Introduction
Retinol binding protein 4 (RBP4) is a member of the
lipocalin family and is synthesized in the liver and adipose tissue
as a retinol translocator (1,2).
Abnormal metabolism of RBP4 can lead to disorders in the synthesis,
transport and distribution of retinol, and affects embryonic growth
and bone tissue differentiation (3–5).
RBP4 has been demonstrated to be significantly decreased in
children with non-syndromic cleft lip and/or palate (NSCLP)
(6), suggesting that RBP4 levels
are associated with the occurrence of NSCLP. Cleft face and palate
have been observed in RBP knockout fetuses in animal models, along
with obvious cranial and maxillofacial bone defects (7). These studies have indicated that RBP4
serves an important role in embryonic development, particularly in
craniofacial growth. It is associated with cleft palate, in which
the absence of RBP4 can lead to cleft palate.
Cleft palate resulting from multigene and
environmental factors is a common malformation (8,9). The
formation of the secondary palate is a critical process in
palatogenesis, including the growth, elevation and fusion of the
palatal shelf. Failed elevation of the palatal processes is widely
considered to be one of the primary causes of cleft palate and is
attributed to growth inhibition of the embryonic palatal mesenchyme
(EPM) (10). During the early
phase of palatogenesis, large numbers of EPM cells proliferate for
growth and elevation. When the proliferation of EPM cells is
suppressed, the palatal processes become dysplastic and fail to
elevate in an appropriate time. Palatal shelves cannot complete the
contact and fusion, resulting in cleft palate.
As a derivative of vitamin A, all-trans retinoic
acid (atRA) is involved in the differentiation, proliferation and
apoptosis of palatogenesis (11).
Previous studies have confirmed that atRA inhibits the
proliferation and induces cell cycle arrest in EPM cells to cause
palatal dysplasia and the development of cleft palate (12,13).
Previous studies have demonstrated that atRA suppresses the
expression of RBP4 in adipose tissues and liver (14). However, the effect of atRA on RBP4
in the embryonic palate requires further elucidation. The present
study aimed to evaluate the function of RBP4 in the development of
the atRA-induced cleft palate.
Materials and methods
Animals and atRA treatment
All animal experiments were approved by the Ethics
Committee for Animal Experiments of Sun Yat-sen University (IACUC:
DB-15-0302). A total of 40 male and 40 female mice were used in the
present study. The animals were housed at 22°C with a 12-h
light/dark cycle, and given pellet food and tap water ad
libitum. Specific pathogen-free C57BL/6J male mice aged 7–8
weeks (weight, 22–25 g) and female mice aged 4–5 weeks (weight,
14–18 g) were mated. Day 0 was defined as the presence of a vaginal
plug. At embryonic day 10 (E10.0), 20 pregnant mice in the
experimental group were administered atRA (100 mg/kg;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) in corn oil by
gavage, and 20 pregnant mice in the control group received the same
volume of corn oil. The mice were euthanized on E12.5, E13.5 and
E14.5 by cervical dislocation to obtain embryonic palates.
Immunohistochemistry
A total of 58 embryonic palates were fixed in 4%
paraformaldehyde for 48 h and dehydrated using an ethanol series.
Next, they were embedded in paraffin in the coronal orientation.
The paraffin blocks were sliced into 4 µm sections. Every section
was deparaffinized and hydrated using an ethanol series. The
sections were retrieved by applying citric acid buffer (pH 6.0) for
30 min in a microwave for antigen retrieval, followed by incubation
with anti-RBP4 (cat. no. ab109193; 1:200; Abcam, Cambridge, UK) for
18 h at 4°C. The sections were washed with phosphate-buffered
saline (PBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and incubated with secondary Ab-BIO (OriGene Technologies,
Inc., Beijing, China) at 37°C for 1 h. Staining was performed with
3,3′-diaminobenzidine (DAB; OriGene Technologies, Inc.), and the
presence of brown granules was considered to indicate positive
staining. Subsequently, the sections were stained with hematoxylin
for 3 sec. Images were obtained using a Zeiss Axioskop 40 (Leica
Microsystems GmbH, Wetzlar, Germany).
Cell culture and treatment with
atRA
Human embryonic palatal mesenchymal (HEPM) cells
were purchased from the American Type Culture Collection (Manassas,
VA, USA). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco, Carlsbad, CA) containing 10% fetal bovine
serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.) in
a 5% CO2 atmosphere at 37°C. To study the effects of
atRA, the cells were exposed to different concentrations of atRA (1
and 3 µM). There were 3 duplicate cultures in each group, and
experiments were performed in triplicate.
Gene silencing using small interfering
RNA (siRNA)
HEPM cells were cultured in 6-well plates
(1×104 cells/well) in DMEM containing 10% FBS. When the
HEPM cells had reached 70% confluency, they were transfected with a
final concentration of 25 pmol RBP4 siRNA (Guangzhou RiboBio Co.,
Ltd., Guangzhou, China) using Lipofectamine RNAiMAX Reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) for 72 h according to
the manufacturer's instructions. An irrelevant siRNA (Guangzhou
RiboBio Co., Ltd.) was used as the control. A total of 3 duplicate
samples were used in each group, and experiments were performed in
triplicate. These cells were collected for analysis of the
associated mRNA and protein expression. Reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting were be used to verify the knockdown efficiency of
the target gene.
Overexpression of RBP4
RBP4 protein was overexpressed in HEPM cells to
assess the function of RBP4 in the palatal mesenchyme according to
a standard transient transfection protocol. The full-length RBP4
gene was cloned into pEZ-M98-GFP (iGeneBio Biotechnology Co., Ltd.,
Guangzhou, China) for the experiments. Cells transfected with
pEZ-M98-GFP (without RBP4) were used as the control group. The HEPM
cells were seeded into 6-well plates (1×105 cells/well)
in DMEM containing 10% FBS. When the HEPM cells had reached 70%
confluency, they were transfected with the above-mentioned
constructs in Opti-MEM medium (Gibco; Thermo Fisher Scientific,
Inc.), and Lipofectamine 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) was used for transfecting nucleic acids (2500
ng/well) into cells according to standard procedures. Following
transfection for 48 h, the medium was refreshed, and the cells were
exposed to atRA (3 µM) for 48 h. A total of 3 duplicate samples
were used in each group, and experiments were performed in
triplicate. The efficiency of the target gene overexpression was
evaluated by western blotting.
RT-qPCR
Total RNA was extracted from mouse embryonic palates
(60 in each group) or HEPM cells using a Qiagen RNeasy Mini kit
(Qiagen GmbH, Hilden, Germany) according to the instructions
provided by the manufacturer. A total of ~20 µl cDNA was
synthesized from 1 µg of total RNA in a 20 µl reaction mixture. The
reaction mixture included total RNA, Anchored-oligo (dT)18 Primer,
Random Hexamer Primer, PCR-grade H2O, Transcriptor RT
Reaction Buffer (5X), Protector RNase Inhibitor, Deoxynucleotide
Mix and Transcriptor Reverse Transcriptase (all Roche Diagnostics
GmbH, Mannheim, Germany). RT-qPCR was conducted using the Light
Cycler 480 System (Roche Diagnostics GmbH) with SYBR Green I Master
Mix (Roche Diagnostics GmbH) to quantitatively measure the mRNA
levels. β-actin and GAPDH were used as reference genes. The
following PCR primers (Generay Biotech Co., Ltd., Shanghai, China)
were used in the experiments: RBP4 (Mus), forward
5′-GACACGGACTACGACACC-3′ and reverse 5′-CACGAGAAAACACAAAGGA-3′;
β-actin (Mus), forward 5′-TCACCCACACTGTGCCCATCTACGA-3′ and reverse
5′-GGATGCCACAGGATTCCATACCCA-3′; RBP4 (homo), forward
5′-GAGGACCCTGCCAAGTTCA-3′ and reverse 5′-GGGAAAACACGAAGGAGTAGC-3′;
P27 (homo), forward 5′-CAAACGTGCGAGTGTCTA-3′ and reverse
5′-CAGTGCTTCTCCAAGTCC-3′; cyclin D1 (homo), forward
5′-TCCTACTACCGCCTCACA-3′ and reverse 5′-ACCTCCTCCTCCTCCTCT-3′;
GAPDH (homo), forward 5′-GGACCTGACCTGCCGTCTAG-3′ and reverse
5′-GTAGCCCAGGATGCCCTTGA-3′.
Western blot analysis
60 mouse embryonic palates in each group were
treated with 1 U/ml dispase II (Roche Diagnostics, Indianapolis,
IN, USA) for 15 min at 37°C to remove epithelia and obtain EPM at
E12.5, E13.5 and E14.5. The palatal mesenchyme was flushed with
cold PBS three times and lysed in cell lysis buffer (10% RIPA, 1%
protease inhibitor, 1% phosphatase inhibitor; Sigma-Aldrich; Merck
Millipore) on ice for 30 min. The protein was collected after the
lysate solution was centrifuged at 4°C and 14,000 × g for 30 min.
The protein (30 µg) was separated by 10% SDS-PAGE (CWBIO, Beijing,
China) and transferred onto polyvinylidene difluoride membranes.
The membrane was blocked in 5% bovine serum albumin (Beyotime
Institute of Biotechnology, Shanghai, China) for 1 h and incubated
overnight at 4°C with the following antibodies: Rabbit monoclonal
anti-RBP4 (cat. no. ab109193; 1:1,000; Abcam), rabbit monoclonal
anti-p27 (cat. no. 3688s; 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA) and rabbit monoclonal anti-cyclin D1 (cat. no.
ab134175; 1:2,000; Abcam) antibodies. The membrane was incubated
with horseradish peroxidase-coupled secondary antibodies for 1 h
following three washes with Tris-buffered saline/Tween 20 (CWBIO).
The membrane was detected using chemiluminescence with Immobilon
western chemiluminescent horseradish-peroxidase substrate (EMD
Millipore, Billerica, MA, USA).
Protein derived from cultured HEPM cells was also
detected by western blotting. The following steps were repeated as
described previously (15). In
addition to the antibodies mentioned above, the following
antibodies were used: Rabbit monoclonal anti-extracellular
signal-related kinase (ERK) 1/2 (cat. no. 4695s; 1:1,000; Cell
Signaling Technology, Inc.), rabbit monoclonal anti-phosphorylated
(p)-ERK1/2 (cat. no. 4370s; Thr202/Tyr204; 1:2,000; Cell Signaling
Technology, Inc.), rabbit monoclonal anti-AKT (cat. no. 4691s;
1:1,000; Cell Signaling Technology, Inc.), rabbit monoclonal
anti-p-AKT (cat. no. 4060s; Ser473, 1:1,000; Cell Signaling
Technology, Inc.), rabbit monoclonal anti-proliferating cell
nuclear antigen (PCNA) (cat. no. 13110s; 1:1,000; Cell Signaling
Technology, Inc.).
Cell counting kit-8 (CCK8) assay
HEPM cells were cultured in a 96-well plate (5,000
cells/well) in DMEM at 37°C for 24 h. The medium was then replaced
with fresh DMEM, and the cultures were exposed to atRA (3 µM) or
the transfection complex. Following 24, 48, 72 and 96 h, 10 µl CCK8
(Dojindo Molecular Technologies, Inc., Tokyo, Japan) was added to
each well for 2 h at 37°C. The absorbance was measured using a
microplate reader at 450 nm.
Statistical analysis
All investigations, including individual
experiments, were performed in triplicate. All data were analyzed
using SPSS software, version 13.0 (SPSS, Inc., Chicago, IL, USA)
with the Student's t-test, or one way analysis of variance followed
by the Bonferroni post hoc test to correct for multiple
comparisons. The results are presented as the mean values ±
standard deviation, and P<0.05 was considered to indicate a
statistically significant difference.
Results
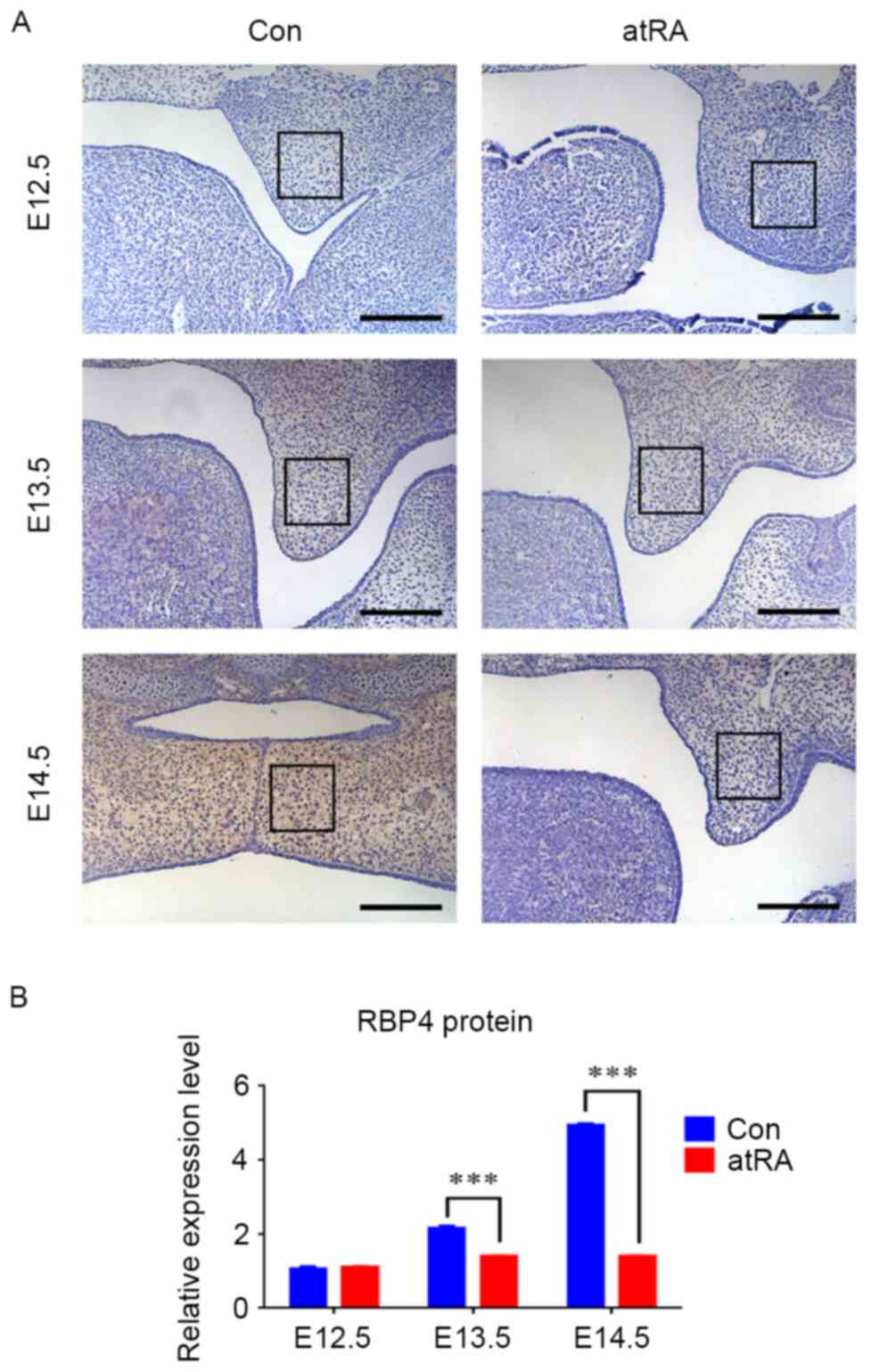
RBP4 was downregulated in the EPM of
cleft palate induced by atRA
At E12.5, the palate grew vertically along the side
of the tongue, and the expression of RBP4 was negative without
apparent differences in the EPM in either group (Fig. 1). At E13.5, the palatal shelves
still grew in a vertical position alongside the tongue, and they
appeared smaller in the atRA-treated group than that in the control
group (Fig. 1A). There were more
RBP4-positive cells in the EPM in the control group compared with
the atRA group (Fig. 1;
P<0.001). At E14.5, the bilateral palates shifted toward a
horizontal position to contact one another in the control group.
During this period, the medial edge epithelium was contacted to
develop into the medial edge epithelial seam, and finally palate
shelves fused. The expression of RBP4 was strongly positive in the
EPM (Fig. 1A). By contrast, the
palatal shelves appeared abnormally small and failed to undergo
elevation and fusion, finally developing into cleft palate in the
atRA-treated group at E14.5 (Fig.
1A). The incidence of cleft palate in the atRA-exposed groups
was 96.3% (78 of 81 embryos). RBP4 was significantly downregulated
in the EPM in atRA treatment compared with the control group
(Fig. 1; P<0.001). The RT-qPCR
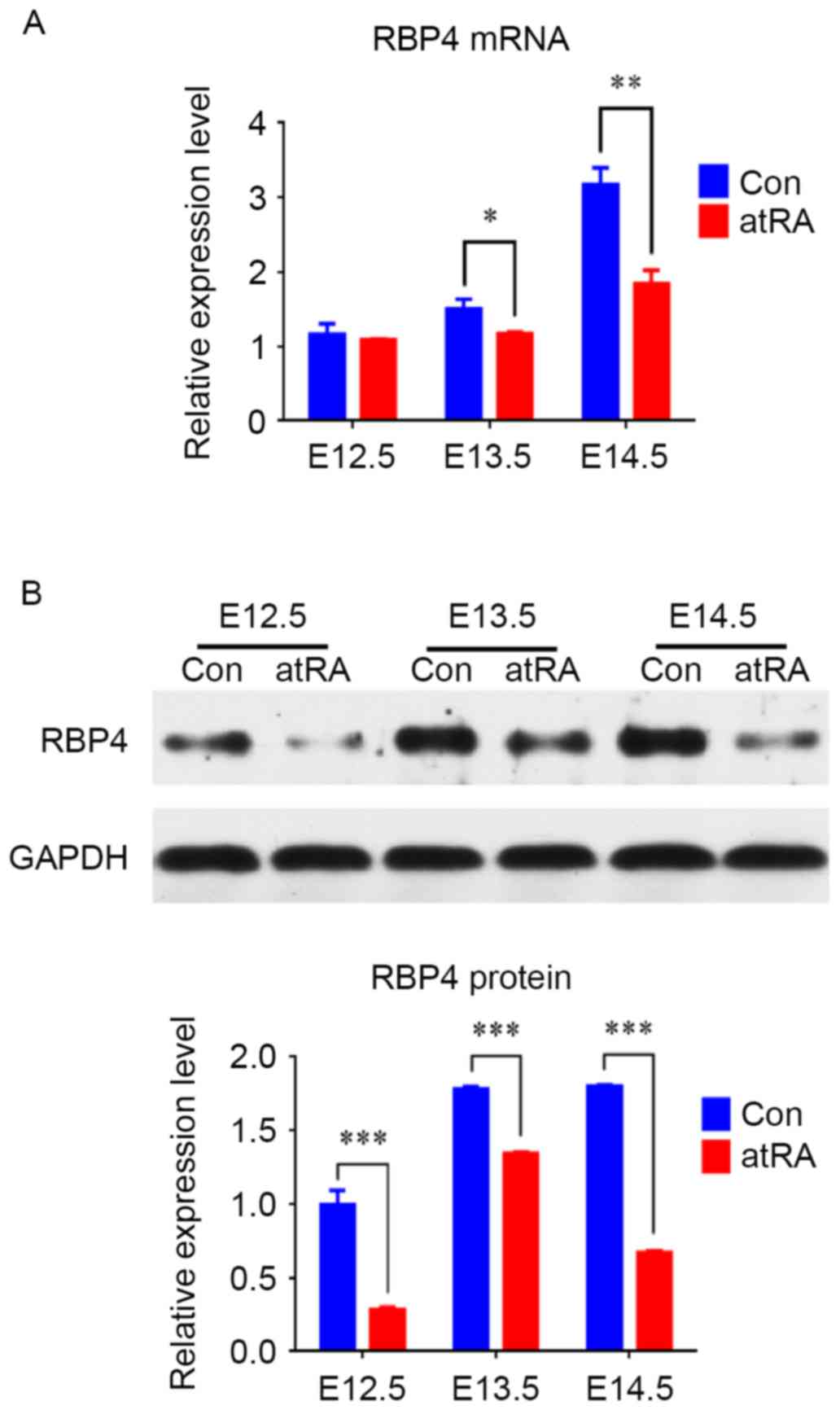
results indicated that the mRNA levels of RBP4 were reduced at
E13.5 (P<0.05) and downregulated at E14.5 in the atRA compared
with the control group (Fig. 2A;
P<0.01). Western blotting results identified that from E12.5 to
E14.5, the protein levels of RBP4 were significantly reduced in the
atRA-exposed vs. the control EPM, particularly at E14.5 (Fig. 2B; P<0.001).
atRA induced RBP4 downregulation and
regulated the expression of p27, cyclin D1, PCNA and the associated
signaling pathway
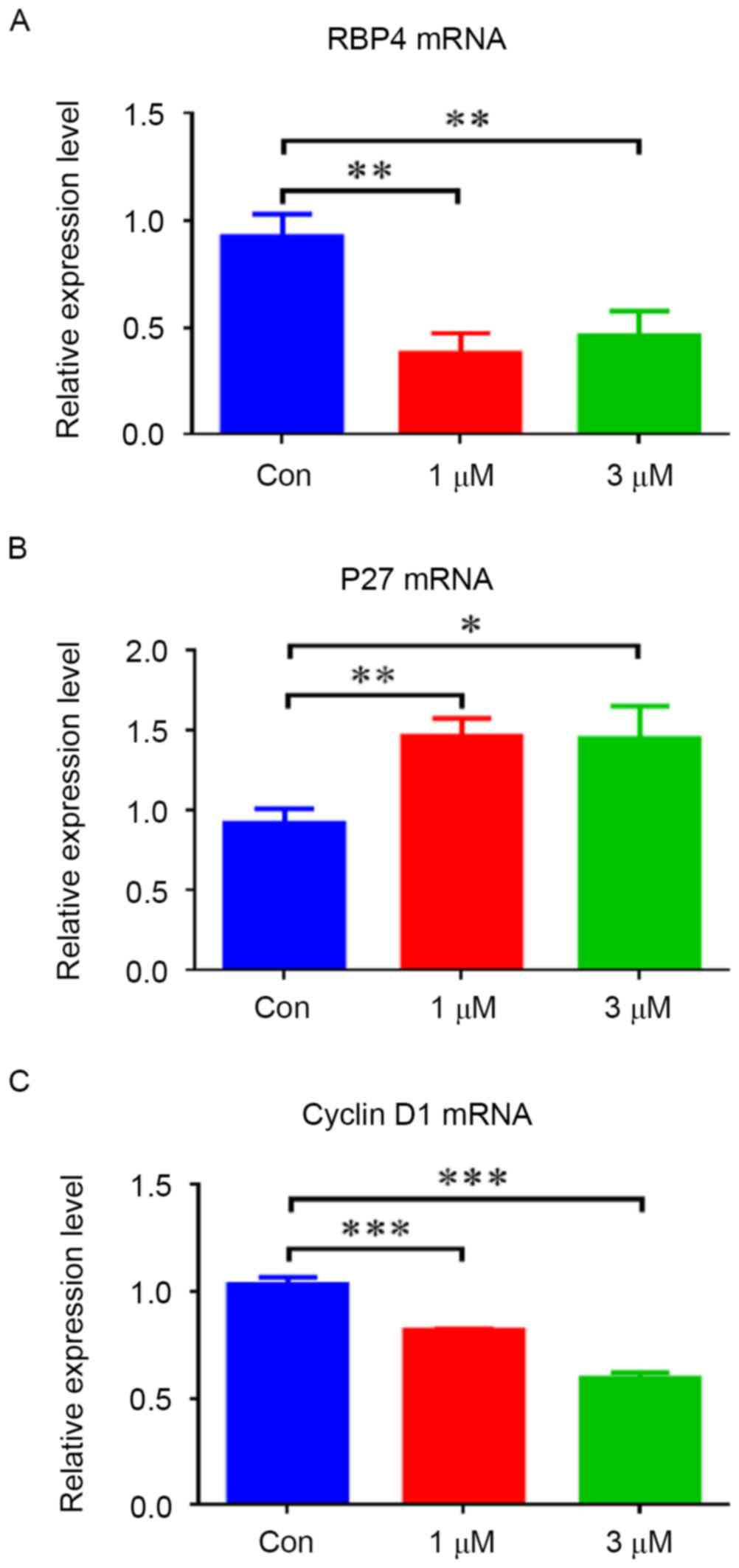
To evaluate the effect of atRA on RBP4, HEPM cells
were exposed to different concentrations of atRA (1 and 3 µM). The
results indicated that the mRNA levels of RBP4 were decreased in
HEPM cells treated with atRA (1 and 3 µM) (Fig. 3A; P<0.01). The protein levels of
RBP4 were also decreased in the atRA-treated groups compared with
the controls (Fig. 4; P<0.001).
In contrast, elevated p27 mRNA levels (Fig. 3B; P<0.05) and protein levels
(Fig. 4; P<0.001) were observed
in HEPM cells exposed to atRA (1 and 3 µM). Cyclin D1 mRNA and
protein levels were suppressed in the atRA-exposed groups compared
with the control group (Figs. 3C
and 4; P<0.001). The protein
levels of PCNA were reduced in the atRA-exposed groups compared
with the controls (Fig. 4).
Western blotting results identified that p-ERK1/2 was reduced in
atRA-treated cells (Fig. 4;
P<0.01), and p-AKT was also reduced compared with the control
group (Fig. 4; P<0.001).
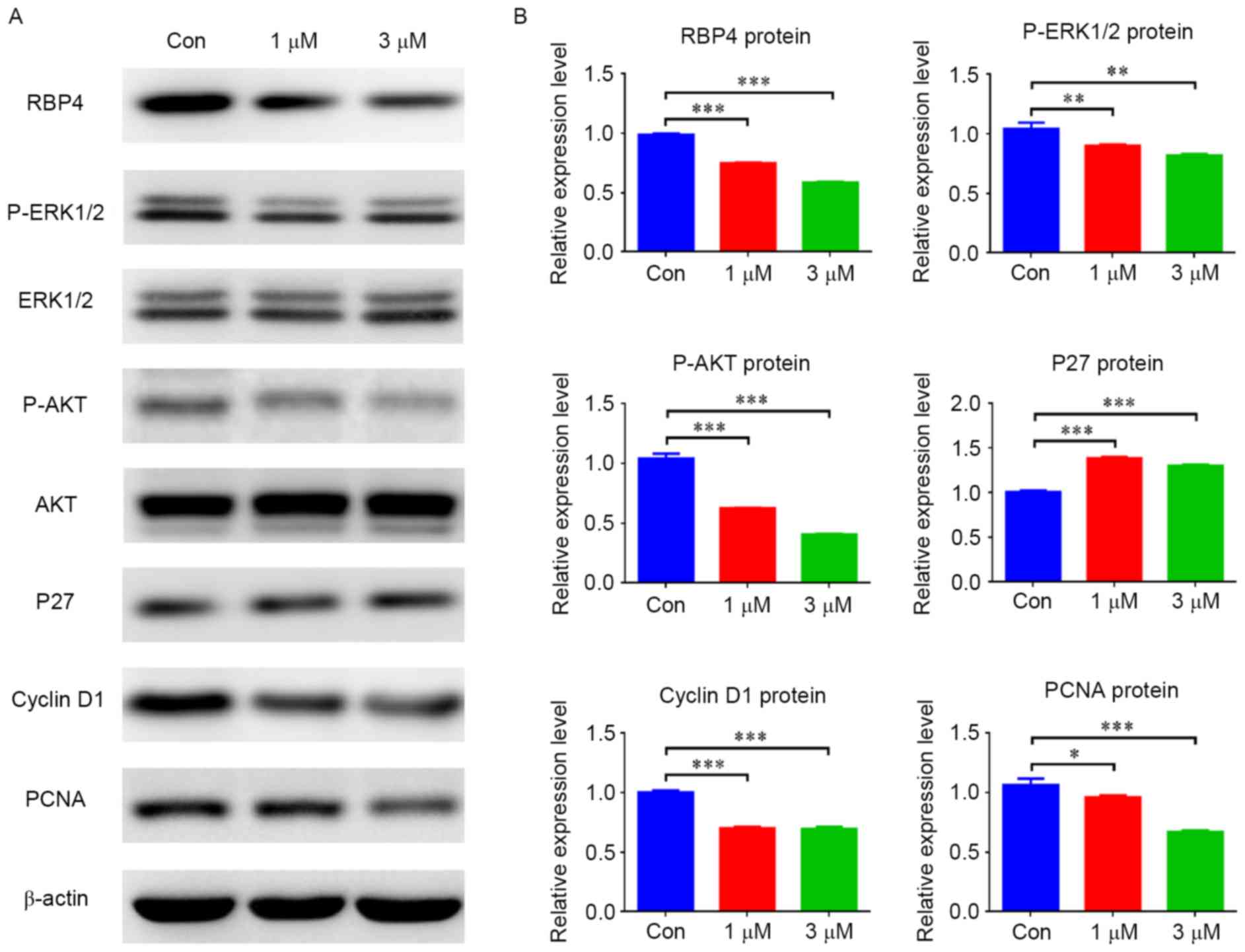 | Figure 4.atRA inhibited RBP4 protein levels and
altered the expression of associated regulators and signaling
pathways in human embryonic palatal mesenchymal cells. (A) Protein
levels of RBP4, cyclin D1, PCNA, p-ERK1/2 and p-AKT were all
decreased in the atRA-treated group, however p27 was increased
compared with the Con group. (B) Data for the protein levels are
presented as the mean + standard deviation. There were 3 duplicate
cultures in each group, and experiments were performed in
triplicate. *P<0.05, **P<0.01 and ***P<0.001. atRA,
all-trans retinoic acid; RBP4, retinol binding protein 4; PCNA,
proliferating cell nuclear antigen; p-, phosphorylated; ERK,
extracellular signal-related kinase; AKT, protein kinase B; Con,
control. |
RBP4 suppressed the expression of p27
and stimulated the expression of cyclin D1 via the AKT and ERK1/2
signaling pathway
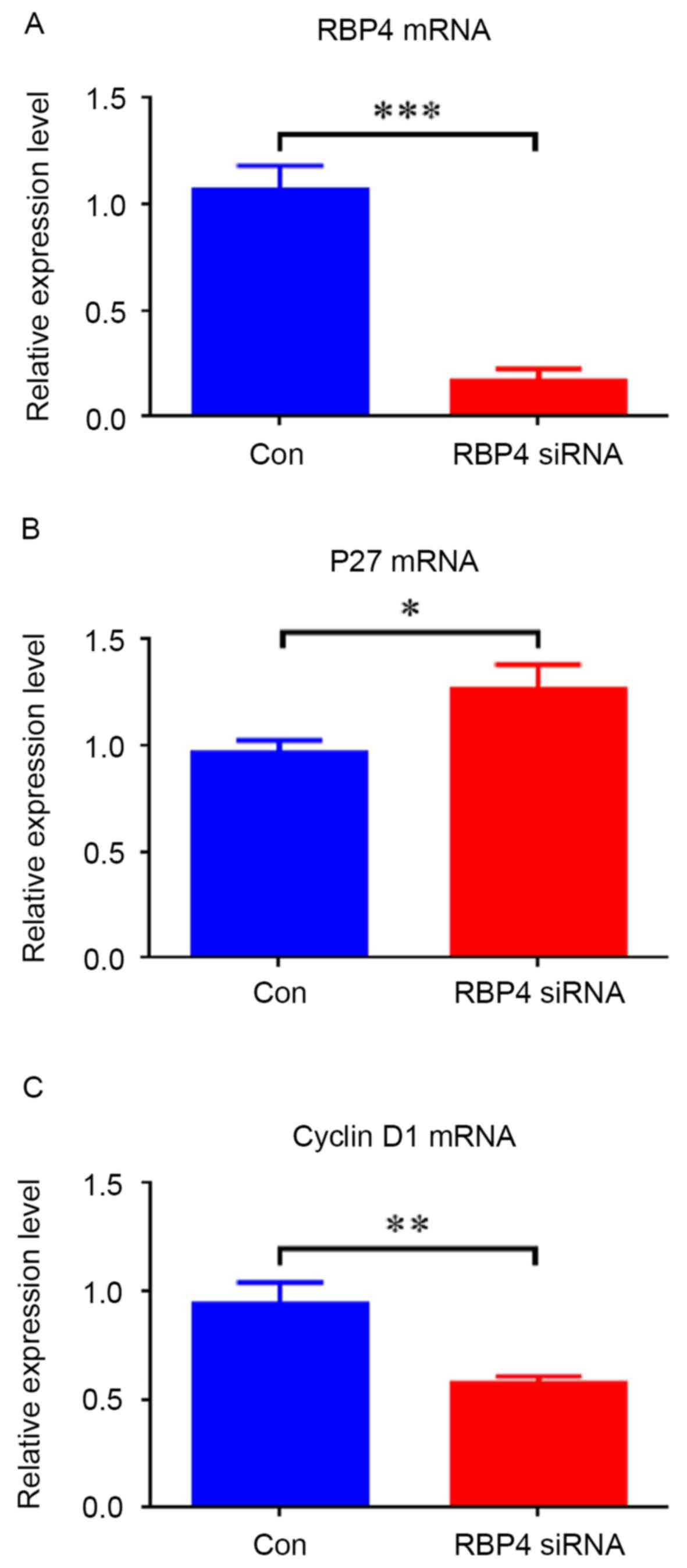
siRNA knockdown of RBP4 was performed to evaluate
its involvement in cell proliferation (Figs. 5 and 6). The RT-qPCR and western blotting
results demonstrated that RBP4 was effectively knocked down by
siRNA both at the mRNA and protein levels; mRNA levels were
repressed by 84.1% (0.17±0.06 vs. 1.07±0.12; P<0.001; Fig. 5A), and protein levels were
repressed by 56.7% (0.45±0.02 vs. 1.04±0.05; P<0.001) (Fig. 6). Downregulation of RBP4 in HEPM
cells transfected with RBP4 siRNA resulted in decreased expression
of cyclin D1 at both mRNA (Fig.
5C; P<0.01) and protein (Fig.
6) levels (P<0.001), while increased p27 mRNA (Fig. 5B; P<0.05) and protein (Fig. 6) levels were observed (P<0.001).
The protein levels of PCNA were reduced in RBP4-siRNA HEPM cells
compared with the control group (Fig.
6; P<0.01). The western blotting results demonstrated that
p-ERK1/2 and p-AKT were reduced along with RBP4 (Fig. 6; P<0.001). The western blotting
results indicated that RBP4 was effectively upregulated in response
to RBP4 overexpression in HEPM cells and the degree of
overexpression was 51.2% (1.50±0.02 vs. 0.99±0.01; P<0.001),
cyclin D1 was also upregulated (P<0.001), while p27 was
downregulated (Fig. 7;
P<0.001). In the atRA-exposed group, RBP4 levels and cyclin D1
were decreased (Fig. 7;
P<0.01), however p27 was increased (Fig. 7; P<0.001), as compared with the
control group. By contrast, RBP4 and cyclin D1 were not
downregulated (Fig. 7; P<0.001)
and p27 levels did not increase (Fig.
7; P<0.001) in the RBP4 overexpression plus atRA-treated
group compared with the control. The western blot results
additionally indicated that p-ERK1/2 and p-AKT were both
upregulated in response to RBP4 overexpression in HEPM cells
(Fig. 7; P<0.001), and they
were not downregulated in the RBP4 overexpression plus atRA-treated
group (P<0.001) compared with the control.
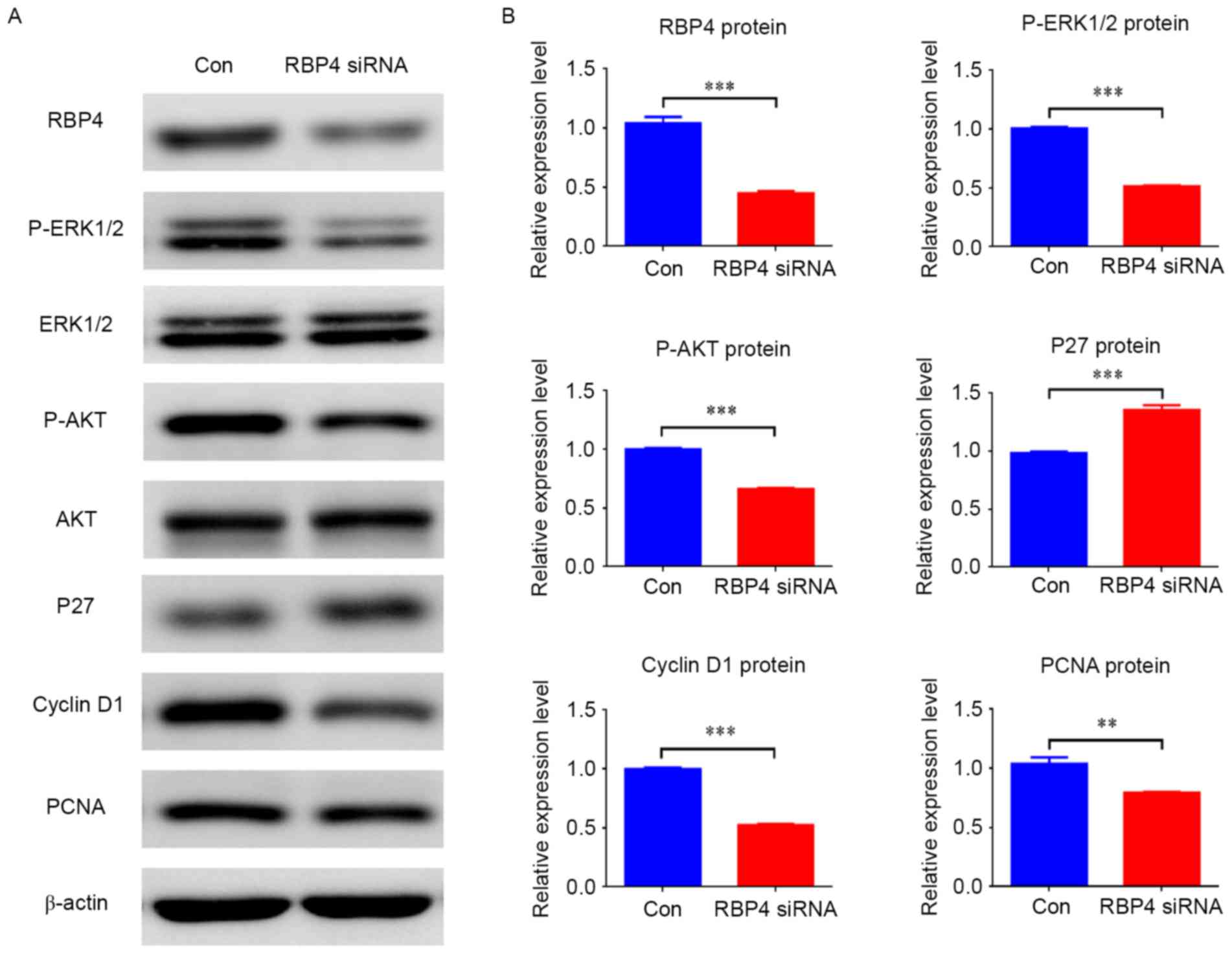 | Figure 6.Knockdown of RBP4 altered the
expression of related regulators and signaling pathways in HEPM
cells. (A) Protein levels of RBP4, cyclin D1, PCNA, p-ERK1/2 and
p-AKT were all downregulated in HEPM cells treated with RBP4 siRNA,
however p27 was upregulated compared with the Con group. (B) Data
for the protein levels are presented as the mean + standard
deviation. A total of 3 duplicate samples were used in each group,
and experiments were performed in triplicate. **P<0.01 and
***P<0.001. RBP4, retinol binding protein 4; HEPM, human
embryonic palatal mesenchymal; PCNA, proliferating cell nuclear
antigen; p-, phosphorylated; ERK, extracellular signal-related
kinase; AKT, protein kinase B; siRNA, small interfering RNA; Con,
control siRNA. |
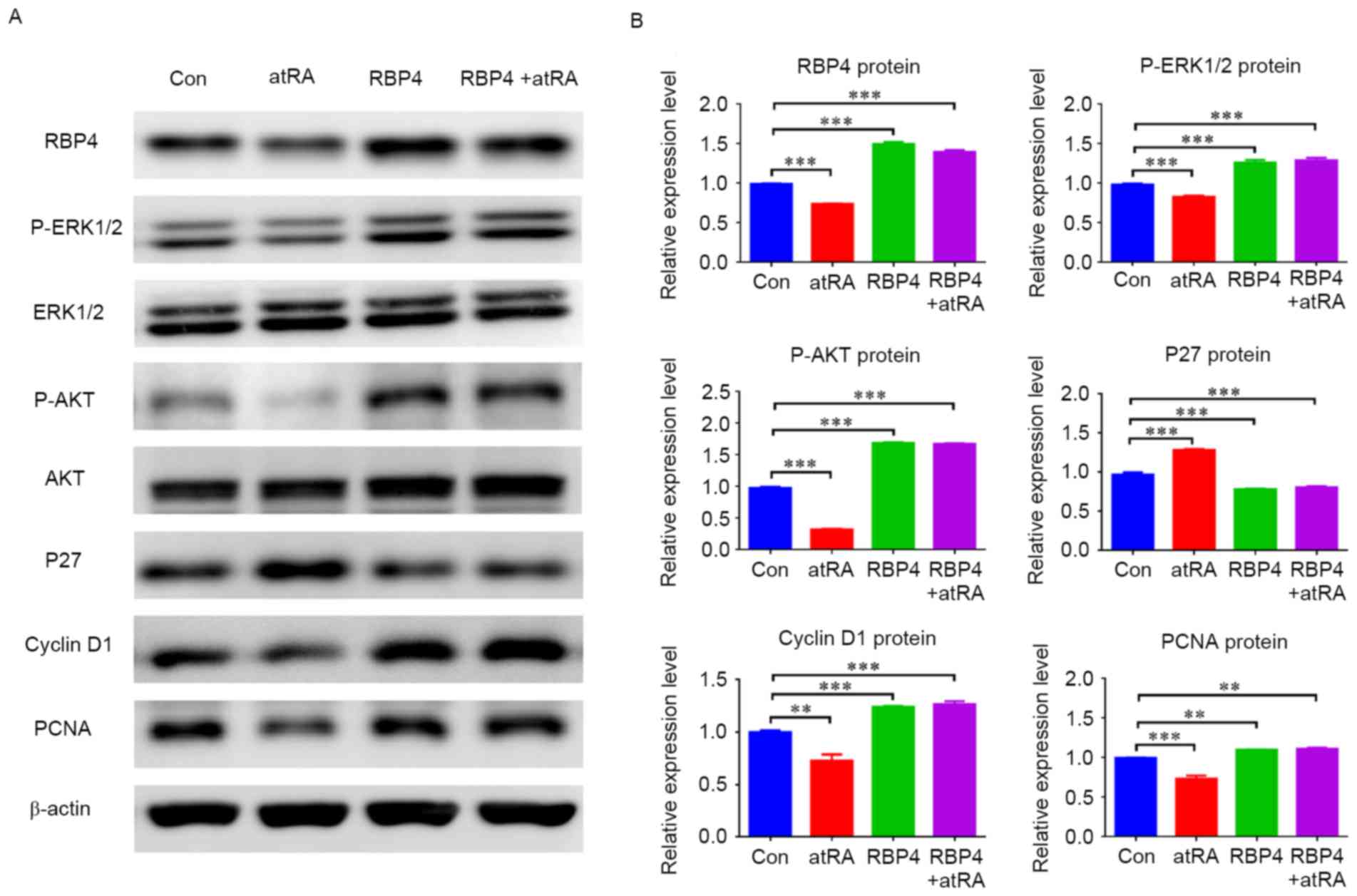 | Figure 7.Overexpression of RBP4 altered the
expression of related regulators and related signal pathways in
HEPM cells. (A) The protein levels of RBP4, cyclin D1, PCNA,
p-ERK1/2 and p-AKT were all upregulated in HEPM cells
overexpressing RBP4, however p27 was downregulated compared with
the Con group. The protein levels of RBP4, cyclin D1, PCNA,
p-ERK1/2 and p-AKT remained upregulated in the RBP4 overexpressing
group treated with atRA, and p27 was downregulated compared with
the Con group. (B) The data for the protein levels are presented as
the mean + standard deviation. A total of 3 duplicate samples were
used in each group, and experiments were performed in triplicate.
**P<0.01 and ***P<0.001. RBP4, retinol binding protein 4;
HEPM, human embryonic palatal mesenchymal; PCNA, proliferating cell
nuclear antigen; p-, phosphorylated; ERK, extracellular
signal-related kinase; AKT, protein kinase B; atRA, all-trans
retinoic acid; Con, control. |
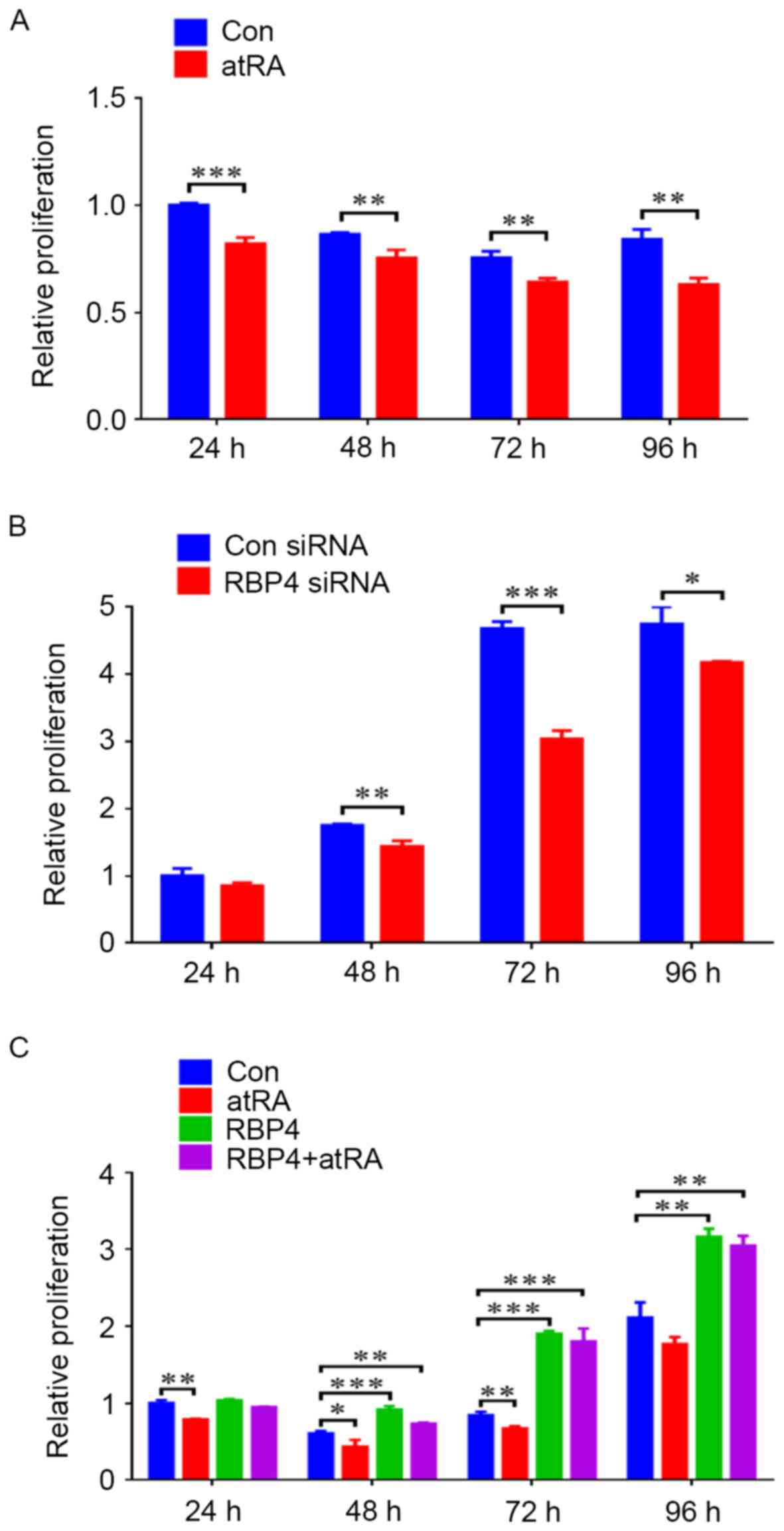
RBP4 promoted the proliferation of
HEPM cells by CCK8 assay
When HEPM cells were exposed to atRA (3 µM), cell
proliferation levels were reduced from 24 to 96 h (Fig. 8A). siRNA knockdown of RBP4 in HEPM
cells resulted in markedly reduced cell proliferation levels from
48 to 96 h (Fig. 8B). By contrast,
RBP4 overexpression in HEPM cells resulted in enhanced cell
proliferation levels from 48 to 96 h, with maximum proliferation
observed at 96 h (Fig. 8C;
P<0.01). Compared with the control group, when the
RBP4-overexpressing HEPM cells were exposed to atRA, cell
proliferation levels were maintained, particularly at 72 h
(Fig. 8C; P<0.001).
Discussion
atRA is essential for the development of embryos, by
regulating morphogenesis, cell proliferation and differentiation
(11,16). Embryos develop normally under low
concentrations of atRA, while when exposed to overdoses of atRA,
they develop defects in both animals and humans, such as cleft
palate (16,17). atRA has been widely used as a
teratogen to establish animal models and cytotoxicity tests to
determine the pathogenesis of cleft palate. Previous studies have
confirmed that in pregnant mice exposed to atRA (100 mg/kg) at
E10.0, the palatal shelves become dysplastic and fail to contact
one another, resulting in cleft palate (17,18).
In the mouse, palate shelves initially grow vertically alongside
the tongue at E12.5 and then elevate to the horizontal position
above the tongue, and contact with each other and fuse at E14.5
(19,13). Normal growth of EPM serves a key
role in this process, a previous study indicated that atRA
suppressed the process of growth and differentiation in the
developing mesenchyme to cause an abnormally small palate and
unfused palatal shelves (20).
atRA has been demonstrated to inhibit the proliferation of EPM
cells to cause cleft palate (13).
The results additionally indicated that the palatal shelves were
abnormally small and dysplastic in the atRA-exposed embryos, and
unable to shift horizontally due to the inhibition effect of
atRA.
RBP4 is a 21 kDa protein that is located in 10q23-24
in humans (21). RBP4 is primarily
synthesized in the liver and transported to target tissues; it can
also be detected in extrahepatic tissues such as adipose tissue,
the kidneys and the lungs (22).
RBP4, which is previously known as RBP, participates in retinol
translocation, lipid metabolism, fetal growth, bone growth and
inflammatory responses (23,24).
A study reported that RBP knockout mouse have morphological
abnormalities, including cleft palate and maxillofacial bone
defects (7). This indicated that
RBP4 serves an important role in normal physiological function and
development, particularly in that of the palate and the
maxillofacial region. It has been observed that the level of serum
RBP4 is significantly lower in children with NSCLP than in those
without (6). However, whether RBP4
can be detected in the EPM and the mechanism of RBP4 in cleft
palate remains unclear. The present study observed that RBP4 was
strongly expressed in the EPM at E13.5 and E14.5 in the normal
embryonic palate, while it was downregulated in cleft palate
induced by atRA. These observations indicated that RBP4 was
involved in the development of the EPM and that the deficiency in
RBP4 was associated with the occurrence of cleft palate.
In order to elucidate the mechanism of RBP4 in the
cleft palate induced by atRA, HEPM cells were used in the in
vitro experiments. The levels of RBP4 mRNA and protein were
downregulated by atRA in HEPM cells. This observation was
consistent with the results of the in vivo animal
experiments, demonstrating that atRA can significantly inhibit the
expression of RBP4. Numerous studies have demonstrated that atRA
can suppress the proliferation via the upregulation of p21/p27 and
the downregulation of cyclin D1 in various cell lines (25–27).
P27 structurally homologous to p21 is known as cyclin-dependent
kinase inhibitor, which serves a negative role in cell
proliferation and cell cycle progression (28,29).
p27 expression can cause cell cycle arrest, while downregulation of
p27 leads to cell cycle progression and cell proliferation
(28,29). Cyclin D1 belongs to the highly
conserved cyclin family, and function as regulator of
cyclin-dependent protein kinases, promoting cell cycle progression
(30). PCNA, a cofactor of DNA
polymerase, is widely used as a proliferation associated marker
(31). The results indicated that
the expression of p27 was increased and cyclin D1 and PCNA were
decreased in HEPM cells exposed to atRA, indicating that atRA
inhibited the proliferation of HEPM cells. The CCK8 assay also
proved that atRA had an inhibitory effect on cell proliferation.
The western blotting results also indicated that p-AKT and p-ERK1/2
were both reduced in HEPM cells treated with atRA. It has been
reported that the AKT and ERK1/2 signaling pathways are involved in
proliferation and survival in different cells (32). Activation of AKT signaling promotes
DNA synthesis and cell cycle progression, regulating cellular
proliferation and survival (28).
ERK1/2 has been demonstrated to be an essential signaling pathway
to modulate cell proliferation and apoptosis (33). The results of the present study
indicated that they may be involved in cell proliferation in HEPM
cells.
Previous reports have confirmed that RBP4 can
enhance proliferation in vascular smooth muscle cells and rat
aortic smooth muscle cells induced by hyperinsulinism (34,35).
However, the effect of RBP4 on HEPM cells remains unclear. In the
current study, the CCK8 assay demonstrated that RBP4 enhanced the
proliferation of HEPM cells, and the growth inhibition induced by
atRA was mitigated by RBP4 overexpression. RBP4 has been previously
observed to promote cell proliferation by upregulating p-ERK1/2 in
rat aortic smooth muscle cells (35). A previous study additionally
confirmed that RBP4 can significantly increase the expression of
p-AKT in vascular endothelial cells (36). In the present study, results
suggested that RBP4 could upregulate the expression of p-ERK1/2 and
p-AKT to activate the AKT and ERK1/2 signaling pathways in HEPM
cells. However, the mechanism of how RBP4 affects p-ERK1/2 and
p-AKT remains unclear, and further research is required. A previous
study observed that activation of the ERK1/2 and AKT signaling
pathways suppresses the expression of cyclin-dependent kinase
inhibitors (p27 and p21) and increases the expression of cell
cyclins (cyclin D1 and cyclin E) to promote cell cycle progression
and cell proliferation (32). The
results of the present study demonstrated that RBP4 could stimulate
cyclin D1 and inhibit p27 to promote cell proliferation via the AKT
and ERK1/2 signaling pathways. When RBP4 was suppressed, it can
cause growth inhibition. The in vivo and in vitro
studies indicated that depression of RBP4 in the palate could
suppress the expression of p-ERK1/2 and p-AKT, and affect the
expression of p27 and cyclin D1 to cause growth inhibition of EPM,
leading to cleft palate. The results demonstrated that RBP4 served
an important role in cleft palate induced by atRA, indicating that
it could be used as detection index or prophylaxis method for cleft
palate.
In summary, the study confirmed that RBP4 is
involved in cleft palate induced by atRA, and it is downregulated
in the EPM treated with atRA. RBP4 can be suppressed by atRA to
cause growth inhibition in the embryonic palate.
Acknowledgements
The current study was funded by grants from the
National Natural Science Foundation of China (grant no. 81100739),
the Project of Health and Family Planning Commission of Shenzhen
Municipality (grant no. 201302202) and the Natural Science
Foundation of Guangdong Province (grant no. S2011040004190).
References
|
1
|
Noy N: Retinoid-binding proteins:
Mediators of retinoid action. Biochem J. 348:481–495. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang Q, Graham TE, Mody N, Preitner F,
Peroni OD, Zabolotny JM, Kotani K, Quadro L and Kahn BB: Serum
retinol binding protein 4 contributes to insulin resistance in
obesity and type 2 diabetes. Nature. 436:356–362. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Munkhtulga L, Nakayama K, Utsumi N,
Yanagisawa Y, Gotoh T, Omi T, Kumada M, Erdenebulgan B, Zolzaya K,
Lkhagvasuren T and Iwamoto S: Identification of a regulatory SNP in
the retinol binding protein 4 gene associated with type 2 diabetes
in Mongolia. Hum Genet. 120:879–888. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan TF, Tsai YC, Wu CH, Lee CH, Wang SH
and Su JH: The positive correlation between cord serum
retinol-binding protein 4 concentrations and fetal growth. Gynecol
Obstet Invest. 72:98–1028. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hatfield JT, Anderson PJ and Powell BC:
Retinol-binding protein 4 is expressed in chondrocytes of
developing mouse long bones: Implications for a local role in
formation of the secondary ossification center. Histochem Cell
Biol. 139:727–734. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang J, Zhou S, Zhang Q, Feng S, Chen Y,
Zheng H, Wang X, Zhao W, Zhang T, Zhou Y, et al: Proteomic analysis
of RBP4/Vitamin A in children with cleft lip and/or palate. J Dent
Res. 93:547–552. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Quadro L, Hamberger L, Gottesman ME, Wang
F, Colantuoni V, Blaner WS and Mendelsohn CL: Pathways of vitamin A
delivery to the embryo: Insights from a new tunable model of
embryonic vitamin A deficiency. Endocrinology. 146:4479–4490. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marazita ML and Mooney MP: Current
concepts in the embryology and genetics of cleft lip and cleft
palate. Clin Plast Surg. 31:125–140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wyszynski DF and Beaty TH: Phenotypic
discordance in a family with monozygotic twins and nonsyndromic
cleft lip and palate: Follow-up. Am J Med Genet. 110:182–183. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Murray JC and Schutte BC: Cleft palate:
Players, pathways, and pursuits. J Clin Invest. 113:1676–1678.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clagett-Dame M and DeLuca HF: The role of
vitamin A in mammalian reproduction and embryonic development. Annu
Rev Nutr. 22:347–381. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Okano J, Suzuki S and Shiota K:
Involvement of apoptotic cell death and cell cycle perturbation in
retinoic acid-induced cleft palate in mice. Toxicol Appl Pharmacol.
221:42–56. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Z, Lin J, Xiao Y, Han J, Zhang X, Jia
H, Tang Y and Li Y: Induction of cell-cycle arrest by all-trans
retinoic acid in mouse embryonic palatal mesenchymal (MEPM) cells.
Toxicol Sci. 83:349–354. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mercader J, Granados N, Bonet ML and Palou
A: All-trans retinoic acid decreases murine adipose retinol binding
protein 4 production. Cell Physiol Biochem. 22:363–372. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang Y, Dong S, Wang W, Wang J, Wang M,
Chen M, Hou J and Huang H: Activation of Notch1 inhibits medial
edge epithelium apoptosis in all-trans retinoic acid-induced cleft
palate in mice. Biochem Biophys Res Commun. 477:322–328. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ross SA, McCaffery PJ, Drager UC and De
Luca LM: Retinoids in embryonal development. Physiol Rev.
80:1021–1054. 2000.PubMed/NCBI
|
|
17
|
Wang M, Huang H and Chen Y: Smad2/3 is
involved in growth inhibition of mouse embryonic palate mesenchymal
cells induced by all-trans retinoic acid. Birth Defects Res A Clin
Mol Teratol. 85:780–790. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abbott BD and Birnbaum LS: Retinoic
acid-induced alterations in the expression of growth factors in
embryonic mouse palatal shelves. Teratology. 42:597–610. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ferguson MW: Palate development.
Development. 103 Suppl:S41–S60. 1988.
|
|
20
|
Campbell JL Jr, Smith MA, Fisher JW and
Warren DA: Dose-response for retinoic acid-induced forelimb
malformations and cleft palate: A comparison of computerized image
analysis and visual inspection. Birth Defects Res B Dev Reprod
Toxicol. 71:289–295. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rocchi M, Covone A, Romeo G, Faraonio R
and Colantuoni V: Regional mapping of RBP4 to 10q23-q24 and RBP1 to
3q21-q22 in man. Somat Cell Mol Genet. 15:185–190. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blaner WS: Retinol-binding protein: The
serum transport protein for vitamin A. Endocr Rev. 10:308–316.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wolf G: Serum retinol-binding protein: A
link between obesity, insulin resistance, and type 2 diabetes. Nutr
Rev. 65:251–256. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vaisbuch E, Romero R, Mazaki-Tovi S, Erez
O, Kim SK, Chaiworapongsa T, Gotsch F, Than NG, Dong Z, Pacora P,
et al: Retinol binding protein 4-a novel association with
early-onset preeclampsia. J Perinat Med. 38:129–139. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Singh AT, Evens AM, Anderson RJ, Beckstead
JA, Sankar N, Sassano A, Bhalla S, Yang S, Platanias LC, Forte TM,
et al: All trans retinoic acid nanodisks enhance retinoic acid
receptor mediated apoptosis and cell cycle arrest in mantle cell
lymphoma. Br J Haematol. 150:158–169. 2010.PubMed/NCBI
|
|
26
|
Radu M, Soprano DR and Soprano KJ: S10
phosphorylation of p27 mediates atRA induced growth arrest in
ovarian carcinoma cell lines. J Cell Physiol. 217:558–568. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang ML, Tao Y, Zhou WQ, Ma PC, Cao YP,
He CD, Wei J and Li LJ: All-trans retinoic acid induces cell-cycle
arrest in human cutaneous squamous carcinoma cells by inhibiting
the mitogen-activated protein kinase-activated protein 1 pathway.
Clin Exp Dermatol. 39:354–360. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wesley UV, Hatcher JF and Dempsey RJ:
Sphingomyelin synthase 1 regulates Neuro-2a cell proliferation and
cell cycle progression through modulation of p27 expression and Akt
signaling. Mol Neurobiol. 51:1530–1541. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fillies T, Woltering M, Brandt B, Van
Diest JP, Werkmeister R, Joos U and Buerger H: Cell cycle
regulating proteins p21 and p27 in prognosis of oral squamous cell
carcinomas. Oncol Rep. 17:355–359. 2007.PubMed/NCBI
|
|
30
|
Dubey RK, Fingerle J, Gillespie DG, Mi Z,
Rosselli M, Imthurn B and Jackson EK: Adenosine attenuates human
coronary artery smooth muscle cell proliferation by inhibiting
multiple signaling pathways that converge on Cyclin D.
Hypertension. 66:1207–1219. 2015.PubMed/NCBI
|
|
31
|
Han YH, Gao B, Huang JH, Wang Z, Guo Z,
Jie Q, Yang L and Luo ZJ: Expression of CD147, PCNA, VEGF, MMPs and
their clinical significance in the giant cell tumor of bones. Int J
Clin Exp Pathol. 8:8446–8452. 2015.PubMed/NCBI
|
|
32
|
Shen H, Zhou E, Wei X, Fu Z, Niu C, Li Y,
Pan B, Mathew AV, Wang X, Pennathur S, et al: High density
lipoprotein promotes proliferation of adipose-derived stem cells
via S1P1 receptor and Akt, ERK1/2 signal pathways. Stem Cell Res
Ther. 6:952015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang XJ and Jia SS: Fisetin inhibits
laryngeal carcinoma through regulation of AKT/NF-κB/mTOR and ERK1/2
signaling pathways. Biomed Pharmacother. 83:1164–1174. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li F, Xia K, Sheikh SA, Cheng J, Li C and
Yang T: Involvement of RBP4 in hyperinsulinism-induced vascular
smooth muscle cell proliferation. Endocrine. 48:472–482. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li F, Xia K, Sheikh MS, Cheng J, Li C and
Yang T: Retinol binding protein 4 promotes hyperinsulinism-induced
proliferation of rat aortic smooth muscle cells. Mol Med Rep.
9:1634–1640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takebayashi K, Sohma R, Aso Y and Inukai
T: Effects of retinol binding protein-4 on vascular endothelial
cells. Biochem Biophys Res Commun. 408:58–64. 2011. View Article : Google Scholar : PubMed/NCBI
|