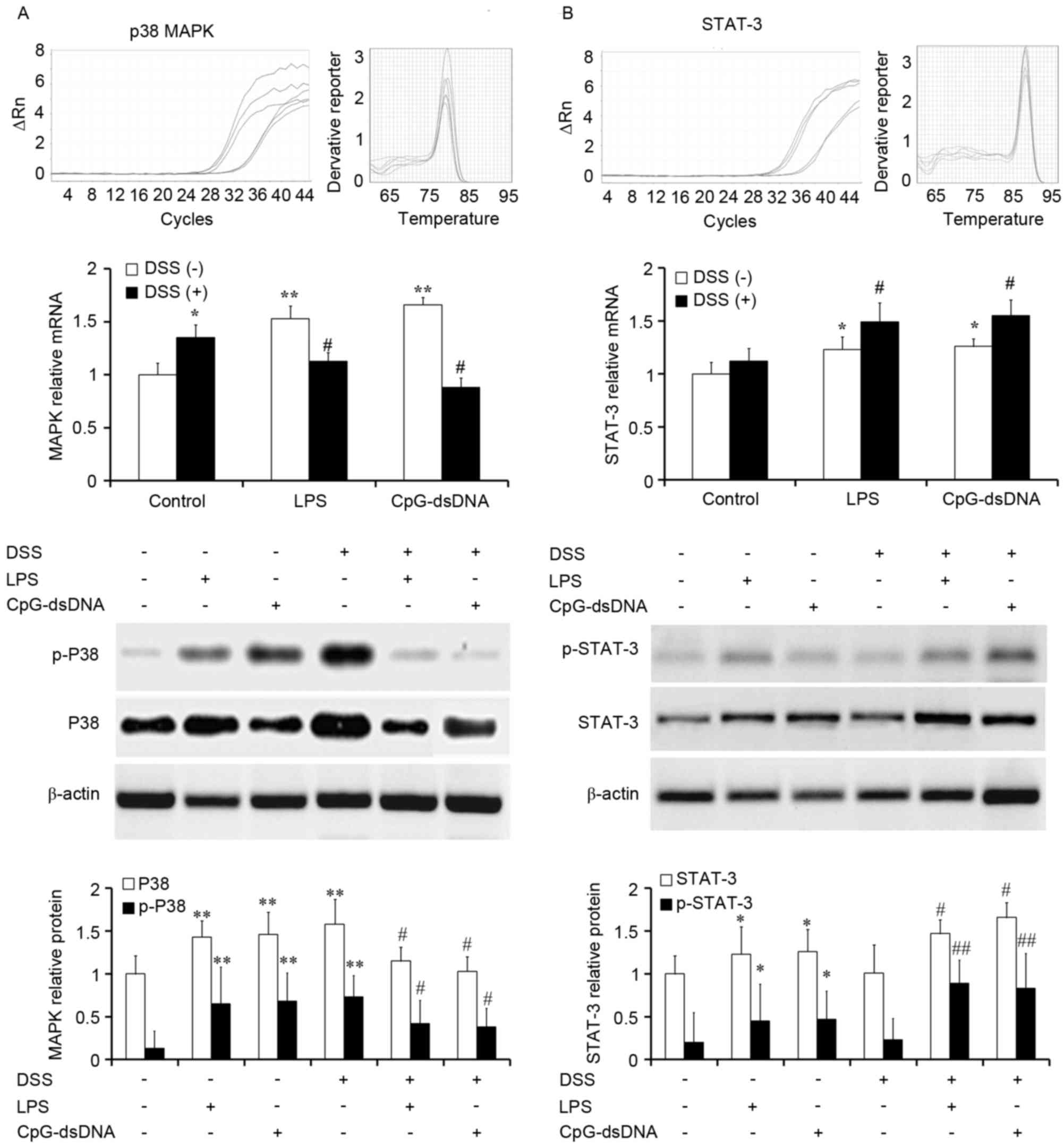 | Figure 4.Expression and activation of p38 MAPK
and STAT-3 determined using reverse transcription-quantitative
polymerase chain reaction and western blot analyses (n=3). (A) LPS
and CpG-dsDNA promoted the expression and activation of MAPK in
normal HT-29 cells. Increased expression and activation of p38 were
observed in the DSS-treated HT-29, which was significantly
inhibited by CpG-dsDNA or LPS pretreatment. (B) LPS and CpG-dsDNA
promoted the expression and activation of STAT-3 in normal HT-29
cells. However, the DSS-treated HT-29 cells pretreated with
CpG-dsDNA or LPS exhibited increased expression and activation of
STAT3, compared with the DSS group. *P<0.05 and **P<0.01, vs.
control group; #P<0.05 and ##P<0.01,
vs. DSS group. LPS, lipopolysaccharide; DSS, dextran sodium
sulfate; MAPK, mitogen-activated protein kinase; STAT-3, signal
transducer and activator of transcription-3; p-,
phosphorylated. |
The present study hypothesized that the LPS- or
CpG-dsDNA-mediated protection of epithelial cells is essentially
through the activation of STAT3. To evaluate the role of the STAT3
cascade in the LPS- or CpG-dsDNA-mediated protective mechanisms,
RT-qPCR analysis and western blot analysis were performed for
STAT3. The data showed that LPS and CpG-dsDNA promoted the mRNA
expression of STAT-3 in normal HT-29 cells (P<0.01). However,
the DSS-stimulated HT-29 cells pretreated with LPS (20 µg/ml) or
CpG-dsDNA (20 µg/ml) led to increased mRNA expression levels of
STAT3, compared with that in the DSS group (Fig. 4B; P<0.05). The protein
expression and activation of STAT-3 exhibited the same tendency as
the mRNA expression. These data suggested that the inhibition of
p38MAPK signaling and activation of STAT3 signaling may be
responsible for the protective effects.
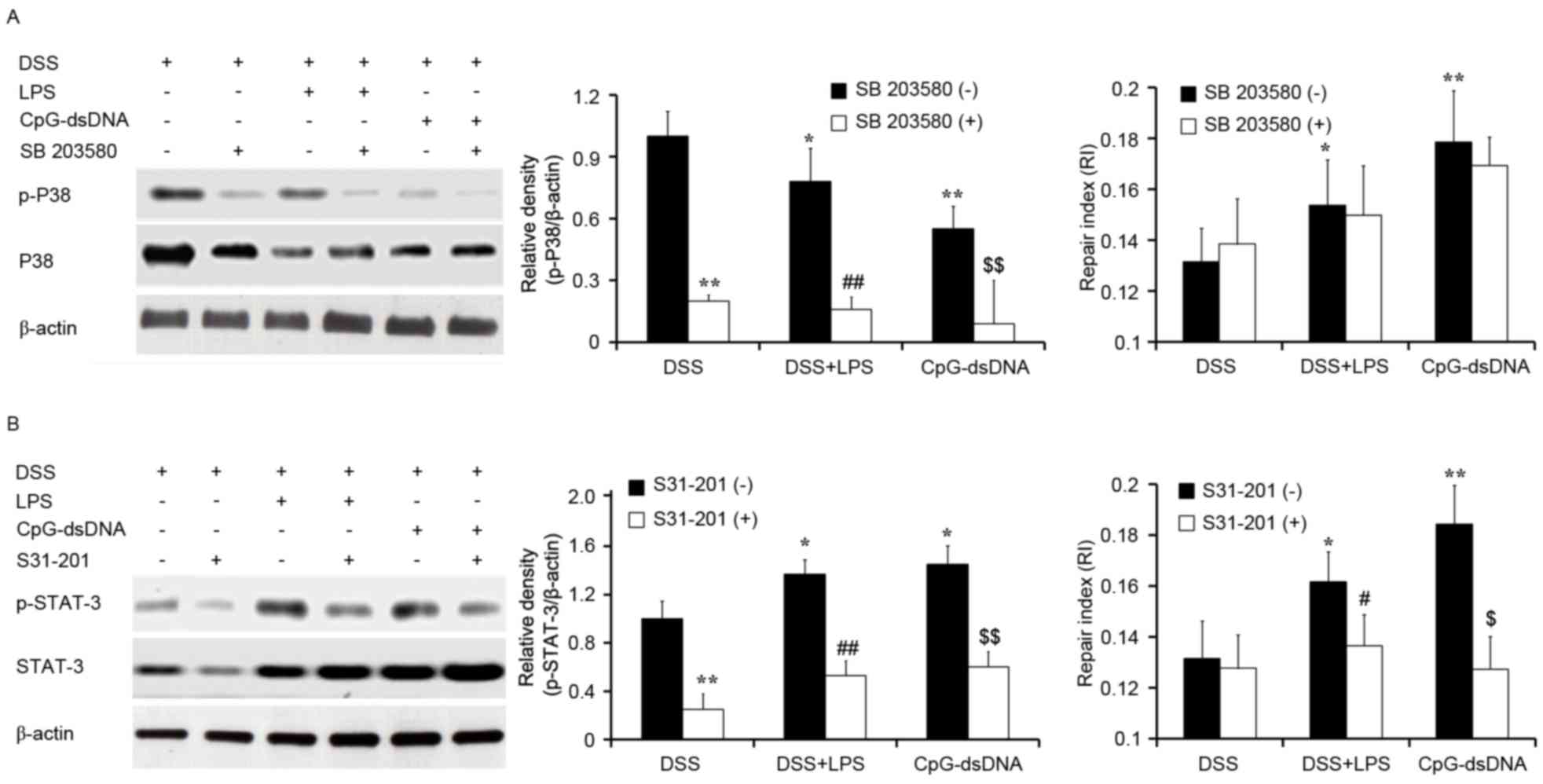
Effects of MAPK and STAT-3 inhibitors
on the protective effects of LPS or CpG-dsDNA pretreatment
In order to determine the effects of MAPK or STAT-3
inhibitors on wound repair of the HT-29 cells, the HT-29 cells were
pretreated with MAPK inhibitor (SB 203580; 500 nM) or STAT-3
inhibitor (S31-201; 100 µM) for 30 min following treatment with DSS
for 30 min. The wound repair was assayed following treatment with
DSS for 24 h. The results of western blot analysis showed that the
MAPK inhibitor inhibited the activation of MAPK, but had no effects
on wound repair (Fig. 5A). The
STAT-3 inhibitor inhibited the activation of STAT-3 and wound
repair in the DSS-treated HT-29 cells (Fig. 5B).
Discussion
Previous studies have shown that defects in the
function and integrity of the intestinal epithelium are important
in the occurrence and development of IBD (18). Wound repair is the primary
mechanism in maintaining structural and functional integrity.
Luminal microbiota may exert important effects on structural and
functional integrity, and microbes may have beneficial effects on
the epithelium or may cause adverse effects towards the epithelium
(19,20). Through the use of TLRs, which
recognize conserved microbial structures, epithelial cells are able
to sense microbes, which lead to the inducible secretion of further
mediators and corresponding T cell responses.
The upregulation of CD40 in the microcirculation of
IBD-affected mucosa is of particular interest as the CD40 pathway
is intimately involved in exaggerated inflammation. The stimulation
of CD40-bearing cells triggers multiple inflammatory signals,
resulting in leukocyte recruitment and amplification of tissue
injury (21,22). CD40 molecules stimulate the
activation of multiple signaling pathways. TNF-receptor associated
factor or Janus kinase, respectively, interact with different
structural domains in the cytoplasm of CD40 and mediate the
activation of NF-κB, STATs and P38MAPK, and DNA dependent protein
kinase (23,24). In the present study, it was
observed that LPS and CpG-dsDNA activated CD40, MAPK/TNFα and
STAT-3/IL-6, and inhibited wound repair in normal HT-29 cells. When
the cells were treated with DSS, there was an absolute upregulation
of CD40 and MAPK/TNF α and inhibiton of wound repair. However, when
the cells were pretreated with LPS or CpG-dsDNA, there was a
downregulation of CD40 and MAPK/TNF α, and an upregulation of
STAT-3/IL-6, compared with the DSS-injured cells. These results
suggested that there was a complex interaction between bacterial
and chemical factors, and that STAT-3/IL-6 mediated the promoting
wound repair induced by pretreatment of bacterial components.
In conclusion, the results of the present study
demonstrated that LPS and CpG-dsDNA can provoke preadaptation to
DSS-induced colitis. This preadaptation was accompanied by the
activation of STAT-3. Bacterial components may offer potential as a
strategy for the therapeutic prevention of IBD.
References
|
1
|
de Mattos BR, Garcia MP, Nogueira JB,
Paiatto LN, Albuquerque CG, Souza CL, Fernandes LG, Tamashiro WM
and Simioni PU: Inflammatory bowel disease: An overview of immune
mechanisms and biological treatments. Mediators Inflamm.
2015:4930122015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chassaing B, Koren O, Carvalho FA, Ley RE
and Gewirtz AT: AIEC pathobiont instigates chronic colitis in
susceptible hosts by altering microbiota composition. Gut.
63:1069–1080. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sahay B, Ge Y, Colliou N, Zadeh M, Weiner
C, Mila A, Owen JL and Mohamadzadeh M: Advancing the use of
Lactobacillus acidophilus surface layer protein A for the treatment
of intestinal disorders in humans. Gut Microbes. 6:392–397. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu J, Wang Y, Yang F, Sang L, Zhai J, Li
S, Li Y, Wang D, Lu C and Sun X: IL-33 alleviates DSS-induced
chronic colitis in C57BL/6 mice colon lamina propria by suppressing
Th17 cell response as well as Th1 cell response. Int
mmunopharmacol. 29:846–853. 2015. View Article : Google Scholar
|
|
5
|
Smyth K, Garcia K, Sun Z, Tuo W and Xiao
Z: TLR agonists are highly effective at eliciting functional memory
CTLs of effector memory phenotype in peptide immunization. Int
Immunopharmacol. 15:67–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saito K, Katakura K, Suzuki R, Suzuki T
and Ohira H: Modulating Toll-like receptor 4 signaling pathway
protects mice from experimental colitis. Fukushima J Med Sci.
59:81–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Berkowitz D, Peri R, Lavy A and Kessel A:
Increased Toll-like receptor 9 expression by B cells from
inflammatory bowel disease patients. Hum Immunol. 74:1519–1523.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koguchi Y, Buenafe AC, Thauland TJ,
Gardell JL, Bivins-Smith ER, Jacoby DB, Slifka MK and Parker DC:
Preformed CD40L is stored in Th1, Th2, Th17, and T follicular
helper cells as well as CD4+ 8-thymocytes and invariant
NKT cells but not in Treg cells. PLoS One. 7:e312962012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iezzi G, Sonderegger I, Ampenberger F,
Schmitz N, Marsland BJ and Kopf M: CD40-CD40L cross-talk integrates
strong antigenic signals and microbial stimuli to induce
development of IL-17-producing CD4+ T cells. Proc Natl
Acad Sci USA. 106:876–881. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Senhaji N, Kojok K, Darif Y, Fadainia C
and Zaid Y: The contribution of CD40/CD40L axis in inflammatory
bowel disease: An update. Front Immunol. 6:5292015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Borcherding F, Nitschke M, Hundorfean G,
Rupp J, von Smolinski D, Bieber K, van Kooten C, Lehnert H,
Fellermann K and Büning J: The CD40-CD40L pathway contributes to
the proinflammatory function of intestinal epithelial cells in
inflammatory bowel disease. Am J Pathol. 176:1816–1827. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arranz A, Reinsch C, Papadakis KA,
Dieckmann A, Rauchhaus U, Androulidaki A, Zacharioudaki V,
Margioris AN, Tsatsanis C and Panzner S: Treatment of experimental
murine colitis with CD40 antisense oligonucleotides delivered in
amphoteric liposomes. J Control Release. 165:163–172. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao D, Wagner AH, Fankhaenel S, Stojanovic
T, Schweyer S, Panzner S and Hecker M: CD40 antisense
oligonucleotide inhibition of trinitrobenzene sulphonic acid
induced rat colitis. Gut. 54:70–77. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JW, Wang P, Kattah MG, Youssef S,
Steinman L, DeFea K and Straus DS: Differential regulation of
chemokines by IL-17 in colonic epithelial cells. J Immunol.
181:6536–6545. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ng HP, Burris RL and Nagarajan S:
Attenuated atherosclerotic lesions in apoE-Fcγ-chain-deficient
hyperlipidemic mouse model is associated with inhibition of Th17
cells and promotion of regulatory T cells. J Immunol.
187:6082–6093. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tan YR, Qi MM, Qin XQ, Xiang Y, Li X, Wang
Y, Qu F, Liu HJ and Zhang JS: Wound repair and proliferation of
bronchial epithelial cells enhanced by bombesin receptor subtype 3
activation. Peptides. 27:1852–1858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frosali S, Pagliari D, Gambassi G,
Landolfi R, Pandolfi F and Cianci R: How the intricate interaction
among toll-like receptors, microbiota and intestinal immunity can
influence gastrointestinal pathology. J Immunol Res.
2015:4898212015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buttó LF, Schaubeck M and Haller D:
Mechanisms of microbe-host interaction in Crohn's disease:
Dysbiosis vs. Pathobiont selection. Front Immunol. 6:5552015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang L, Wu G, Qin X, Ma Q, Zhou Y, Liu S
and Tan Y: Expression of nodal on bronchial epithelial cells
influenced by lung microbes through DNA methylation modulates the
differentiation of T-Helper cells. Cell Physiol Biochem.
37:2012–2022. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Greene JA, Portillo JA, Corcino Y Lopez
and Subauste CS: CD40-TRAF signaling upregulates CX3CL1 and TNF-α
in human aortic endothelial cells but not in retinal endothelial
cells. PLoS One. 10:e01441332015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang W, Bai L, Qiao H, Lu Y, Yang L, Zhang
J, Lin R, Ren F, Zhang J and Ji M: The protective effect of
fenofibrate against TNF-α-induced CD40 expression through
SIRT1-mediated deacetylation of NF-κB in endothelial cells.
Inflammation. 37:177–185. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seibold K and Ehrenschwender M: p62
regulates CD40-mediated NFκB activation in macrophages through
interaction with TRAF6. Biochem Biophys Res Commun. 464:330–335.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chakraborty S, Srivastava A, Jha MK, Nair
A, Pandey SP, Srivastava N, Kumari S, Singh S, Krishnasastry MV and
Saha B: Inhibition of CD40-induced N-Ras activation reduces
leishmania major infection. J Immunol. 194:3852–3860. 2015.
View Article : Google Scholar : PubMed/NCBI
|