Introduction
Reperfusion therapy, which ensures the return of
blood supply as soon as possible to the ischemic region of the
myocardium, was originally the main treatment of acute myocardial
ischemia. However, Peuhkurinen (1)
described reperfusion as a ‘double edged sword’, due to the fact
that reperfusion itself can accelerate myocardial damage and lead
to additional myocardial injury, which is termed
ischemia/reperfusion (I/R) injury. I/R injury can be induced by
organ transplant dysfunction, stroke, myocardial infarction and
shock, and reperfusion leads to myocardial biochemical, structural
and functional changes and may determine myocardial cell survival
and death (2–4). Therefore, it is important to
elucidate how to improve myocardial function, reduce the
arrhythmogenesis, attenuate myocardial cell apoptosis or necrosis
and decrease the infarct size during I/R injury.
Rho-kinase (ROCK), a member of the serine threonine
protein kinase family, is ~160 kDa in size and expressed
ubiquitously in several tissues (5). ROCKs implicated in numerous types of
vital movement, including the regulation of cellular contraction,
growth, division, metabolism, migration, apoptosis and gene
expression (6,7). According to previous studies, ROCK is
closely associated with the development of a wide range of diseases
including coronary heart disease, atherosclerosis, hypertension,
pulmonary hypertension, heart failure, diabetes, stroke and cancer
(8–10). PARP is predominantly expressed in
the nucleus and can be activated under conditions including
radiation, inflammation and sepsis. In the present years,
accumulating data has shown that DNA strand is appear nick and
breaks in I/R, at the same time, PARP is up-regulated and utilized
its enzymes activation to repair these impaired DNA strands and
ensure the fidelity of genomic DNA replication (11). But excessive PARP expression and
activation will consumes profuse NAD+ and ATP and lead
to cardiomyocyte apoptosis (12).
Therefore, to explore the role and relationship of ROCK and PARP in
I/R will benefit us to deal with it inducing injury.
In the present study, we demonstrate that the
expression of phosphorylated (p-) myosin phosphatase target (MYPT;
the major effector of ROCK) and PARP are raised during I/R. In
addition, 3-aminobenzamide (3-AB), which is a PARP inhibitor, is
able to significantly diminish PARP expression in I/R. Notably,
Y-27632, which is a ROCK inhibitor, attenuates the phosphorylation
of MYPT in addition to the expression of PARP. Furthermore, these
two inhibitors can rescue myocardial infarction size and
cardiomyocyte apoptosis. Notably, the inhibitory role of Y-27632
was observed to be superior to 3-AB. It was verified that the
mitogen-activated protein kinase (MAPK)/extracellular
signal-related kinase (ERK) signaling pathway may serve a pivotal
role in Y-27632 or 3-AB protecting cardiomyocyte apoptosis induced
by I/R.
Materials and methods
Antibodies and reagents
Antibodies for phosphorylated (p-)MYPT-1 (BS4114;
dilution, 1:1,500) was from Bioworld Technology, Inc. (Minneapolis,
MN, USA). Antibodies for PARP (#9542; dilution, 1:300), caspase-3
(#9668; dilution, 1:1,000), B-cell lymphoma 2 (Bcl-2; #2870;
dilution, 1:1,000), Bcl-2-associated X protein (Bax; #2772;
dilution, 1:1,000) and p-ERK (#9101; dilution, 1:1,000) were from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Antibodies for
β-actin (sc-47778; dilution, 1:2,000) and horseradish peroxidase
(HRP)-conjugated secondary antibodies (sc-2004 and sc-2005;
dilution, 1:5,000) were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Y-27632 (ROCK inhibitor) was purchased
from Calbiochem (Merck Millipore, Darmstadt, Germany). 3-AB (PARP
inhibitor) was obtained from Sigma-Aldrich (Merck Millipore).
Culture and I/R of the cell line
H9C2
Rat ventricular cell line H9C2 was obtained from the
American Type Culture Collection (Manassas, VA, USA). The cells
were cultured at 37°C with 5% CO2 in DMEM containing 10%
(vol/vol) FBS and 100 U/ml penicillin and 100 μg/ml streptomycin
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). H9C2
cells were subjected to I/R. Specifically, these cells were
pretreated with corresponding inhibitors and then placed into a
hypoxic chamber at 37°C for 60 min, then were re-oxygenated for 120
min with DMEM containing 10% FBS (13).
Hoechst staining
In order to detect myocardial apoptosis, chromatin
morphology was observed with fluorescence microscopy after DNA
staining with 0.5 mg/l Hoechst 33258 (Nanjing KeyGen Biotech. Co.,
Ltd., Nanjing, China). The percentage of apoptotic cells was
calculated as the number of apoptotic cells compared with the
number of total cells counted in 10 randomly selected fields.
Images from Hoechst-stained samples were acquired by using a Leica™
fluorescence microscope equipped with a Leica™ camera (Leica
Microsystems GmbH, Wetzlar, Germany).
Immunoblot analyses
For western blot analysis, cells were lysed with
mammalian protein extraction reagent (CelLytic™ Cell Lysis Reagent;
Sigma-Aldrich; Merck Millipore) supplemented with the protease
inhibitor cocktail (#5872; Cell Signaling Technology, Inc.). Equal
amounts of protein were separated by 10% SDS-PAGE. Blots were
probed with the corresponding antibodies and were incubated
overnight at 4°C. Following subsequent washing with Tris-buffered
saline with Tween-20 (T1081; Beijing Solarbio Science &
Technology, Co., Ltd., Beijing, China), the membranes were
incubated with the corresponding secondary antibodies conjugated
with HRP for 1 h at room temperature. Then the membranes were
visualized using a SuperSignal West Pico substrate
chemiluminescence detection kit (Pierce, Rockford, IL, USA). In
order to confirm uniform loading, membranes were stripped and
re-incubated with anti-β-actin antibodies.
Preparation and grouping of the rat
I/R model
A total of 60 female Wistar rats (12–16 weeks old;
body weight 250–300 g) were purchased from the Experimental Animal
Center of Shandong University (Shandong, China) and were housed
individually and provided with standard lighting (12 h light and
dark cycles), temperature (22±0.5°C) and humidity (60±10%) for at
least 1 week prior to the experiment. All experiments were approved
by the Institution Animal Care and Use Committee of Shandong
Provincial Hospital Affiliated to Shandong University and were
performed in line with the National Institutes of Health
Guidelines. Female Wistar rats were anesthetized with urethane (25
mg/kg; intraperitoneal injection). The left thoracotomy was made in
the fourth intercostal space. Subsequent to occlusion of the left
anterior branch of the descending coronary artery (LAD) for 60 min,
the ligation was loosened for 120 min. Following this, these rats
were sacrificed and their hearts were obtained for the following
experiments. The control rats underwent this procedure, however the
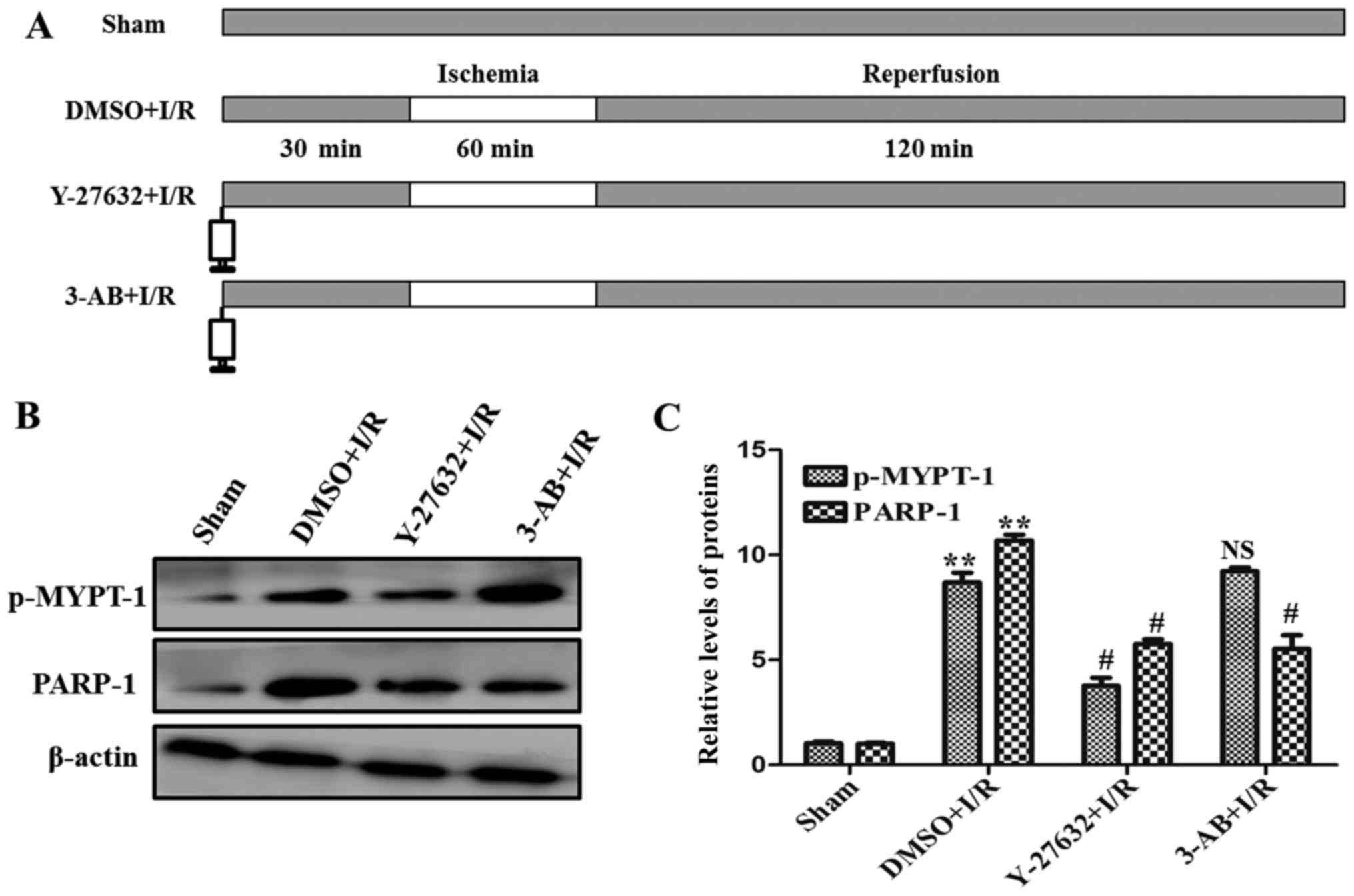
LAD was not ligated. As presented in Fig. 1A, the rats were divided randomly
into four groups: i) Sham operation (n=15); ii) after
administration of dimethyl sulfoxide (DMSO), LAD was occluded for
60 min and reperfused for 120 min (n=15); iii) after administration
of Y-27632 (inhibitor of ROCK), LAD was occluded for 60 min and
reperfused for 120 min (n=15); iv) after administration of 3-AB
(inhibitor of PARP), LAD was occluded for 60 min and reperfused for
120 min (n=15).
Evaluation of myocardial infarct size
and area at risk
Following I/R, the LAD was reoccluded and 3 ml Evans
blue was injected into the left ventricle to delineate myocardial
area at risk. Ischemic area (also termed area at risk, AAR) was not
stained and the nonischemic area was stained blue. Subsequently,
the heart was obtained and washed with normal saline. The great
vessels, right ventricle and atria were removed. The tissue was
frozen in a −20°C freezer for 30 min in order to facilitate
slicing. After this, the left ventricle was separated and cut into
slices approximately 1 mm in thickness. The AAR was isolated from
the area not at risk and then was incubated with nitro blue
tetrazolium (NBT) solution (1% w/v) to distinguish between the
ischemic or infarcted area at 37°C for 15 min. The parts stained
blue were designated as the ischemic area, while those parts
without blue staining were designated as the infarct area. Finally,
the different parts of the ventricle were weighed respectively.
Myocardial AAR was indicated with the percentage of the left
ventricle. Infarct size was demonstrated as the percentage of the
AAR (12).
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
Myocardial apoptosis was analyzed with the TUNEL
technique (Roche Diagnostics GmbH, Mannheim, Germany) according to
the manufacturer's instructions. Nuclei were labeled with
hematoxylin and the number of TUNEL-positive cells were observed
using light microscopy and calculated by randomly counting in 10
fields in the section and were demonstrated with percentage of
normal nuclei.
Enzyme-linked immunosorbent assay
(ELISA)
The cell culture supernatants were collected, and
the concentration of creatine kinase-MB isoenzyme (CK-MB;
CSB-E14403r; Cusabio Biotech Co., Ltd., College Park, MD, USA),
IL-6 (Invitrogen; Thermo Fisher Scientific, Inc.) and TNF-α
(Invitrogen; Thermo Fisher Scientific, Inc.) were measured using a
commercially available ELISA kit in accordance with the
manufacturer's instructions.
Assay of LDH and NO release
Myocardial damage was examined by measuring lactate
dehydrogenase (LDH) activity in culture media with the
LDH-Cytotoxicity Assay Kit (C0016; Beyotime Institute of
Biotechnology, Haimen, China) according to the manufacturer's
recommendations. NO in the serum of rats was measured indirectly
using the Griess reagent kit (Beyotime Institute of Biotechnology)
according to the manufacturer's recommendations. The optical
density was detected at 490 nm (for LDH) or 540 nm (for NO) using
the Multiskan GO microplate reader (Thermo Fisher Scientific,
Inc.). The LDH activity and NO concentration were determined using
a standard curve.
Measurement of NAD+
levels
Left ventricles were washed with cold
phosphate-buffered saline and were ground into a fine powder with a
pestle and mortar in liquid nitrogen. The powder was transferred
into 1.5 ml Eppendorf tube and added 400 μl nicotinamide adenine
dinucleotide phosphate (NADP)/NADPH extraction buffer to
homogenize. The samples were centrifuged at 16,000 × g for 5 min at
4°C. Subsequently, the extracted samples were transferred into new
tubes. Samples were heated to 60°C for 30 min in a water bath and
then were cooled on ice. Following this, 50 µl of the above
solution was added into a 96-well bottom assay plate in duplicate,
NADP cycling mix (K337-100; BioVision, Inc., Milpitas, CA, USA) was
added to each well, and the plate was incubated at room temperature
for 5 min. Finally, 10 μl developer was added and the samples were
incubated for 2 h. The absorbance was measured at 450 nm.
Statistical analysis
All data was presented as a result of three or four
independent experiments. All data were expressed as the mean ±
standard deviation, and was analyzed via one-way analysis of
variance and two-tailed Student's t-test. In all cases, values of
P<0.05 were considered to indicate a statistically significant
difference.
Results
Inhibition of the ROCK signaling
pathway diminishes PARP expression in I/R-induced injury
To identify the role of ROCK and PARP in the
myocardial I/R injury, the activation of ROCK and the expression of
PARP by were analyzed by western blotting. As presented in Fig. 1B and C, compared with the sham
control, a significant increase in expression of p-MYPT-1, a
well-known ROCK specific substrate, and PARP were detected during
I/R. At the same time, in order to confirm the associations of ROCK
and PARP, the ROCK and PARP specific inhibitors Y-27632 and 3-AB
were used in I/R. As presented in Fig.
1B and C, the expression levels of p-MYPT-1 and PARP were
significantly decreased in the presence of the two inhibitors,
respectively. Notably, the expression of PARP was additionally
abrogated by the ROCK inhibitor Y-27632. Taken together, these data
indicate that ROCK and PARP were upregulated and ROCK may be an
upstream signaling molecule of PARP in myocardial I/R injury.
ROCK-mediated regulation of PARP
expression promotes myocardial cell apoptosis in I/R-induced
injury
To further investigate the effect of ROCK and PARP
for myocardial cell function, the I/R-induced cell apoptosis was
detected with Hoechst staining assay. As presented in Fig. 2A and B, the percentage of apoptotic
cells in the DMSO plus I/R group was significantly increased
compared with that of the sham group. The nuclear edge of the sham
control group was clear and integrated. Nevertheless, apoptotic
salient features including chromatin condensation and DNA
fragmentation etc. were identified in the DMSO plus I/R group. The
percentage of apoptotic cells was significantly decreased in the
presence of Y-27632 or 3-AB. The number of apoptotic cells in the
Y-27632 inhibitor group was relatively less compared with the 3-AB
group. In addition, the expression of the apoptosis-associated
molecules, including Bcl-2, Bax and caspase-3 were assessed by
western blot analysis. As presented in Fig. 2C and D, the expression of
apoptosis-promoting proteins, including Bax and caspase-3, were
markedly increased in the DMSO plus I/R group compared with the
sham group. In contrast, the expression of the anti-apoptotic
protein Bcl-2 was markedly decreased in DMSO plus I/R group.
Otherwise, pretreatment of Y-27632 or 3-AB can significantly
increase Bcl-2 expression and reduce Bax and caspase-3 expression.
Taken together, these results indicated that ROCK and PARP served
an important role in I/R-induced myocardial cell apoptosis and the
effect of Y-27632, a ROCK inhibitor, in reversing cell apoptosis
was more significant than in the 3-AB group.
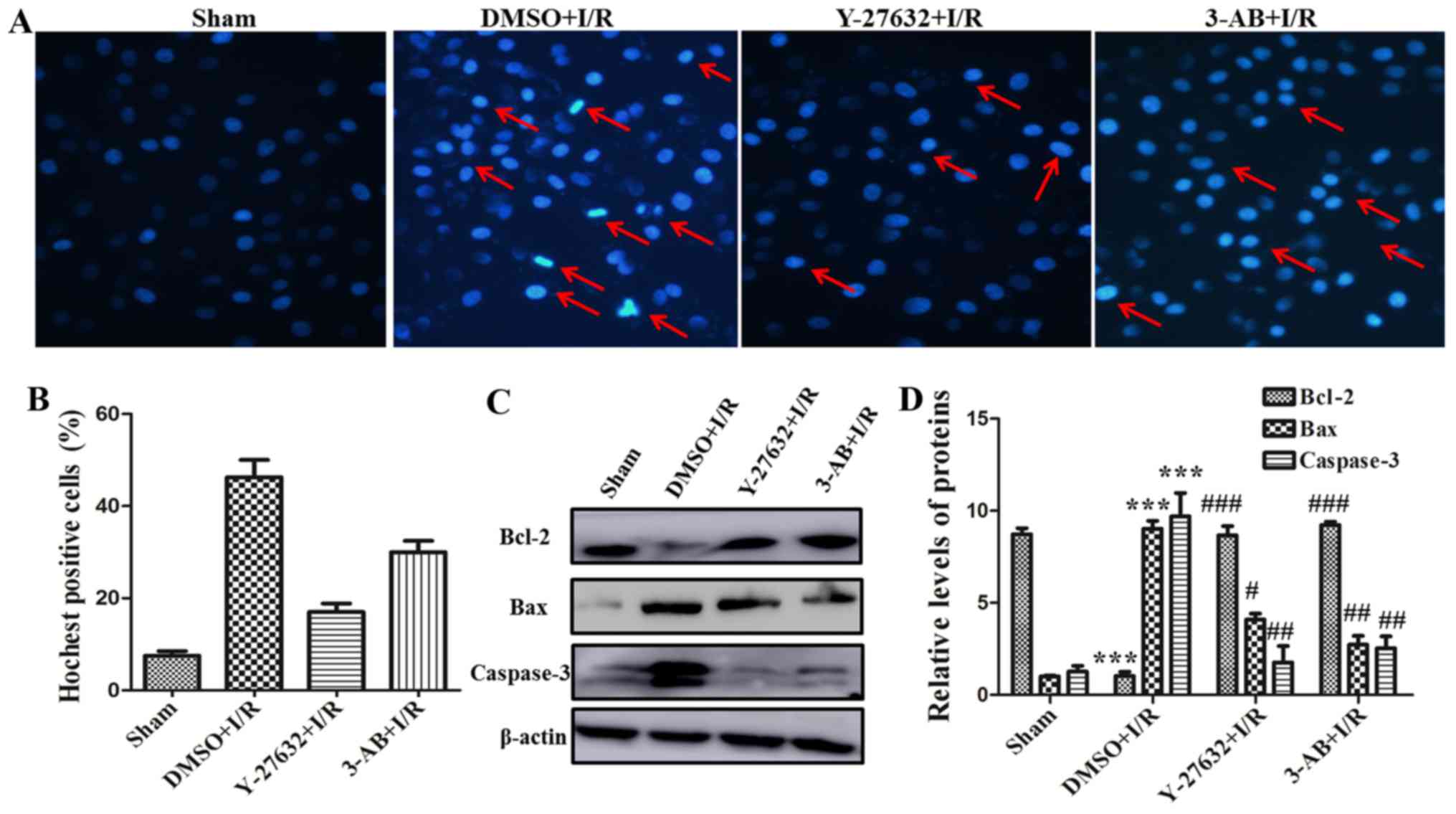 | Figure 2.ROCK regulating PARP expression
promotes myocardial cell apoptosis in I/R-inducing injury. (A)
Hoechst staining was completed (magnification, ×200; red arrows,
chromatin condensation, DNA fragmentation and chromatin
margination). (B) The histogram present the rates of
Hoechst-positive cells. (C) The expression levels of Bcl-2, Bax and
caspase-3 were measured in different groups of H9C2 cells by
western blotting. (D) Relative quantitative analysis of Bcl-2, Bax
and caspase-3 expression. ***P<0.001 vs. Sham;
#P<0.05, ##P<0.01 and
###P<0.001 vs. DMSO + I/R; $P<0.05, vs.
Y-27632 + I/R. Similar observations were obtained in three
independent experiments. ROCK, Rho-kinase; PARP, poly adenosine
diphosphate-ribose polymerase; I/R, ischemia/reperfusion; Bcl-2,
B-cell lymphoma 2; Bax, Bcl-2-associated X protein; DMSO, dimethyl
sulfoxide. |
ROCK and PARP inhibition during I/R
attenuated myocardial cell apoptosis and infarct size in vivo
In order to evaluate the effect of ROCK and PARP
inhibition in vivo, DMSO or corresponding inhibitors were
pre-injected and then established as rat models of I/R. As
presented in Fig. 3A and B, the
AAR and infarct size of the hearts were 57.67±2.52 and 60.67±2.52%,
respectively in the I/R plus DMSO group. Administration of Y-27632
or 3-AB caused significant reduction of AAR and infarct size. The
AAR and infarct size of the hearts were 29.67±4.73 and 30.02±4.10%,
respectively in the I/R plus Y-27632 group. The AAR and infarct
sizes of the hearts were 44.67±7.57 and 46.33±11.50%, respectively
in the I/R plus 3-AB group. In addition, it was observed that the
AAR and infarct sizes of the heart in the I/R plus Y-27632 group
were always lower than in the I/R plus 3-AB group. Furthermore,
myocardial apoptosis was observed with the TUNEL assay. As
presented in Fig. 3F and G,
TUNEL-positive cells were significantly increased in I/R
(37±6.24%). However, the numbers were markedly decreased to 19±2.08
and 32±3.61% in the presence of the ROCK and PARP inhibitor,
respectively. In addition, the content of CK-MB, LDH and NO were
measured in the serum. As presented in Fig. 3C-E, it was identified that the
content of CK-MB and LDH was significantly enhanced and NO was
reduced in the I/R plus DMSO group. Y-27632 or 3-AB were able to
markedly downregulate the content of CK-MB and LDH and rescued NO
in rat serum, and the effect of Y-27632 inhibition was superior to
that of 3-AB. Taken together, these results indicate that
inhibition of ROCK activity and PARP reduces myocardial infarct
size and cell apoptosis in I/R injury. In summary, the effects of
Y-27632 were superior to 3-AB.
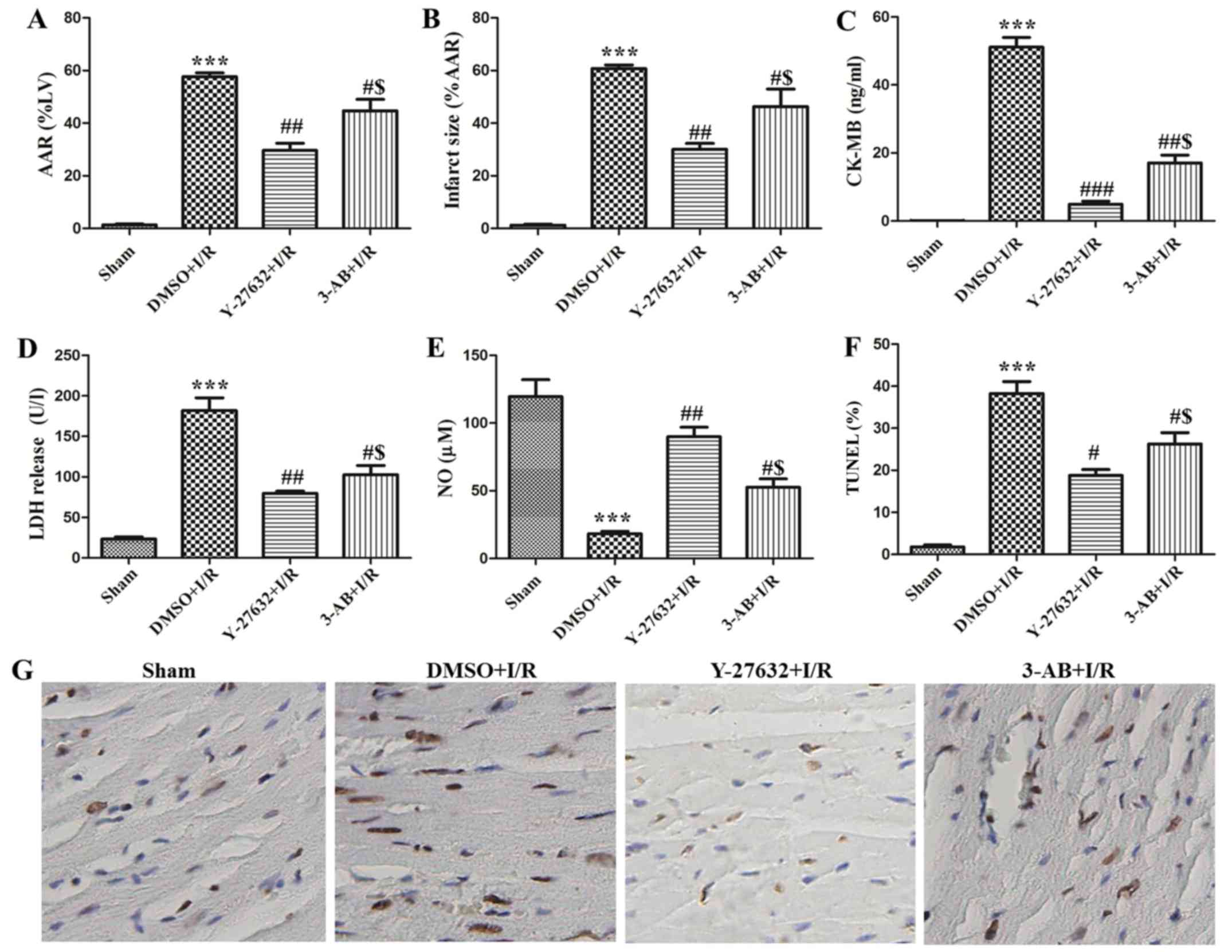 | Figure 3.ROCK and PARP inhibition during I/R
attenuated myocardial cell apoptosis and infarct size in
vivo. Changes in (A) myocardial AAR and (B) infarct size of
different groups (Sham, DMSO + I/R, Y-27632 + I/R, 3-AB + I/R) in
the rat hearts. The content of (C) CK-MB, (D) LDH release and (E)
NO levels were detected in the serum of rats. (G) Representative
photomicrographs of ventricular tissue stained for TUNEL for DNA
breaks in different groups. (F) The histogram shows the percentage
of TUNEL positive cells. n=15; ***P<0.001 vs. Sham;
#P<0.05, ##P<0.01 and
###P<0.001 vs. DMSO + I/R; $P<0.05, vs.
Y-27632 + I/R. ROCK, Rho-kinase; PARP, poly adenosine
diphosphate-ribose polymerase; I/R, ischemia/reperfusion; DMSO,
dimethyl sulfoxide; 3-AB, 3-aminobenzamide; TUNEL, terminal
deoxynucleotidyl transferase dUTP nick end labeling; AAR, area at
risk; CK-MB, creatine kinase-MB; LDH, lactate dehydrogenase; NO,
nitric oxide. |
ROCK/PARP/ERK signaling pathway served
a vital role in I/R-inducing injury
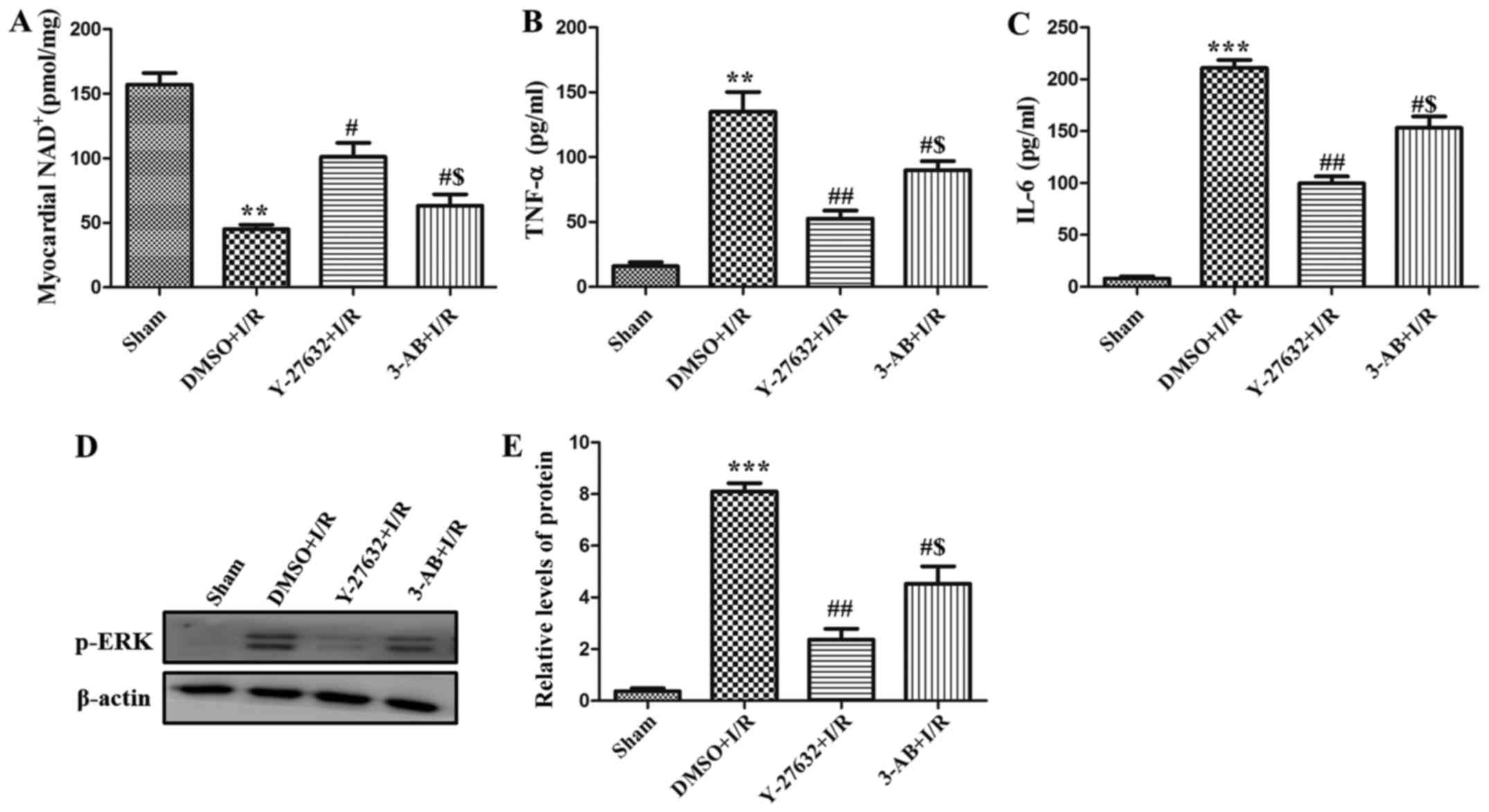
In order to analyze the unique role of PARP during
I/R-inducing injury, the NAD+ level in the left
ventricle was measured. As presented in Fig. 4A, the level of NAD+ was
reduced in the I/R group compared with the sham group. However,
Y-27632 or 3-AB significantly augmented the level of
NAD+ and the effect of Y-27632 was more marked. In
addition, as presented in Fig. 4B and
C, the level of TNF-α and IL-6 in rat serum was observed and it
was identified that the content of the two cytokines were also
increased in I/R. Pre-treatment with Y-27632 or 3-AB decreased
their levels in rat serum. In addition, the phosphorylation levels
of ERK were measured using western blot analysis. As presented in
Fig. 4D and E, it was demonstrated
that phosphorylation of ERK was clearly increased in the hearts of
the I/R group and decreased in the pre-treatment of Y-27632 or 3-AB
groups. The expression of p-ERK in the Y-27632 group was lower than
that of the 3-AB group. Taken together, these data indicate that
the MAPK/ERK signaling pathway serves an important role in the
regulation of I/R injury by ROCK and PARP.
Discussion
In the current study, the activation of ROCK and the
expression of PARP were demonstrated to be increased in the
hypoxia-reoxygenation injury model in the H9C2 cell line, and they
were observed to aggravate the apoptosis of myocardial cells. In
addition, it was identified that there was an association between
ROCK and PARP via a corresponding inhibitors pretreatment assay.
PARP may be located downstream of ROCK and regulated myocardial
cell apoptosis in heart I/R injury. Furthermore, it was validated
that the MAPK/ERK signaling pathway served a prominent role in
heart I/R injury by effecting production of proinflammatory
cytokines, including TNF-α and IL-6. The results of the present
study suggest that ROCK increases myocardial apoptosis via the
PARP/ERK signaling pathway.
Ischemic heart disease has become one of the leading
factors of death worldwide (14).
Protection of the heart against injury in I/R injury remains a
challenge, thus it is particularly important to clarify the
pathogenesis and role of important molecules during I/R. ROCK is a
major regulator of the execution phase of apoptosis, including cell
contraction, dynamic membrane blebbing, nuclear disintegration and
fragmentation of apoptotic cells into apoptotic bodies (15,16).
Specifically, when DNA strand-breaks happen, PARP, a key protein
participating in DNA repair (17),
binds rapidly to the DNA strand-breaks and converts NAD into long
branched polymers to attach onto a variety of nuclear proteins
(12,18). Nevertheless, the activation of
excessive PARP may cause a negative effect via a
caspase-independent pathway-mediated cell apoptosis (19). Previously, although it has been
reported that ROCK and PARP are involved in I/R injury and they
aggravate cell apoptosis and augment infarct size (8,9,12),
the correlation and functional mechanism of ROCK and PARP remain to
be fully understood. In the present study, ROCK was demonstrated to
be located upstream of PARP to induce myocardial apoptosis.
Previous studies have investigated the function of
the the MAPK signaling pathways in I/R, including ERK, c-Jun
N-terminal kinase (JNK) and p38 pathways. For example, Yang et
al (20) identified that CRGP
effectively improved I/R injury of the brain tissue through a
reduction in JNK and p38 phosphorylation and an increase in ERK
phosphorylation in the MAPK pathway. Zhang et al (9) demonstrated that ROCK enhanced
cardiomyocyte apoptosis in the heart I/R via promotion of
JNK-mediated apoptosis-inducing factor translocation. Nevertheless,
it was demonstrated in the current study that inhibition of ROCK
and PARP can reduce the phosphorylation of ERK and the resulting
production of proinflammatory factors.
In conclusion, the present study elucidated a novel
pathway for ROCK and PARP in the regulation of heart I/R injury.
ROCK promotes the expression of PARP to accelerate myocardial
apoptosis through raising ERK phosphorylation and the release of
TNF-α and IL-6. Given the pathological role of ROCK and PARP in I/R
injury, ROCK/PARP/ERK may aid in the understanding of the mechanism
of myocardial cell apoptosis and provide novel suggestions for the
therapy of I/R.
Acknowledgements
The present study was supported in part by grants
from the Promotive Research Fund for Excellent Young and
Middle-aged Scientists of Shandong Province (grant no.
BS2014YY037), the Project Funded by China Postdoctoral Science
Foundation (grant no. 2013M541926), the Postdoctoral Innovation
Project Special Foundation of Shandong Province (grant no.
201302031) and the Science and Technology Development Plans of
Shandong Province (grant no. 2012GSF21807).
References
|
1
|
Peuhkurinen K: Myocardial reperfusion-a
double-edged sword? Duodecim. 105:822–830. 1989.(In Finnish).
PubMed/NCBI
|
|
2
|
Kurian GA, Rajagopal R, Vedantham S and
Rajesh M: The role of oxidative stress in myocardial ischemia and
reperfusion injury and remodeling: Revisited. Oxid Med Cell Longev.
2016:16564502016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Patil KD, Halperin HR and Becker LB:
Cardiac arrest: Resuscitation and reperfusion. Circ Res.
116:2041–2049. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsushima S, Tsutsui H and Sadoshima J:
Physiological and pathological functions of NADPH oxidases during
myocardial ischemia-reperfusion. Trends Cardiovasc Med. 24:202–205.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Julian L and Olson MF: Rho-associated
coiled-coil containing kinases (ROCK): Structure, regulation, and
functions. Small GTPases. 5:e298462014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Minambres R, Guasch RM, Perez-Aragó A and
Guerri C: The RhoA/ROCK-I/MLC pathway is involved in the
ethanol-induced apoptosis by anoikis in astrocytes. J Cell Sci.
119:271–282. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chang J, Xie M, Shah VR, Schneider MD,
Entman ML, Wei L and Schwartz RJ: Activation of Rho-associated
coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays
an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci
USA. 103:14495–14500. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Bian HJ, Li XX, Liu XB, Sun JP,
Li N, Zhang Y and Ji XP: ERK-MAPK signaling opposes rho-kinase to
reduce cardiomyocyte apoptosis in heart ischemic preconditioning.
Mol Med. 16:307–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Li XX, Bian HJ, Liu XB, Ji XP and
Zhang Y: Inhibition of the activity of Rho-kinase reduces
cardiomyocyte apoptosis in heart ischemia/reperfusion via
suppressing JNK-mediated AIF translocation. Clin Chim Acta.
401:76–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abe K, Shimokawa H, Morikawa K, Uwatoku T,
Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K
and Takeshit A: Long-term treatment with a Rho-kinase inhibitor
improves monocrotaline-induced fatal pulmonary hypertension in
rats. Circ Res. 94:385–393. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Harris JL, Jakob B, Taucher-Scholz G,
Dianov GL, Becherel OJ and Lavin MF: Aprataxin, poly-ADP ribose
polymerase 1 (PARP-1) and apurinic endonuclease 1 (APE1) function
together to protect the genome against oxidative damage. Hum Mol
Genet. 18:4102–4117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song ZF, Ji XP, Li XX, Wang SJ, Wang SH
and Zhang Y: Inhibition of the activity of poly (ADP-ribose)
polymerase reduces heart ischaemia/reperfusion injury via
suppressing JNK-mediated AIF translocation. J Cell Mol Med.
12:1220–1228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li B, Li R, Zhang C, Bian HJ, Wang F, Xiao
J, Liu SW, Yi W, Zhang MX, Wang SX, et al: MicroRNA-7a/b protects
against cardiac myocyte injury in ischemia/reperfusion by targeting
poly (ADP-ribose) polymerase. PLoS One. 9:e900962014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bolli R, Becker L, Gross G, Mentzer R Jr,
Balshaw D and Lathrop DA: NHLBI Working Group on the Translation of
Therapies for Protecting the Heart from Ischemia: Myocardial
protection at a cross roads: The need for translation into clinical
therapy. Circ Res. 95:125–134. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Erwig LP, McPhilips KA, Wynes MW, Ivetic
A, Ridley AJ and Henson PM: Differential regulation of phagosome
maturation in macrophages and dendritic cells mediated by Rho
GTPases and ezrin-radixin-moesin (ERM) proteins. Proc Natl Acad Sci
USA. 103:12825–12830. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tosello-Trampont AC, Nakada-Tsukui K and
Ravichandran KS: Engulfment of apoptotic cells is negatively
regulated by Rho-mediated signaling. J Biol Chem. 278:49911–49919.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng X, Song W, Deng B, Xing Z and Zhang
W: 3-aminobenzamide, one of poly(ADP-ribose)polymerase-1
inhibitors, rescues apoptosis in rat models of spinal cord injury.
Int J Clin Exp Pathol. 8:12207–12215. 2015.PubMed/NCBI
|
|
18
|
Wang SJ, Wang SH, Song ZF, Liu XW, Wang R
and Chi ZF: Poly(ADP-ribose) polymerase inhibitor is
neuroprotective in epileptic rat via apoptosis-inducing factor and
Akt signaling. Neuroreport. 18:1285–1289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meli E, Pangallo M, Picca R, Baronti R,
Moroni F and Pellegrini-Giampietro DE: Differential role of
poly(ADP-ribose) polymerase-1 in apoptotic and necrotic neuronal
death induced by mild or intense NMDA exposure in vitro. Mol Cell
Neurosci. 25:172–180. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang SI, Yuan Y, Jiao S, Luo QI and Yu J:
Calcitonin gene-related peptide protects rats from cerebral
ischemia/reperfusion injury via a mechanism of action in the MAPK
pathway. Biomed Rep. 4:699–703. 2016.PubMed/NCBI
|