Introduction
Cerebral malaria is a devastating infectious
disease, and its symptoms at an advanced stage include coma,
neurological dysfunction, inflammation, severe anemia and acidosis.
Mortality in patients with cerebral malaria primarily occurs due to
the lack of or delay in administration of suitable treatments. Even
when proper intervention is received, the mortality rate of
cerebral malaria is 15–20% (1).
Furthermore, out of the children who survive cerebral malaria, ~15%
exhibit permanent neurological impairment (1,2).
In last few decades, multiple reports have revealed
that hydrogen sulfide (H2S) is a bioactive molecule,
exerting multiple functions in physiological and pathological
processes. One of the important physiological functions of
H2S is that it serves as an endogenous anti-inflammatory
agent (3–5). Several H2S-releasing drugs
are currently being developed for the treatment of many types of
inflammatory diseases (6,7). For instance, in a rat model of
carrageenan-induced hind paw edema, it was demonstrated that the
H2S donor, sodium hydrosulfide, provided protection
against inflammation and edema formation, potentially due to its
strong inhibitory effects on leukocyte adherence to the vascular
endothelium, resulting in a significant decrease of paw volume
(8).
Furthermore, H2S has been demonstrated to
be an important neuroprotectant by its ability to enhance NMDA
receptor-mediated responses and induce hippocampal long-term
potentiation (9,10). In addition, previous reports
demonstrated that H2S serves as a neuronal defense
against hypochlorous acid- or lipopolysaccharide-induced
neuroinflammation (11,12).
Despite its involvement in the regulation of
inflammation and neuro-protection, the involvement of
H2S in cerebral malaria pathogenesis remains poorly
understood. The primary aim of the present study was to investigate
the effects of H2S in the development of cerebral
malaria.
Materials and methods
Plasmodium berghei ANKA (PbA)
infection
C57BL/6 mice (6–8 weeks; 21–25 g; male, n=80; and
female, n=80) were purchased from the Guangdong Medical
Laboratorial Animals Center (Guangzhou, China). All protocols were
approved and followed the guidelines of the Animal Use Committee of
Sun Yat-Sen University (Guangzhou, China). Animals were maintained
at room temperature (24±0.5°C) in a 12-h light/dark cycle and were
allowed ad libitum access to food and water. PbA was thawed and
inoculated in one C57BL/6 mouse. Parasitaemia of the infected mouse
was detected by Giemsa-stain of a blood smear. Briefly, a drop of
blood from tails was placed on a glass slide and another glass
slide was used to form a layer of film on the glass. Giemsa A
solution was added to blood film and incubated for 10 min at room
temperature, Giemsa A solution was washed off with water and Giemsa
B solution was added and incubated for 10 min, followed by a
further wash with water. The number of infected red blood cells
(iRBCs) out of a total of 1,000 RBCs was counted under an oil
immersion lens (x100) of a light microscope. Giemsa-staining was
performed on the infected mouse every day following infection, on
three individual glass slides each time. When the infection rate
reached 50%, the iRBCs from this infected mouse were subsequently
used to infect all other mice (13). Parasitaemia of all other mice was
detected by Giemsa-staining on the day before infection, and at 2,
3 and 6 days after infection.
Cumulative survival
Each mouse was infected with 1×106 iRBCs. A total of
40 infected mice were randomly divided into saline, NaHS,
L-cysteine and aminooxyacetic acid (AOAA) groups (n=10 mouse per
group). Saline, NaHS (an exogenous donor of H2S; 1
mg/kg), L-cysteine (an endogenous donor of H2S; 0.3
mg/kg), or AOAA [cystathionine-β-synthase (CBS) inhibitor; 10
mg/kg] was injected intraperitoneally into mice three times a day.
The survival rate was confirmed by quantitative assessment using a
rapid murine coma and behavior scale of cerebral malaria (14).
Extravasation of Evans blue
A total of 50 mice were randomly divided into two
groups: Uninfected (n=10) and infected (n=40). Following infection
with 1×106 iRBCs, the 40 mice were randomized into four groups:
Infection group treated with saline for 2 days (D2+saline; n=10),
infection group treated with NaHS for 2 days (D2+NaHS; n=10),
infection group treated with saline for 6 days (D6+saline; n=10),
and infection group treated with NaHS for 6 days (D6+NaHS; n=10).
Mice were subsequently anesthetized by a subcutaneous injection of
40 mg/kg sodium pentobarbital (Guangzhou Qiyun Biotechnology Co.,
Ltd., Guangzhou, China). The right caudal vein was cannulated with
polyethylene tubing and 200 nnu Evans blue (2% in saline) was
administered via the catheter. This procedure was followed by an
injection of 200 µl 0.9% saline. After 30 min, all mice were killed
by dislocating cervical vertebra. The hair and skin of mice heads
were removed by surgical scissors, the top of skull was split along
the gap with scissors and the brain was gently removed from the
skull. Extracted brains were placed in 1 ml N,N-dimethylformamide
(Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) for 48 h to
extract the Evans blue dye (15),
and the absorbance of the Evans blue dye solution was measured at a
wavelength of 630 nm using a spectrophotometer.
Acquisition of plasma, brain, liver
and spleen
A total of 70 mice were randomly divided into the
uninfected group, infection group treated with saline for 2 days
(D2+saline), infection group treated with NaHS for 2 days
(D2+NaHS), infection group treated with saline for 3 days
(D3+saline), infection group treated with NaHS for 3 days
(D3+NaHS), infection group treated with saline for 6 days
(D6+saline), and infection group treated with NaHS for 6 days
(D6+NaHS), n=10 per group. Subsequently, all mice were killed by
dislocating cervical vertebra. Following administration, the
spleen, brain and liver were carefully excised and weighed. The
brain tissues were incised into four parts. One was placed in
pH-balanced 10% formalin, embedded by paraffin, sectioned (8 µm)
and examined by hematoxylin and eosin (H&E) staining, two were
used for total RNA and protein measurement, respectively, and the
rest were ground into homogenate for measurement of H2S
by ELISA (cat. no. 10355371; Shanghai Institute of Biotechnology
Co., Ltd., Shanghai, China). The plasma was drawn out for
measurement of interleukin-18 (IL-18; cat. no. 7625), matrix
metalloproteinase-9 (MMP-9; cat. no. MMPT90) and serum cluster of
differentiation 40 (sCD40; cat. no. MCCD40) by ELISA (R&D
Systems, Inc., Minneapolis, MN, USA), following the manufacturer's
protocol.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was isolated from the brain tissue using
TRIzol® (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). RT-PCR was performed using the Prime Script® RT-PCR kit
(Takara Biotechnology Co., Ltd., Dalian, China) following the
manufacturer's protocol. The PCR conditions involved initialization
at 94°C for 3 min, followed by 40 cycles of denaturation at 94°C
for 30 sec, annealing at 60°C for 30 sec and polymerization at 72°C
for 30 sec, final elongation was performed at 72°C for 10 min. The
primer sequences for CBS were as follows: Forward,
5′-CGTGAGCAGACCCAGACAT-3′ and reverse,
5′-GCTACTCGGGCATAGAGGATT-3′.
Western blot analysis
Proteins were extracted from brain tissue, briefly,
brain tissue was triturated in a tissue mortar with RIPA lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China) at 4°C
for 10 min, the brain lysate solution was centrifuged at 15,000 × g
and 4°C for 10 min. Following centrifugation, the supernatant was
collected. Protein (80 µg) was loaded onto each lane, separated by
10% SDS-PAGE and subsequently transferred to a polyvinylidene
difluoride membrane. The membrane was blocked in 5% nonfat milk
powder for 30 min and subsequently probed with rabbit polyclonal
CBS (1:1,000; cat. no. H00000875-D01P; Abnova, Taipei, Taiwan) and
rabbit monoclonal GAPDH (1:1,000; cat. no. 2118S; Cell Signaling
Technology, Inc., Danvers, MA, USA) primary antibodies for 2 h at
room temperature, followed by incubation with horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(1:20,000; cat. no. E030120; EarthOx Life Sciences, Millbrae, CA,
USA) for 1 h at room temperature. Membranes were developed with an
Enhanced Chemiluminescence reagent (LumiGold; SignaGen
Laboratories, Rockville, MD, USA) and a Tanon 5200 Gel Imaging
System.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Database construction and statistical analyses were
performed using Origin 8.5 software (OriginLab Corporation,
Northampton, MD, USA). Data were analyzed by analysis of variance
followed by Bonferonni's post hoc test, or two-tailed Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
ECM model validation
Following infection with PbA, the amount of
parasitized red blood cells was detected by Giemsa staining of a
blood smear (Fig. 1A). Hemorrhage
into the brain on day 6 of ECM in saline-treated mice was
additionally demonstrated by H&E-staining (Fig. 1B). Similarly, the hemorrhage was
visibly reduced in the H2S donor administration group
(Fig. 1B), while no apparent
hemorrhage was observed on day 2 or 3 following infection with or
without NaHS treatment (Fig. 1B).
In contrast, significant hemorrhaging in the brain was observed on
day 6 of ECM (Fig. 1B). In
addition, the indexes of spleen, brain and liver/10 g weight
increased gradually in a time-dependent manner (Fig. 1C).
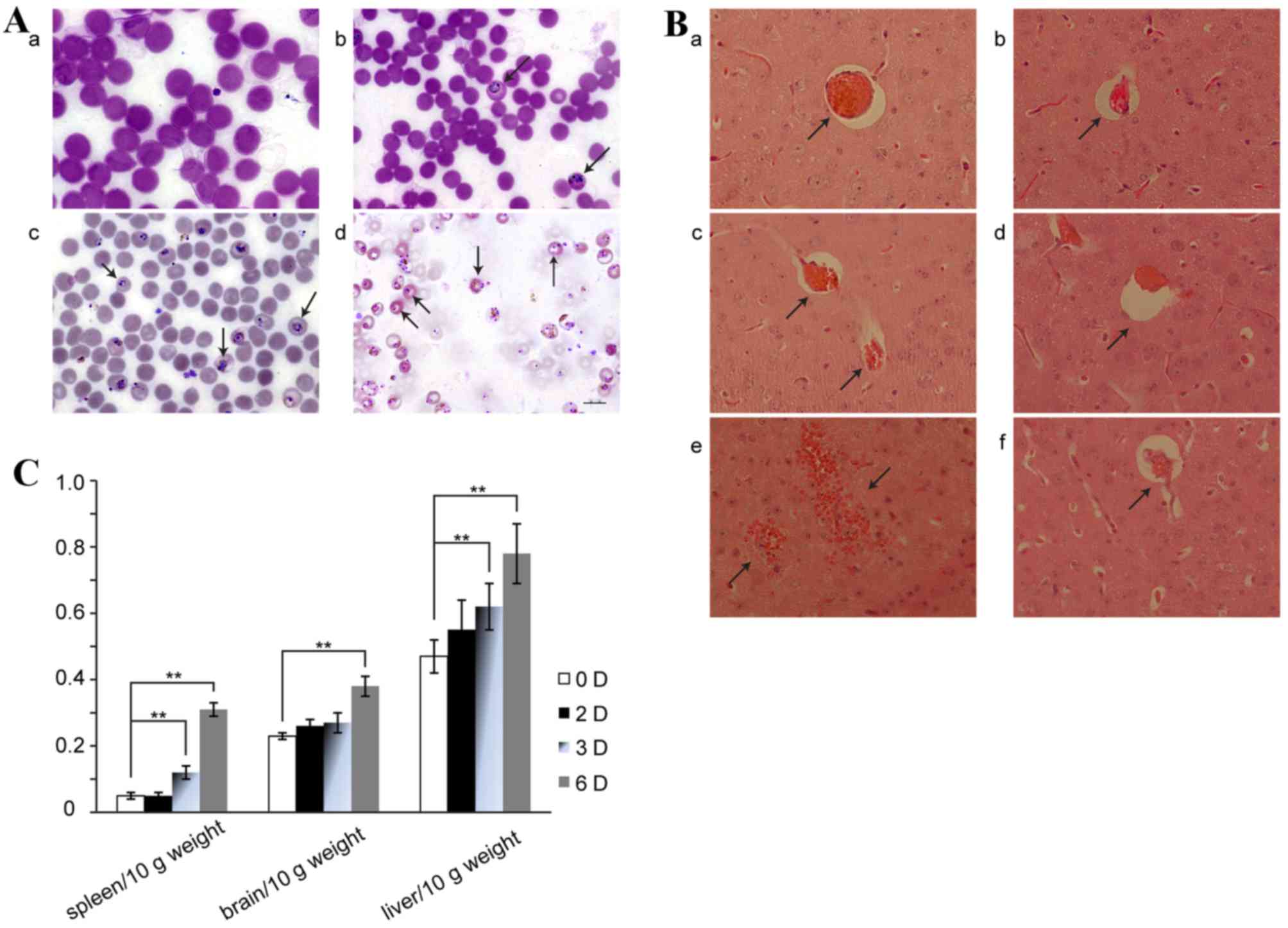 | Figure 1.Validation of the experimental
cerebral malaria model. (A) Results of Giemsa staining. Parasitized
red blood cells are indicated by black arrows. The number of
parasitized red blood cells increased with time. a, uninfected
group; b, D2+saline group; c, D3+saline group; d, D6+saline group.
(Scale bar=10 µm). (B) Cerebral micro-vessels as viewed by
hematoxylin and eosin staining. a, D2+saline; b, infected group
treated with NaHS (1 mg/kg) for 2 days; c, D3+saline; d, infected
group treated with NaHS (1 mg/kg) for 3 days; e, D6+saline; f,
infected group treated with NaHS (1 mg/kg) for 6 days. Black arrows
indicate hemorrhages or blood vessels. Magnification, ×40. (C)
Alterations in spleen, brain and liver indexes. Data are presented
as the mean ± standard error of the mean, n=10. 0 D, uninfected
group; 2 D, D2+saline; 3D, D3+saline; 6 D, D6+saline. D2+saline,
infected group treated with saline for 2 days; D3+saline, infected
group treated with saline for 3 days; D6+saline, infected group
treated with saline for 6 days. **P<0.01. |
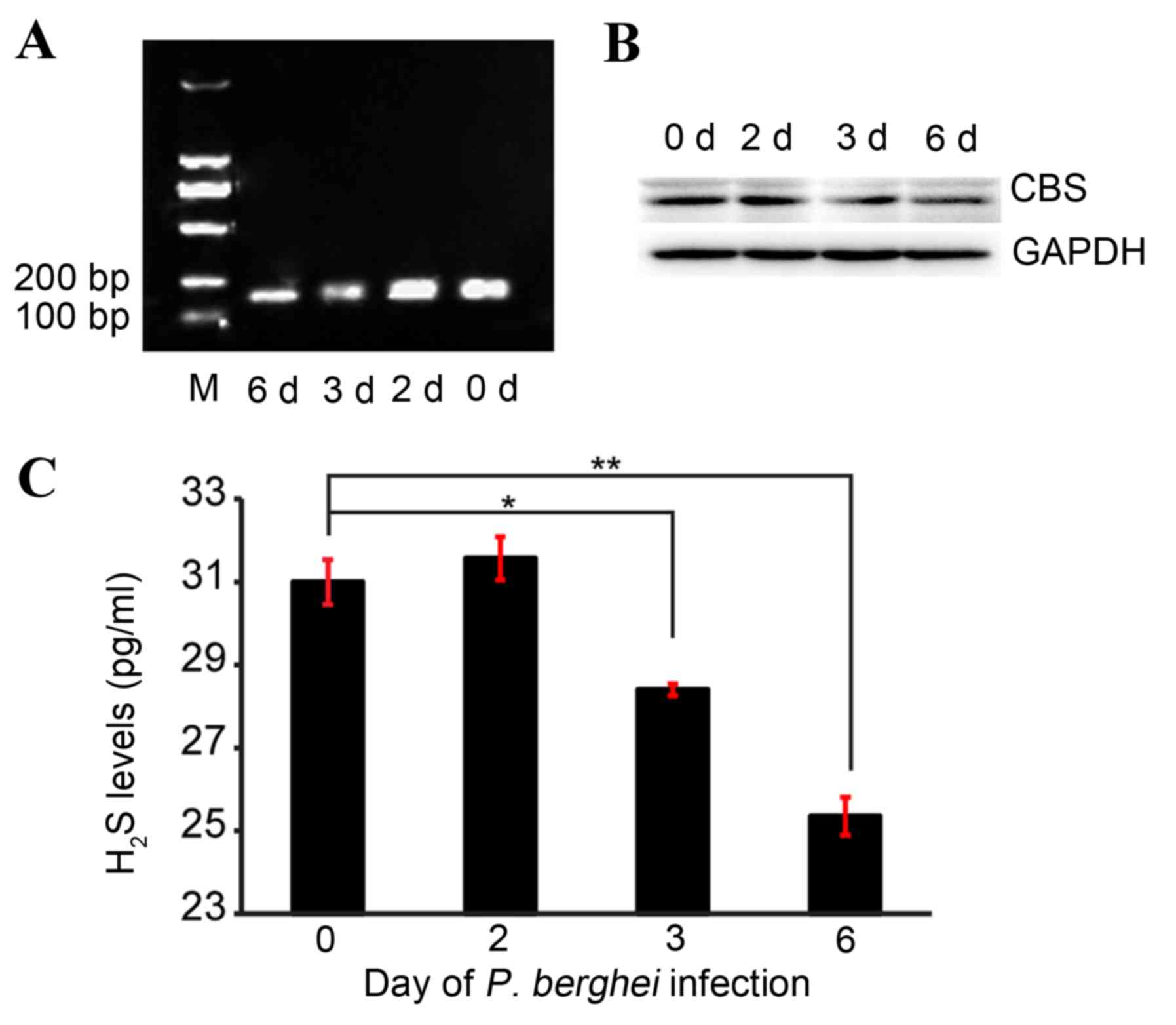
CBS levels of ECM
Alterations in CBS levels during ECM were assessed
using RT-PCR and western blotting. The RT-PCR results revealed that
CBS mRNA levels decreased gradually with saline treatment time
(Fig. 2A). Compared with day 0,
CBS mRNA expression levels visibly decreased following saline
treatment for 3 and 6 days (Fig.
2A). Consistently, CBS protein expression levels demonstrated
similar reduction patterns (Fig.
2B).
H2S levels of ECM
The concentration of H2S in the brain was
measured by ELISA. The levels of H2S in the brain
decreased gradually with saline treatment time, and were
significantly reduced on day 3 (28.40±0.14 vs. 31.00±0.54;
P<0.05; Fig. 2C) and day 6 of
saline treatment (25.35±0.46 vs. 31.00±0.54, P<0.05; Fig. 2C) compared with day 0.
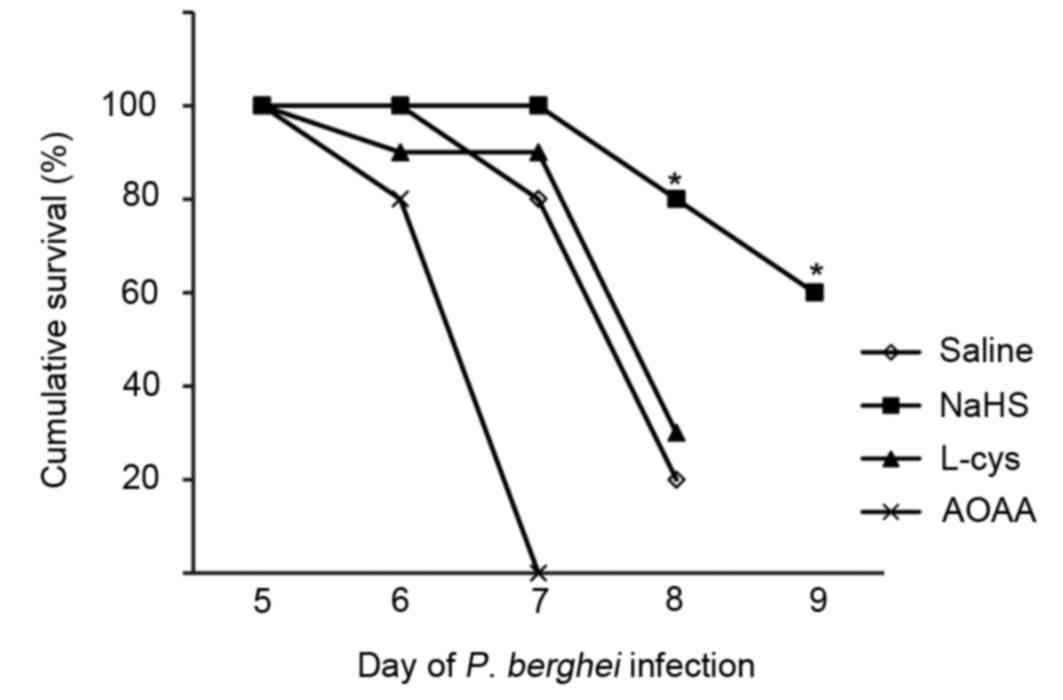
Survival of ECM
Following treatment with NaHS, L-cysteine or AOAA,
the survival of mice with ECM was evaluated by quantitative
assessment of coma and behavior scales of ECM. The results revealed
that only 2/10 saline-treated mice survived 8 days following
infection. Mice treated with NaHS survived significantly longer
than saline-treated mice; 8/10 NaHS-treated mice were alive at day
8 following infection. However, the mice treated with L-cys
demonstrated no significant difference from mice treated with
saline, and all mice treated with AOAA, a CBS inhibitor, died by
day 7 following infection. The difference of survival rate between
the saline and NaHS-treated groups at days 8 and 9 was significant
(P<0.05; Fig. 3).
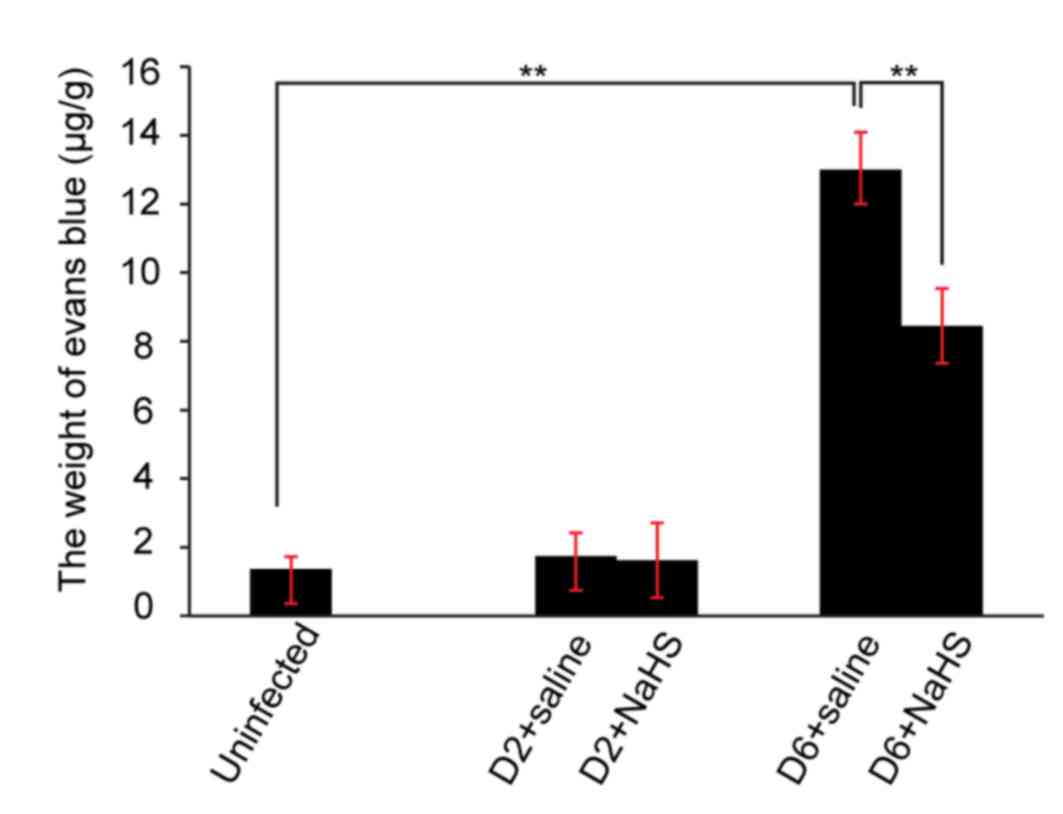
Integrity of the blood brain barrier
(BBB) during ECM
The integrity of the BBB was assessed by Evans blue
extravasation, H&E staining and measurement of MMP-9. On day 2
of ECM there was no significant increase in Evans blue
extravasation in the ECM brain (Fig.
4). In contrast, on day 6 of ECM, a significant increase in
Evans blue in the brains of saline-treated mice was observed
compared with uninfected mice (Fig.
4). Furthermore, administration of H2S donor
significantly decreased Evans blue extravasation into the brain
compared with saline-injected mice (n=10 per group, P<0.05;
Fig. 4).
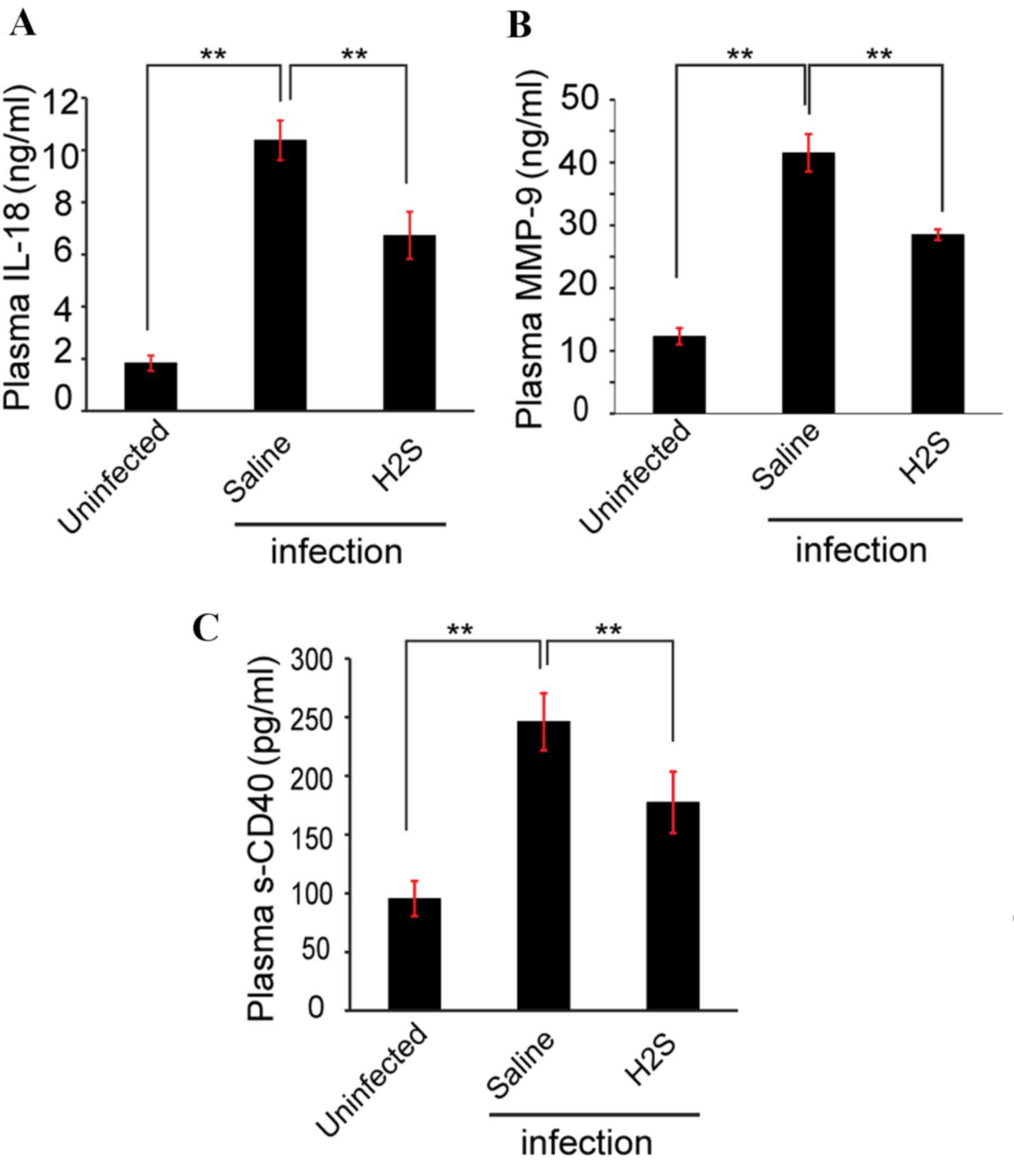
MMP-9 protein expression levels in the infection
group treated with saline (41.52±2.99 ng/ml) was ~3 times increased
compared with the uninfected group (12.32±1.30 ng/ml) as measured
by ELISA (Fig. 5A). Compared with
saline-treated group, MMP-9 protein levels in the NaHS-treated
group (28.48±0.87 ng/ml) were significantly decreased (P<0.05;
Fig. 5A).
Inflammation of ECM
ELISA was used to evaluate the concentration of
inflammatory factors in the plasma during ECM. On day 6, the levels
of the proinflammatory cytokine IL-18 (Fig. 5B) and the inflammatory marker sCD40
(Fig. 5C) were significantly
increased in the plasma of saline-treated mice compared with
uninfected mice, and administration of the H2S donor
significantly reduced the concentration of IL-18 and sCD40 compared
with saline-treated mice (P<0.05; Figs. 5B and C).
Discussion
The present study used a PbA-infected C57BL/6
mouse model to examine human cerebral malaria (13). Similar to the classical symptoms of
humans with cerebral malaria, the infected C57BL/6 mice also
manifest parasitemia, inflammation and vascular leak (16). Collectively, the findings of the
present study demonstrated that mice infected with PbA had
pathophysiological markers of ECM. Sequential increased amounts of
parasitized red blood cells indicated that successful invasion and
proliferation of malaria parasite occurred in the C57BL/6 mice.
Significant hemorrhaging into the brain was observed by H&E
staining on day 6 of ECM, suggesting that invasion and
proliferation of the malaria parasite resulted in disruption of the
BBB. In addition, the increased levels of inflammatory factors
(IL-18 and sCD40) indicated that inflammation occurred following
infection. Increased indexes of the spleen, brain and liver were
additionally observed in the infection group, demonstrating that
hepatomegaly and encephaledema appeared in the infected mice. These
impairments are also observed in humans infected with cerebral
malaria, providing support for the fact that this ECM model
accurately reflected the clinical pathophysiology of ECM.
H2S has been demonstrated by DellaValle
et al (17) to prevent
P. falciparum growth and metabolism in vitro, and the
presence of endogenous H2S in the human and rat brain
has been established (18).
However, the involvement of H2S in cerebral malaria
pathogenesis remains to be clarified. In order to evaluate whether
H2S is involved in ECM development, H2S
concentration was tested by ELISA. Consistent with previous
reports, the levels of H2S derived from the brain
gradually decreased with time. For instance, patients with coronary
heart disease have significantly decreased plasma sulphide levels,
from 50 to 25 µM (19). It is
possible to mimic the decrease of plasma sulphide levels in rodent
models, whereby spontaneously hypertensive rats (20 µM) have
substantially lower plasma sulphide levels than normotensive
control Wistar Kyoto rats (48 µM) (20). The present study reported that
decreased H2S in the brain may be involved in the
development of ECM.
However, there are no data that explain how
H2S is decreased in the brain following infection with
PbA. It is well known that endogenous H2S is
generated from L-cysteine by three enzymes: CBS,
cystathionine-synthase and 3-mercaptopyruvate sulfurtransferase. In
the brain, the production of endogenous H2S is initially
attributed to the actions of CBS (21–23).
RT-PCR and western blotting were used to determine CBS mRNA and
protein levels, respectively. RT-PCR analysis indicated that the
level of CBS mRNA in the brain progressively decreased following
infection with PbA. This decrease in transcriptional
regulation additionally manifested at the CBS protein level.
Collectively, these results suggested that low H2S
bioavailability contributed to the development of ECM, that the
primary reason for reduced H2S concentrations during ECM
may be inhibition of CBS transcription and translation caused by
PbA infection, and that the beginning of inhibition occurs
three days following infection.
As low levels of H2S in the brain
contributes to the progression of ECM, it was hypothesized that
administration of H2S donors may provide protection
against ECM. NaHS was used as an exogenous donor of H2S,
L-cysteine was used as an endogenous donor of H2S and
AOAA was used as an inhibitor of CBS, which inhibits endogenous
H2S generation from L-cysteine. Quantitative assessment
using a rapid murine coma and behavior scale was performed to
evaluate the survival rate of ECM. The NaHS-treated groups
demonstrated a longer survival time than the saline or
L-cysteine-treated group, and the L-cysteine treated group had a
similar survival time as the saline-treated group, indicating that
the exogenous H2S donor NaHS provided protection against
PbA-induced death, but the endogenous H2S donor
L-cysteine did not protect against PbA infection. These
findings provided evidence to demonstrated that the ability of
endogenous H2S generation from L-cysteine is deficient
during ECM, as confirmed by the decreased levels of CBS measured by
RT-PCR and western blotting. Furthermore, the AOAA treated group
had the same survival time as the saline-treated group and a
shorter survival time than the NaHS-treated group, which further
confirmed the involvement of H2S in the development of
ECM.
For two main reasons, the present study aimed to
further investigate the effects of H2S on inflammation.
First, it is well known that P. falciparum erythrocyte
membrane protein 1 is a parasite protein that is expressed on the
infected erythrocyte surface and mediates parasite binding to
receptors, including CD36 and intercellular adhesion molecule-1
(16,24–26).
This leads to conglutination of infected erythrocytes on the brain
micro-vascular endothelial cells and accompanying inflammation.
Furthermore, the sequestration of parasitized erythrocytes within
the brain blood vessels is worsened by pro-inflammatory responses
(27). Activated T cell-mediated
immune responses have been demonstrated to be involved in the
generation of cerebral malaria (28–30).
Secondly, H2S decreases inflammation and
leukocyte-endothelial cell interactions in vivo via
inhibition of leukocyte rolling and adhesion, as well as ensuing
diapedesis (8). The present study
used ELISA to investigate whether administration of the
H2S donor decreased proinflammatory biomarkers during
ECM. Compared with saline-treated mice, the observed decreases of
IL-18 (a proinflammatory cytokine) and sCD40 (an inflammatory
marker) in NaHS-treated mice on day 6 of ECM suggested that
H2S donor administration reduced inflammation of
ECM.
Notably, the high mortality of ECM is mainly
attributed to the sequestration of infected red blood cells in the
endothelial cells, which further disrupts the integrity of the BBB
(31–34), with flaws in the BBB being
associated with the advance of cerebral disease (34,35).
Three methods were used by the present study to assess the
integrity of the BBB during ECM: H&E staining of brain tissue
to test cerebrovascular hemorrhage, extravasation of Evans blue in
the brain to test cerebrovascular permeability, and levels of MMP-9
in the plasma, which is a protease that contributes to the
disruption of endothelial barrier integrity (36). Compared with the saline-treated
group, decreased extravasation of Evans blue, and reduced and
hemorrhage and MMP-9 levels, were observed in the NaHS
administration group, indicating that NaHS protected against
disruption of the BBB induced by PbA infection. The
underlying mechanisms by which H2S protected against
disruption of the BBB merit further investigation.
In conclusion, the present study demonstrated that
low levels of brain H2S contributes to the progression
of ECM. Furthermore, administration of an exogenous H2S
donor protected against disruption of the BBB induced by PbA
infection, and decreased proinflammatory biomarkers in the brain.
Therefore, exogenous H2S may be a useful complementary
therapy for the treatment of ECM.
References
|
1
|
Mung'Ala-Odera V, Snow RW and Newton CR:
The burden of the neurocognitive impairment associated with
Plasmodium falciparum malaria in sub-saharan Africa. Am J Trop Med
Hyg. 71 2 Suppl:S64–S70. 2004.
|
|
2
|
Van Hensbroek MB, Palmer A, Jaffar S,
Schneider G and Kwiatkowski D: Residual neurologic sequelae after
childhood cerebral malaria. J Pediatr. 131:125–129. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wallace JL, Blackler RW, Chan MV, Da Silva
GJ, Elsheikh W, Flannigan KL, Gamaniek I, Manko A, Wang L, Motta JP
and Buret AG: Anti-inflammatory and cytoprotective actions of
hydrogen sulfide: Translation to therapeutics. Antioxid Redox
Signal. 22:398–410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wallace JL, Caliendo G, Santagada V,
Cirino G and Fiorucci S: Gastrointestinal safety and
anti-inflammatory effects of a hydrogen sulfide-releasing
diclofenac derivative in the rat. Gastroenterology. 132:261–271.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang C, Yang Z, Zhang M, Dong Q, Wang X,
Lan A, Zeng F, Chen P, Wang C and Feng J: Hydrogen sulfide protects
against chemical hypoxia-induced cytotoxicity and inflammation in
HaCaT cells through inhibition of ROS/NF-κB/COX-2 pathway. PLoS
One. 6:e219712011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Rossoni G, Sparatore A, Lee LC, Del
Soldato P and Moore PK: Anti-inflammatory and gastrointestinal
effects of a novel diclofenac derivative. Free Radic Biol Med.
42:706–719. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wallace JL: Hydrogen sulfide-releasing
anti-inflammatory drugs. Trends Pharmacol Sci. 28:501–505. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zanardo RC, Brancaleone V, Distrutti E,
Fiorucci S, Cirino G and Wallace JL: Hydrogen sulfide is an
endogenous modulator of leukocyte-mediated inflammation. FASEB J.
20:2118–2120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kimura H: Hydrogen sulfide induces cyclic
AMP and modulates the NMDA receptor. Biochem Biophys Res Commun.
267:129–133. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kimura H: Hydrogen sulfide as a
neuromodulator. Mol Neurobiol. 26:13–19. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Whiteman M, Cheung NS, Zhu YZ, Chu SH,
Siau JL, Wong BS, Armstrong JS and Moore PK: Hydrogen sulphide: A
novel inhibitor of hypochlorous acid-mediated oxidative damage in
the brain? Biochem Biophys Res Commun. 326:794–798. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Whiteman M, Li L, Rose P, Tan CH,
Parkinson DB and Moore PK: The effect of hydrogen sulfide donors on
lipopolysaccharide-induced formation of inflammatory mediators in
macrophages. Antioxid Redox Signal. 12:1147–1154. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
de Oca MM, Engwerda C and Haque A:
Plasmodium berghei ANKA (PbA) infection of C57BL/6J mice: A model
of severe malaria. Methods Mol Biol. 1031:203–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Carroll RW, Wainwright MS, Kim KY, Kidambi
T, Gómez ND, Taylor T and Haldar K: A rapid murine coma and
behavior scale for quantitative assessment of murine cerebral
malaria. PLoS One. 5:pii: e131242010. View Article : Google Scholar
|
|
15
|
Baccarella A, Huang BW, Fontana MF and Kim
CC: Loss of Toll-like receptor 7 alters cytokine production and
protects against experimental cerebral malaria. Malar J.
13:3542014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang WL, Jones SP, Lefer DJ, Welbourne T,
Sun G, Yin L, Suzuki H, Huang J, Granger DN and van der Heyde HC:
CD8(+)-T-cell depletion ameliorates circulatory shock in Plasmodium
berghei-infected mice. Infect Immun. 69:7341–7348. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
DellaValle B, Staalsoe T, Kurtzhals JA and
Hempel C: Investigation of hydrogen sulfide gas as a treatment
against P. falciparum, murine cerebral malaria, and the importance
of thiolation state in the development of cerebral malaria. PLoS
One. 8:e592712013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goodwin LR, Francom D, Dieken FP, Taylor
JD, Warenycia MW, Reiffenstein RJ and Dowling G: Determination of
sulfide in brain tissue by gas dialysis/ion chromatography:
Postmortem studies and two case reports. J Anal Toxicol.
13:105–109. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang HL, Wu HC, Li ZL, Geng B and Tang
CS: Changes of the new gaseous transmitter H2S in patients with
coronary heart disease. Di Yi Jun Yi Da Xue Xue Bao. 25:951–954.
2005.(In Chinese). PubMed/NCBI
|
|
20
|
Du J, Yan H and Tang C: Endogenous H2S is
involved in the development of spontaneous hypertension. Beijing Da
Xue Xue Bao. 35:1022003.(In Chinese). PubMed/NCBI
|
|
21
|
Porter PN, Grishaver MS and Jones OW:
Characterization of human cystathionine beta-synthase. Evidence for
the identity of human L-serine dehydratase and cystathionine
beta-synthase. Biochim Biophys Acta. 364:128–139. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ichinohe A, Kanaumi T, Takashima S,
Enokido Y, Nagai Y and Kimura H: Cystathionine beta-synthase is
enriched in the brains of Down's patients. Biochem Biophys Res
Commun. 338:1547–1550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Enokido Y, Suzuki E, Iwasawa K, Namekata
K, Okazawa H and Kimura H: Cystathionine beta-synthase, a key
enzyme for homocysteine metabolism, is preferentially expressed in
the radial glia/astrocyte lineage of developing mouse CNS. FASEB J.
19:1854–1856. 2005.PubMed/NCBI
|
|
24
|
van der Heyde HC, Nolan J, Combes V,
Gramaglia I and Grau GE: A unified hypothesis for the genesis of
cerebral malaria: Sequestration, inflammation and hemostasis
leading to microcirculatory dysfunction. Trends Parasitol.
22:503–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Razakandrainibe R, Pelleau S, Grau GE and
Jambou R: Antigen presentation by endothelial cells: What role in
the pathophysiology of malaria? Trends Parasitol. 28:151–160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Franke-Fayard B, Janse CJ, Cunha-Rodrigues
M, Ramesar J, Büscher P, Que I, Löwik C, Voshol PJ, den Boer MA,
van Duinen SG, et al: Murine malaria parasite sequestration: CD36
is the major receptor, but cerebral pathology is unlinked to
sequestration. Proc Natl Acad Sci USA. 102:11468–11473. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nie CQ, Bernard NJ, Schofield L and Hansen
DS: CD4+ CD25+ regulatory T cells suppress CD4+ T-cell function and
inhibit the development of Plasmodium berghei-specific TH1
responses involved in cerebral malaria pathogenesis. Infect Immun.
75:2275–2282. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hearn J, Rayment N, Landon DN, Katz DR and
de Souza JB: Immunopathology of cerebral malaria: Morphological
evidence of parasite sequestration in murine brain
microvasculature. Infect Immun. 68:5364–5376. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nitcheu J, Bonduelle O, Combadiere C,
Tefit M, Seilhean D, Mazier D and Combadiere B: Perforin-dependent
brain-infiltrating cytotoxic CD8+ T lymphocytes mediate
experimental cerebral malaria pathogenesis. J Immunol.
170:2221–2228. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pais TF and Chatterjee S: Brain macrophage
activation in murine cerebral malaria precedes accumulation of
leukocytes and CD8+ T cell proliferation. J Neuroimmunol.
163:73–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Miller LH, Ackerman HC, Su XZ and Wellems
TE: Malaria biology and disease pathogenesis: Insights for new
treatments. Nat Med. 19:156–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wautier JL and Wautier MP: Molecular basis
of erythrocyte adhesion to endothelial cells in diseases. Clin
Hemorheol Microcirc. 53:11–21. 2013.PubMed/NCBI
|
|
33
|
Combes V, El-Assaad F, Faille D, Jambou R,
Hunt NH and Grau GE: Microvesiculation and cell interactions at the
brain-endothelial interface in cerebral malaria pathogenesis. Prog
Neurobiol. 91:140–151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Medana IM and Turner GD: Human cerebral
malaria and the blood-brain barrier. Int J Parasitol. 36:555–568.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brown H, Hien TT, Day N, Mai NT, Chuong
LV, Chau TT, Loc PP, Phu NH, Bethell D, Farrar J, et al: Evidence
of blood-brain barrier dysfunction in human cerebral malaria.
Neuropathol Appl Neurobiol. 25:331–340. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zheng M, Wei J, Tang Y, Yang C, Wei Y, Yin
X and Liu Q: ApoE-deficient promotes blood-brain barrier disruption
in experimental autoimmune encephalomyelitis via alteration of
MMP-9. J Mol Neurosci. 54:282–290. 2014. View Article : Google Scholar : PubMed/NCBI
|