Introduction
Systemic lupus erythematosus (SLE) is a complex
systemic autoimmune disease, characterized as a loss of tolerance
to nuclear antigens, the deposition of pathogenic autoantibodies
and the formation of immune complexes leading to inflammation in
multiple organs (1,2). Dysregulated T cells and their
associated mechanisms are important in the pathogenesis of this
complicated disease. Epigenetic modifications including DNA
methylation, histone tail modifications and microRNAs may
additionally serve roles in SLE.
The ubiquitin-proteasome and deubiquitination
systems are important cellular mechanisms of protein degradation
and stabilization, which may influence gene expression and alter
cellular functions without modifying the genomic sequence.
Understanding the molecular mechanisms that are involved in the
pathophysiology of autoimmune diseases is essential for the
introduction of effective, target-directed and accepted therapies
(3,4).
One notable feature in SLE is the continuous
activation of the type I interferon (IFN) system, shared by the
majority of SLE patients (5–7). The
vascular damage, endothelial progenitor cell misbalance and induced
expression of broad signature gene transcripts that reflect
induction are primarily due to the central involvement of IFNα
(8,9). The type I interferon receptor complex
consisting of the human IFN α-2 receptor (IFNAR)1 and IFNAR2
subunits, and cellular responses to IFNα, require adequate
expression levels of IFNAR1 (10).
It has been reported that IFNAR1 may be ubiquitinated by the
Skp1-Cullin1-HOS-Roc1 ubiquitin ligase in vitro (11), however, whether the protein level
of IFNAR1 is regulated by the deubiquitination system and whether
these deubiquitinating enzymes exhibit a role in the clinical
outcomes of SLE patients, remains to be elucidated. The
deubiquitinases contain two major groups: the ubiquitin C-terminal
hydrolase families and the ubiquitin-specific protease (USP)
families. Of the USP families, USP7 is an evolutionarily conserved
protease that was first reported in 1997 (12) and was revealed to possess various
substrates, including Ci/Gli (13), phosphatase and tensin homolog
(14), forkhead box protein O4
(15), histone H2B (16) and tumor protein p53 (17), indicating that USP7 exhibits a role
in multiple cellular processes. However, the expression pattern and
function of USP7 in SLE progression remains to be elucidated.
The present study demonstrated that IFNAR1 acted as
a substrate for USP7, and that USP7 functioned to stabilize IFNAR1,
which was responsible for greater disease activity in SLE.
Materials and methods
Patient samples
A total of 210 patients with SLE were recruited from
the Chinese Medicine Hospital of Zhejiang (Hangzhou, China) between
January 2010 and December 2014; all SLE patients fulfilled the
American College of Rheumatology criteria for the disease (18), and gave written informed consent. A
further 210 control samples were recruited from the outpatient
clinics of the Chinese Medicine Hospital of Zhejiang between
January 2010 and December 2014, including those diagnosed as
normally healthy or with an unrelated condition. Written informed
consent was also obtained from control patients. Patients with
other autoimmune diseases including celiac disease, autoimmune
hepatitis, sarcoidosis, or autoimmune thyroid disease were excluded
from the present study. All procedures were approved by the
institutional review board of Jiangsu University. Peripheral blood
(5 ml) samples were drawn from healthy donors and SLE patients. The
clinical disease activity was measured and assessed according to
the SLE disease activity index (DAI) 2000; SLEDAI scores ≥10 were
defined as active SLE, while SLEDAI scores <10 were defined as
stable disease (19).
Samples of peripheral blood mononuclear cells
(PBMCs) for isolating T cells were purified using the Rosette Sep T
cell isolation kit by negative selection (cat no. HY2015; HaoYang
Biosciences, Tianjin, China). Serum complement 3 and serum
complement 4, double stranded (ds)DNA and anti-nuclear antibodies
(ANA) were measured in blood samples in the Department of
Laboratory Medicine using a EUROIMMUN ANA profile kit (cat no. DL
1590) and the Sprinter XL Immunofluorescence system (both from
EUROIMMUN AG, Luebeck, Germany), according to the manufacturer's
protocol, at the Chinese Medicine Hospital of Zhejiang.
Reagents and antibodies
The short interfering (si)RNA transfection was
performed using Lipofectamine RNAiMAX (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and cells were transfected at
70% confluence, according to the manufacturer's protocol. The 2
USP7 siRNA oligonucleotide sequences that were used were as
follows: siUSP7 1,
5′-CCGGTGTATCTATTGACTGCCCTTTCTCGAGAAAGGGCAGTCAATAGATACATTTTT-3′;
siUSP7 2,
5′-CCGGCCTGGATTTGTGGTTACGTTACTCGAGTAACGTAACCACAAATCCAGGTTTTT-3′.
USP7-targeting siRNA and non-targeting siRNA
(5′-CCGGUUCUCCGAACGUCACGUTTTTTTTT-3′) were obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Antibodies used were as follows: anti-USP7 antibody
(cat no. 4833; 1:1,000; Cell Signaling Technology, Danvers, MA,
USA), anti-IFNAR1 (cat no. SAB1406003-50UG; 1:1,000; Sigma-Aldrich;
Merck KGaA) and anti-β-actin (cat no. 612656; 1:2,000; BD
Biosciences, Franklin Lakes, NJ, USA). Horseradish
peroxidase-conjugated goat anti-mouse immunoglobulin (Ig)G (cat no.
sc-2005; 1:3,000) and goat and anti-rabbit IgG (cat no. sc-2004;
1:3,000) secondary antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA).
Cell culture
HEK-293T cells was purchased from American Type
Culture Collection (Mannasas, VA, USA) and were cultured in
Dulbecco's modified Eagle's medium (Invitrogen; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum
(Hyclone; GE Healthcare Life Sciences, Logan, UT, USA), 100 U/ml of
penicillin, and 100 U/ml of streptomycin (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells were maintained at 37°C in a humidified 5%
CO2 atmosphere.
RNA isolation and reverse
transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol® (Invitrogen;
Thermo Fisher Scientific, Inc.). cDNA was synthesized from 1 µg of
total RNA using reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.) and the cDNA (2 µg) was amplified using the
TransStart Top Green qPCR SuperMix kit (cat no. AQ132-23; TransGen,
Beijing, China) on an ABI 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). GAPDH was used as an
internal normalization control. Thermocycling conditions were as
follows: Initial denaturation at 95°C for 5 min, followed by 40
cycles of denaturation at 95°C for 10 sec, and annealing and
extension at 60°C for 30 sec. The primers used were as follows:
GAPDH, forward GAG AAG TAT GAC AAC AGC CTC-3′, reverse
5′-ATGGACTGTGGTCATGAGTC-3′; IFNAR1, forward GAC TCA TTT ACA CCA TTT
CGC A-3′, reverse 5′-TCAATCCTTTCTTCTACACCTG-3′; and USP7 forward
ATT CCT AAC ATT GCC ACC AG-3′ and reverse
5′-ATTTACACCATTTGCCATCC-3′. Relative gene expression was calculated
according to the 2−∆∆Cq method (20) and normalized to GAPDH. All
experiments were performed at least three times.
Co-immunoprecipitation (co-IP)
assay
Cells were lysed using cold lysis buffer (50 mM
Tris-Cl, pH 7.4; 1% NP-40; 150 mM NaCl; 1 mM EDTA; and, 0.5% sodium
deoxycholate) and a protease inhibitor cocktail (BD Biosciences)
was used to protect cells from degradation. The supernatants of the
lysates were incubated with primary antibodies against USP7 and
IFNAR1. A total of 2 µg normal rabbit and mouse immunoglobin IgG
(cat nos. M8645 and G7402; Sigma-Aldrich; Merck KGaA) were used as
negative control and Pierce™ Protein A/G Magnetic Beads (cat no.
88802; Pierce; Thermo Fisher Scientific, Inc.) were added to the
immune complexes and incubated for 2 h at 4°C. The immune complexes
were washed 5 times, subjected to SDS-PAGE and detected by western
blot analysis.
Western blot analysis
Protein extracts were lysed with a
radioimmunoprecipitation assay buffer (Sigma-Aldrich; Merck KGaA)
containing phenylmethane sulfonyl fluoride and a protease inhibitor
cocktail for 30 min at 4°C, and following centrifugation at 12,000
× g for 15 min at 4°C, the supernatants were collected. Equal
amounts of extracted protein samples (25 µg) were resolved by 10%
SDS-PAGE and transferred onto nitrocellulose membranes, followed by
blocking with 5% non-fat milk for 30 min at room temperature and
incubation at 4°C overnight with anti-USP7, anti-IFNAR1 and
anti-β-actin primary antibodies. Membranes were then incubated with
horseradish peroxidase-conjugated secondary antibodies for 2 h at
room temperature. Protein bands were visualized using an enhanced
chemiluminescence assay system (Sigma Aldrich; Merck KGaA). The
experiments were repeated at least three times.
Glutathione S-transferase (GST) pull
down analysis
To detect in vitro binding between USP7 and
IFNAR1, the GST fusion construct GST-USP7 was purified from BL21
Escherichia coli cells (TransGen), as previously described
(21). The in vitro
transcription and translation of FLAG-tagged IFNAR1 was obtained
from the rabbit reticulocyte lysate (TNT systems, Promega
Corporation, Madison, WI, USA) and pulled down with
glutathione-Sepharose beads (GE Healthcare Life Sciences, Little
Chalfont, UK), according to the manufacturer's protocol.
Statistical analysis
Statistical analysis was performed using SPSS
software version 17.0 (SPSS, Inc., Chicago, IL, USA). All data were
presented as the mean ± standard deviation, unless otherwise
stated, of one representative of three experiments. Spearman's rank
correlation was used to measure correlations between patient
variables and SLE presence or activity. A paired Student's t-test
was used to perform the analysis. P<0.05 was considered to
indicate a statistically significant difference.
Results
The clinical characteristics in
patients with SLE and healthy controls
As demonstrated in Table I, no significant differences were
observed in age or sex distribution between the SLE patients and
healthy controls. The 210 SLE patients were positive for ANA,
whereas the controls were negative; the proportion of lymphocytes
was significantly lower in patients compared with the control,
while the difference in the average number of total white blood
cells was not deemed significant. As mentioned, an SLEDAI score ≥10
was considered as active SLE and there were 103 (49.05%) active
patients and 107 (50.95%) stable patients. As demonstrated in
Table II, no significant
differences were observed in age or sex distribution between these
two groups. The differences in SLEDAI score, dsDNA positivity and
levels of complement 3 and 4 were significant between the two
groups (P<0.05).
 | Table I.Clinicopathological characteristics of
the SLE samples and healthy controls. |
Table I.
Clinicopathological characteristics of
the SLE samples and healthy controls.
| Characteristic | SLE | Healthy controls |
|---|
| Number | 210 | 210 |
| Age, years | 35.8±13.5 | 35.2±13.8 |
| Sex |
| Female
(%) | 202 (96.2%) | 203 (96.67%) |
| Male
(%) | 8 (3.8%) | 7 (3.33%) |
| SLEDAI score | 8.2±5.7 | – |
| WBCs,
×109/l | 5.38±2.07 | 5.82±2.44 |
| Lymphocytes, % |
14.54±9.73a | 20.56±5.23 |
| ANA+ (%) | 210
(100%)a | 0 |
| Serum complement
3 | 0.81±0.42 | – |
| Serum complement
4 | 0.22±0.18 | – |
| Table II.Clinicopathological characteristics of
the active SLE and stable SLE samples. |
Table II.
Clinicopathological characteristics of
the active SLE and stable SLE samples.
| Characteristic | Active SLE | Stable SLE |
|---|
| Number | 103 | 107 |
| Age, years | 35.3±14.2 | 34.9±14.9 |
| Sex |
|
|
| Female
(%) | 100 (97.09%) | 102 (95.33%) |
| Male
(%) | 3 (2.91%) | 5 (4.67%) |
| SLEDAI score | 14.2±3.1a | 4.9±2.2 |
| dsDNA + (%) | 83
(80.59%)a | 21 (19.63%) |
| ANA+ (%) | 103 (100%) | 107 (100%) |
| Serum complement
3 |
0.51±0.22a | 0.82±0.28 |
| Serum complement
4 |
0.19±0.18a | 0.27±0.19 |
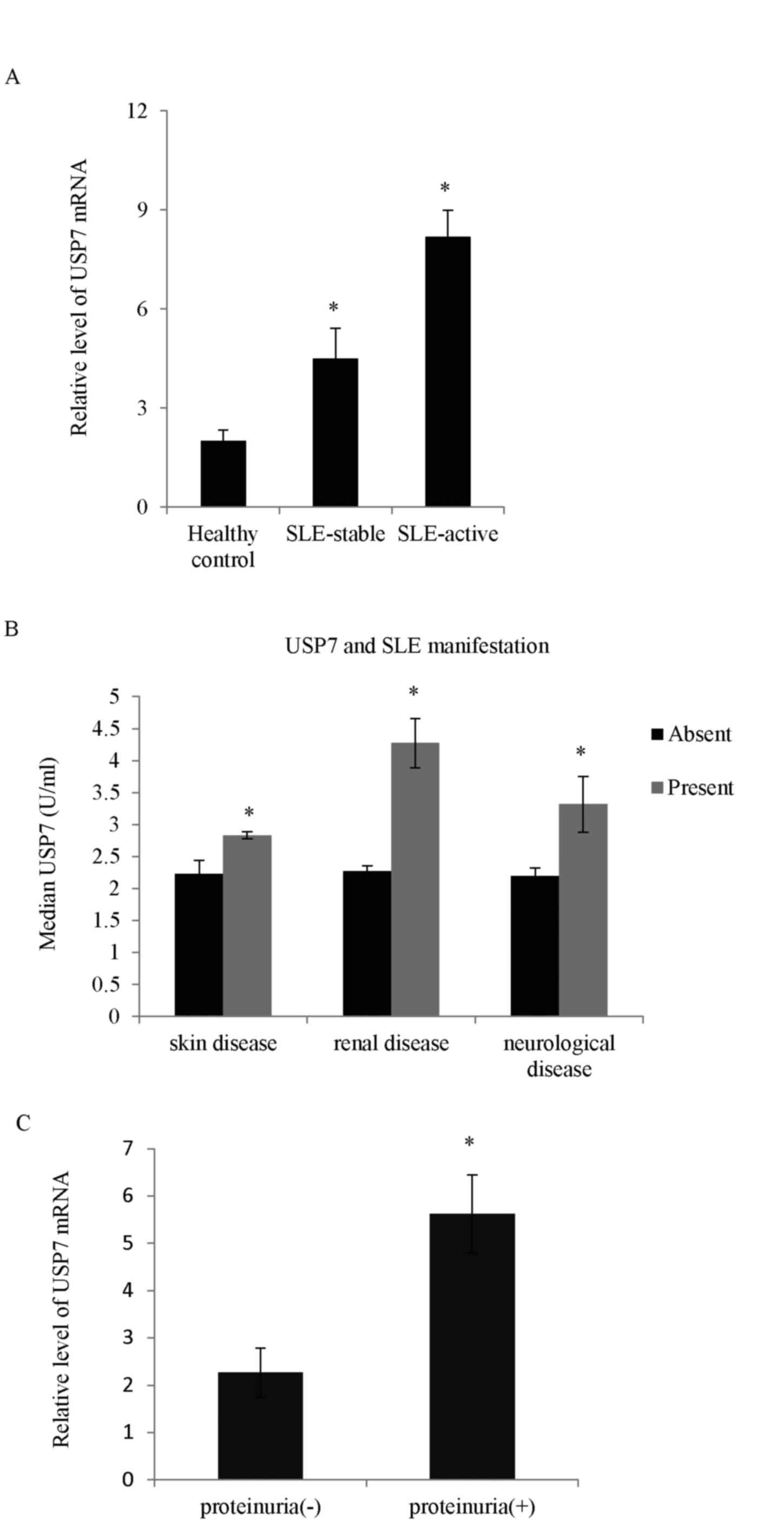
Higher USP7 expression is observed in
patients with SLE
The deubiquitinases take part in numerous cellular
processes, but very little is known concerning the role of USPs in
immune cells and specifically in T cells. In the present study, the
mRNA expression level of different USP proteins between SLE and the
healthy controls was screened. The expression levels of three USP
proteins (USP7, USP10 and USP21) were higher in SLE patients
compared with controls (data not shown). Among the USP proteins,
the expression of USP7 in samples obtained from the 210 SLE
patients was the most significantly upregulated compared with the
normal controls (Fig. 1A). The
role of USP7 in SLE was explored and analysis performed to measure
whether there was any correlation between USP7 levels and the
clinical features of SLE. As demonstrated, the high USP7 expression
correlated positively with SLE cutaneous manifestations including
the presence of skin, renal and neurological diseases (Fig. 1B). SLE patients with concurrent
proteinuria had higher USP7 levels compared with those without
proteinuria (Fig. 1C).
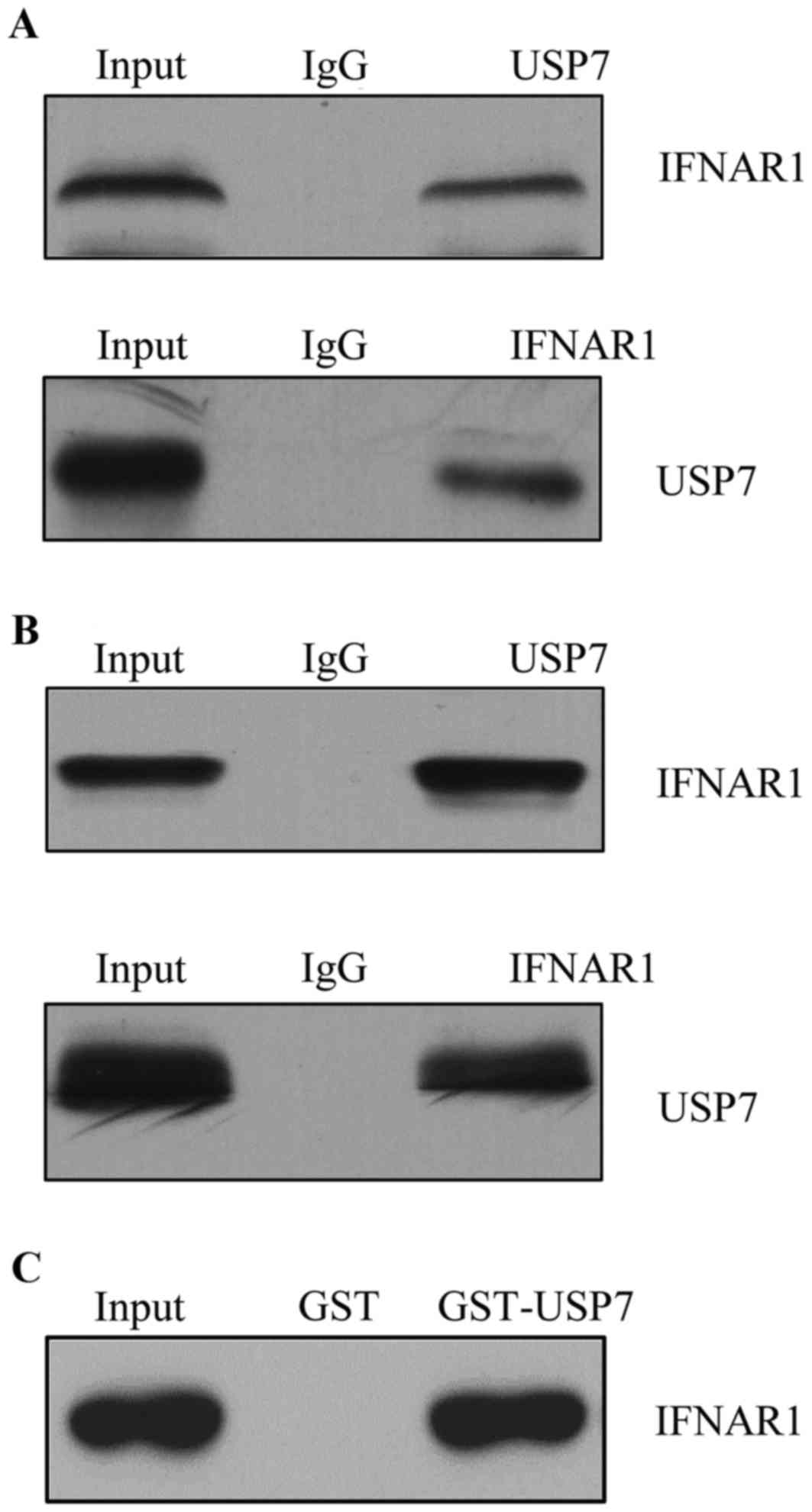
IFNAR1 identifies as a
USP7-interacting protein
One of the biggest challenges in studying USP
families is to identify their substrate and to correlate their
dysregulation with pathogenesis. Affinity purification and mass
spectrometry were used to detect the USP7 association proteins
in vivo (data not shown). IFNAR1 was identified as a
USP7-interacting protein. To further confirm the interaction
between USP7 and IFNAR1 in vivo, a co-IP assay was performed
in T cells and total protein extracted, IP with USP7 antibodies
followed by immunoblotting (IB) with the antibodies against IFNAR1
indicated that USP7 co-immunoprecipitated with IFNAR1; normal
rabbit IgG was used as a negative control (Fig. 2A). Reciprocally, IP was performed
with anti-IFNAR1 followed by IB with anti-USP7 (Fig. 2A bottom panel). To further support
the in vivo interaction between USP7 and IFNAR1, endogenous
proteins from HEK-293T cells were used to confirm the interaction
(Fig. 2B).
Other USP proteins including USP10 and USP21 were
additionally detected; neither had any interaction with IFNAR1, and
data was not shown. To further confirm the interaction between
IFNAR1 and USP7 in vitro, GST pull down assay was performed
and incubation of GST-fused USP7 with in vitro
transcribed/translated IFNAR1 revealed that USP7 interacted with
IFNAR1 directly (Fig. 2C).
USP7 inhibits IFNAR1 ubiquitination
and stabilizes IFNAR1 in vivo
In SLE patients, it was hypothesized that USP7
regulated IFNAR1 expression in T cells and higher USP7 expression
contributed to the observed elevated IFNAR1 expression. Therefore,
human primary T cells were transfected with USP7 siRNAs, control
siRNAs, USP7 overexpression lentivirus or vector lentivirus. The
successful knockdown efficiency (Fig.
3A) and ectopic expression of USP7 (Fig. 3B) in primary T cells was verified
by RT-qPCR and western blot analysis, which demonstrated that the
knockdown of USP7 following transfection resulted in a significant
decrease in USP7 expression, while the overexpression of USP7
lentivirus resulted in a clear increase in USP7 expression.
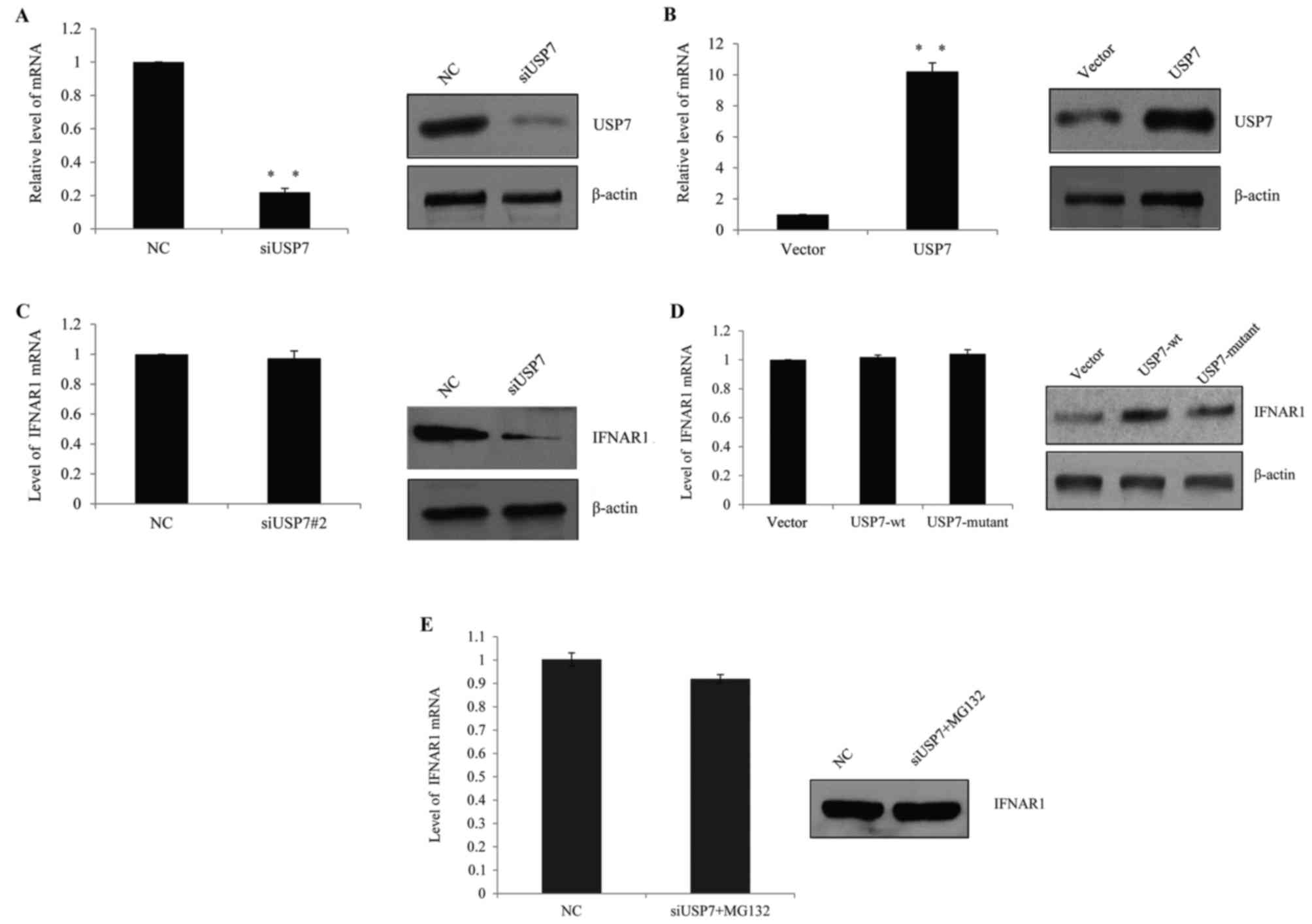 | Figure 3.USP7 inhibited IFNAR1 ubiquitination
and stabilized IFNAR1 in vivo. (A) The knockdown efficiency
of USP7 was confirmed by RT-qPCR (left panel) and western blotting
(right panel). Human primary T cells were transfected with control
siRNA, siUSP7#1 or siUSP7#2. **P<0.01, as indicated. (B) The
overexpression efficiency of USP7 was confirmed by RT-qPCR (left
panel) and western blotting (right panel). Human primary T cells
were transfected with vector, or USP7 overexpression lentivirus.
**P<0.01, as indicated. (C) T cells were transfected with
control siRNA or siUSP7 and the mRNA and protein level of IFNAR1
was detected. (D) T cells were transfected with vector, wt-USP7 or
USP7 mutant lentivirus, mRNA and the protein level of IFNAR1 was
detected. (E) Following USP7 knockdown in T cells, cells were
incubated with the proteasome-specific inhibitor MG132 prior to
harvesting, and RT-qPCR and western blot analysis were used to
measure IFNAR1 expression. USP7, ubiquitin-specific-processing
protease 7; IFNAR1, human interferon α-2 receptor; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; si, short
interfering; wt, wild type; NC, negative control. |
Following USP7 knock down, the expression of IFNAR1
was measured and although the IFNAR1 mRNA level was not changed
(Fig. 3C, left), the IFNAR1
protein level was markedly reduced (Fig. 3C, right). Consistently, when
wild-type (wt) USP7 lentivirus or catalytically inactive USP7
(USP7/C223A) mutant lentivirus was transfected into the cells, the
protein level of IFNAR1 increased in wt-USP7 transfected groups,
but not in the catalytically inactive USP7 (USP7/C223A) mutant
groups (Fig. 3D); the two groups
demonstrated no remarkable change in the mRNA level. This indicated
that USP7 may influence IFNAR1 through post-transcription
modification.
To determine whether the effect of USP7 was
dependent on IFNAR1 protein deubiquitination, USP7 was knocked down
by siRNA and cells were harvested following MG132 (a
proteasome-specific inhibitor) treatment; it was demonstrated that
MG132 was successful in saving IFNAR1 protein from degradation in
USP7 knockdown groups compared with the control groups (Fig. 3E). Based on the above, it was
hypothesized that USP7 stabilized IFNAR1, as it protected the
protein from ubiquitination.
Effects of USP7 on activation of the
IFNα pathway
Whether sustained overexpression of USP7 was able to
induce the activation of the type I IFN pathway in SLE patients was
explored. The activation of STAT proteins was a response to type I
IFN, since the IFN-stimulated transcription factor 3 complex
contained three core subunits that activated STAT-1 and STAT-2 in
addition to interferon regulatory factor (IRF)-9, the function of
which is to initiate transcription of IFN-inducible genes. It was
next considered whether USP7 regulated downstream target
transactivation following stimulation with type I IFN. As
demonstrated from mRNA and protein levels in Fig. 4A and B, normal PBMCs were
transfected and stimulated with type I IFN, and knockdown of USP7
consistently reduced the expression of STAT-1 and STAT-2.
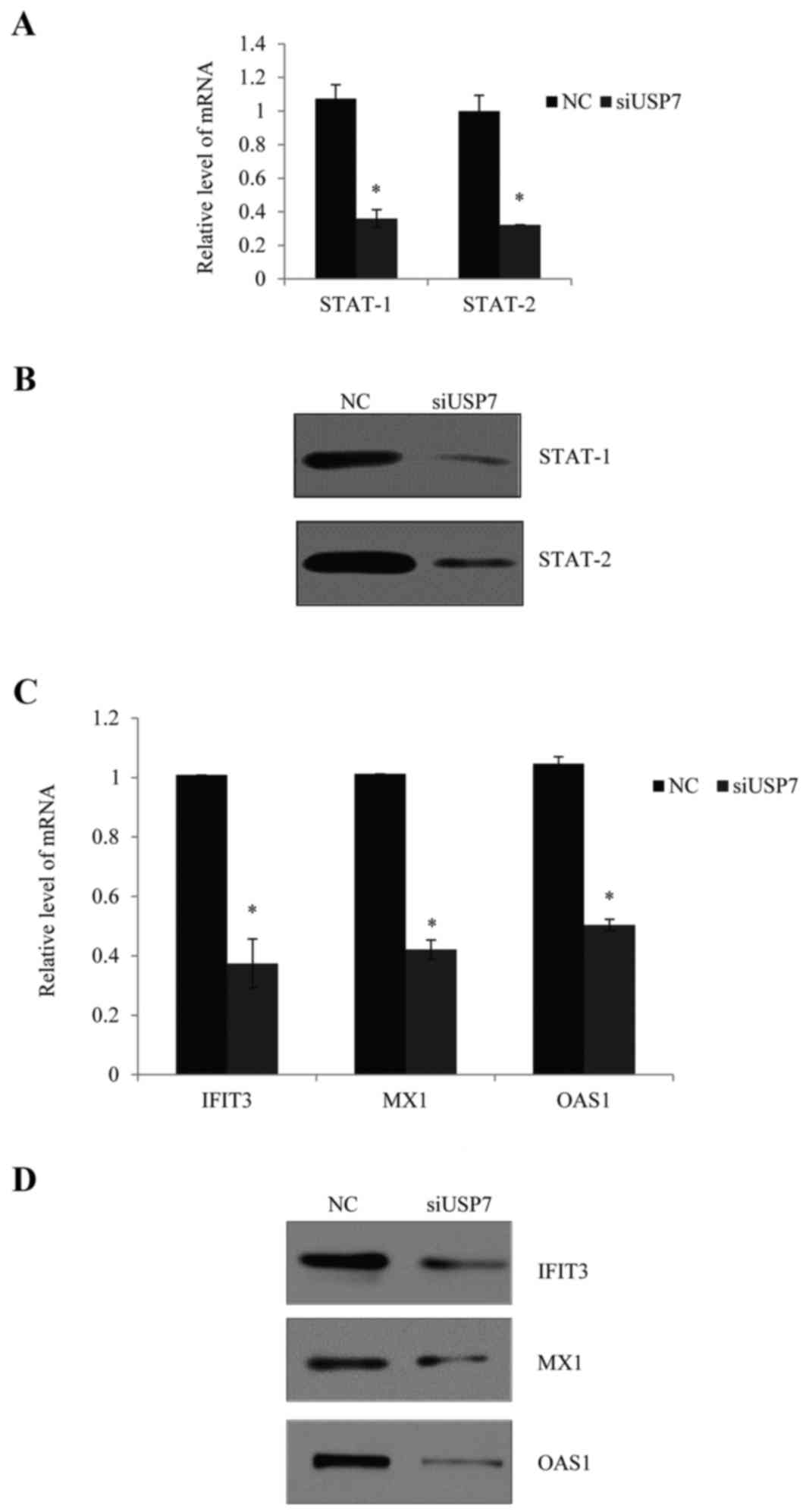 | Figure 4.Effects of USP7 on the activation of
IFNα pathway. (A) PBMCs were transfected with control siRNA and
siUSP7, followed by incubation with type I IFN for 6 h. The mRNA
levels of STAT-1 and STAT-2 were measured, GAPDH was used as an
internal normalization control. Error bars represent standard error
of the mean. *P<0.05, as indicated. (B) Following the above
treatment, the protein levels of STAT-1, STAT-2 and IRF-9 were
measured by western blotting and β-actin was used as an internal
control. Following the above treatment, the (C) mRNA level and (D)
protein level of three IFN-inducible genes, IFIT3, MX1 and OAS1
were quantified. Values are the mean + standard error. USP7,
ubiquitin-specific-processing protease 7; IFN, interferon; PBMCs,
peripheral blood mononuclear cells; si, short interfering; STAT,
signal transducer and activator of transcription; IRF, interferon
regulatory factor; IFIT3, IFN-induced protein with
tetratricopeptide repeats 3; MX1, myxovirus resistance 1; OAS1,
2′,5′-oligoadenylate synthetase 1; NC, negative control. |
Furthermore, knockdown of USP7 notably reduced the
mRNA (Fig. 4C) and protein
expression levels (Fig. 4D) of
selected IFN-inducible genes, including IFN-induced protein with
tetratricopeptide repeats 3 (IFIT3), myxovirus resistance 1 (MX1)
and 2′,5′-oligoadenylate synthetase 1 (OAS1).
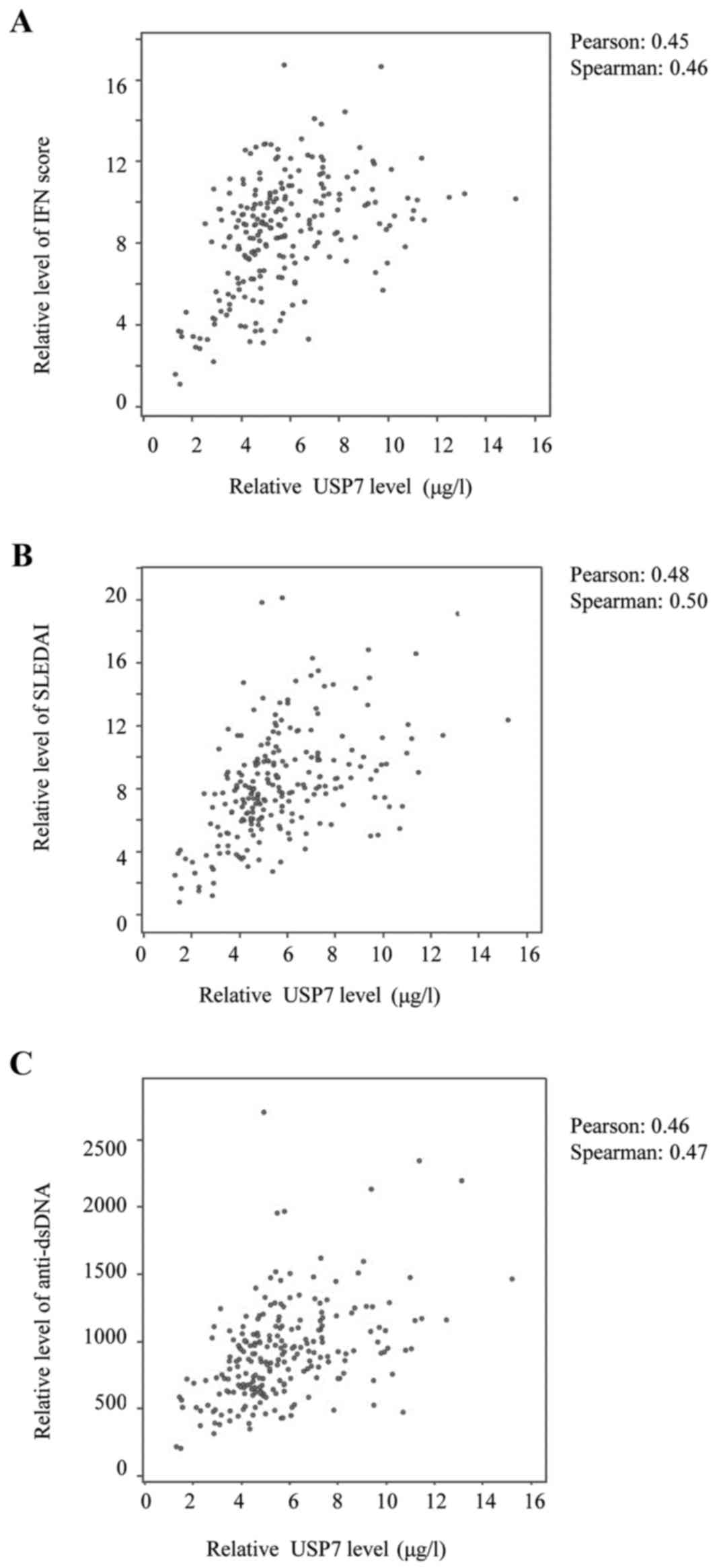
Correlation between USP7 expression
and SLE disease activity
It has been reported by several groups (22,5)
that type I IFN exhibits a key etiological role in SLE since, by
stabilizing IFNAR1, USP7 may reflect defects in positive regulation
of the immune response. The associations were therefore analyzed
and, as expected, a positive correlation was identified between
USP7 levels and IFN scores (Fig.
5A).
A direct positive correlation was observed between
USP7 levels and the SLEDAI scores (Fig. 5B), and between the USP7 levels and
the anti-dsDNA levels (Fig. 5C).
Furthermore, the IFN level also correlated positively with SLEDAI
scores and the anti-dsDNA levels (data not shown); the study
conducted by Dall'era et al (23) reported a similar tendency. It was
concluded that USP7 expression levels correlate positively with SLE
disease activity via stabilization of IFNAR1.
Discussion
Increasing evidence has indicated that IFNα is
associated with the progression of several diseases and may serve
as a target for therapy. The role of the USP family in autoimmunity
is only just beginning to be explored.
The results of the present study established that
USP7 is upregulated in patients with SLE, and that greater
expression of USP7 was associated with skin, renal and neurological
diseases. SLE patients with concurrent proteinuria had higher USP7
levels than those without, which indicated that greater USP7 levels
may result in organ damage.
Furthermore, investigation provided insight into the
mechanism of the regulation by USP7; bioinformatics tools were used
to search for its potential association with the key components
involved in SLE (5), and this led
to the identification of IFNAR1 as a potential USP7 interaction
protein. As it was hypothesized that USP7 interacted with IFNAR1
in vivo and in vitro, co-IP and GST-pull down assays
were used to confirm that USP7 disassembled IFNAR1-dependent
poly-ubiquitin chains and stabilized IFNAR1 in vivo. As the
type I IFN pathway has been reported as a significant contributor
to the pathogenesis of SLE (5),
the activation effects of USP7 on the IFNα pathway were
investigated. When USP7 was knocked down, the signaling downstream
of IFN, including the expression of STAT-1, STAT-2 and its
predicted IFN-inducible genes (IFIT3, MX1 and OAS1), were
downregulated. For the first time, to the best of the authors'
knowledge, it has been demonstrated that USP7 is significantly
overexpressed in SLE patients compared with healthy controls.
Furthermore, a positive correlation was observed between USP7
levels, IFN scores, SLEDAI scores and anti-dsDNA, which indicated
that the activation of the type I IFN pathway in SLE patients is
due to USP7 over-activation in the pathogenesis of autoimmune
conditions. In conclusion, the results of the present study
demonstrated that the overexpression of USP7 is relevant to the
biologic and clinical behavior of SLE. The findings suggested that
the USP family may serve as therapeutic targets via regulation of
IFNAR1 for the treatment of SLE. In the future, knockout and
transgenic animal models will be required to further identify the
role of USP7 in autoimmune diseases.
References
|
1
|
Tsokos GC: Systemic lupus erythematosus. N
Engl J Med. 365:2110–2121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gillis JZ, Panopalis P, Schmajuk G,
Ramsey-Goldman R and Yazdany J: Systematic review of the literature
informing the systemic lupus erythematosus indicators project:
Reproductive health care quality indicators. Arthritis Care Res
(Hoboken). 63:17–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hedrich CM and Tsokos GC: Epigenetic
mechanisms in systemic lupus erythematosus and other autoimmune
diseases. Trends Mol Med. 17:714–724. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qu B and Shen N: miRNAs in the
pathogenesis of systemic lupus erythematosus. Int J Mol Sci.
16:9557–9572. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Crow MK: Advances in understanding the
role of type I interferons in systemic lupus erythematosus. Curr
Opin Rheumatol. 26:467–474. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thacker SG, Zhao W, Smith CK, Luo W, Wang
H, Vivekanandan-Giri A, Rabquer BJ, Koch AE, Pennathur S, Davidson
A, et al: Type I interferons modulate vascular function, repair,
thrombosis, and plaque progression in murine models of lupus and
atherosclerosis. Arthritis Rheum. 64:2975–2985. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee PY, Li Y, Richards HB, Chan FS, Zhuang
H, Narain S, Butfiloski EJ, Sobel ES, Reeves WH and Segal MS: Type
I interferon as a novel risk factor for endothelial progenitor cell
depletion and endothelial dysfunction in systemic lupus
erythematosus. Arthritis Rheum. 56:3759–3769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Denny MF, Thacker S, Mehta H, Somers EC,
Dodick T, Barrat FJ, McCune WJ and Kaplan MJ: Interferon-alpha
promotes abnormal vasculogenesis in lupus: A potential pathway for
premature atherosclerosis. Blood. 110:2907–2915. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reynier F, Petit F, Paye M, Turrel-Davin
F, Imbert PE, Hot A, Mougin B and Miossec P: Importance of
correlation between gene expression levels: Application to the type
I interferon signature in rheumatoid arthritis. PLoS One.
6:e248282011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Müller U, Steinhoff U, Reis LF, Hemmi S,
Pavlovic J, Zinkernagel RM and Aguet M: Functional role of type I
and type II interferons in antiviral defense. Science.
264:1918–1921. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar KG, Tang W, Ravindranath AK, Clark
WA, Croze E and Fuchs SY: SCF(HOS) ubiquitin ligase mediates the
ligand-induced down-regulation of the interferon-alpha receptor.
EMBO J. 22:5480–5490. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Everett RD, Meredith M, Orr A, Cross A,
Kathoria M and Parkinson J: A novel ubiquitin-specific protease is
dynamically associated with the PML nuclear domain and binds to a
herpesvirus regulatory protein. EMBO J. 16:1519–1530. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou Z, Yao X, Li S, Xiong Y, Dong X, Zhao
Y, Jiang J and Zhang Q: Deubiquitination of Ci/Gli by Usp7/HAUSP
regulates hedgehog signaling. Dev Cell. 34:58–72. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song MS, Salmena L, Carracedo A, Egia A,
Lo-Coco F, Teruya-Feldstein J and Pandolfi PP: The
deubiquitinylation and localization of PTEN are regulated by a
HAUSP-PML network. Nature. 455:813–817. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van der Horst A, de Vries-Smits AM,
Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM
and Burgering BM: FOXO4 transcriptional activity is regulated by
monoubiquitination and USP7/HAUSP. Nat Cell Biol. 8:1064–1073.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
van der Knaap JA, Kumar BR, Moshkin YM,
Langenberg K, Krijgsveld J, Heck AJ, Karch F and Verrijzer CP: GMP
synthetase stimulates histone H2B deubiquitylation by the
epigenetic silencer USP7. Mol Cell. 17:695–707. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li M, Chen D, Shiloh A, Luo J, Nikolaev
AY, Qin J and Gu W: Deubiquitination of p53 by HAUSP is an
important pathway for p53 stabilization. Nature. 416:648–653. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hochberg MC: Updating the American College
of Rheumatology revised criteria for the classification of systemic
lupus erythematosus. Arthritis Rheum. 40:17251997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gladman DD, Ibañez D and Urowitz MB:
Systemic lupus erythematosus disease activity index 2000. J
Rheumatol. 29:288–291. 2002.PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Einarson MB, Pugacheva EN and Orlinick JR:
GST Pull-down. CSH Protoc. 2007:pdb prot47572007.PubMed/NCBI
|
|
22
|
Zhang ZM, Rothbart SB, Allison DF, Cai Q,
Harrison JS, Li L, Wang Y, Strahl BD, Wang GG and Song J: An
allosteric interaction links USP7 to deubiquitination and chromatin
targeting of UHRF1. Cell Rep. 12:1400–1406. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dall'era MC, Cardarelli PM, Preston BT,
Witte A and Davis JC Jr: Type I interferon correlates with
serological and clinical manifestations of SLE. Ann Rheum Dis.
64:1692–1697. 2005. View Article : Google Scholar : PubMed/NCBI
|