Introduction
Endothelial cell injury has been demonstrated to be
linked with angiocardiopathy-associated diseases, including
atherosclerosis, hypertension and diabetic vasculopathy (1). Angiotensin (Ang) II induces injuries
of endothelial cells and exerts substantial effects on the
renin-angiotensin-aldosterone system, which is closely associated
with regulation of the above-mentioned cardiovascular diseases
(2,3). Under the pressure of Ang II,
production of certain defensive substances [such as nitric oxide
(NO)] would be stimulated in endothelial cells (4). Up to now, however, the method by
which endothelial cells initiate defense mechanisms against the
injuries induced by Ang II remains unknown.
Numerous physiological functions, including those
relevant to immune and cardiovascular systems, are mediated by the
known gaseous messenger, hydrogen sulfide (H2S), which
is endogenously generated by cystathionine β-synthase, which is
highly expressed in the brain, and cystathionine γ-lyase (CSE),
which is concentrated in the vasculature (5). Furthermore, as H2S may
induce various vascular effects that are similar to NO, including
angiogenesis and smooth vascular muscle relaxation, it is
hypothesized that certain internal connections may be present
between H2S and NO (6).
Furthermore, inhibition of H2S exhibits negative impacts
on angiogenesis in the case of ischemic myocardium, therefore,
identifying the pro-angiogenic mechanism of H2S may
serve as a promising strategy for treating myocardial disorders
(7).
Additionally, sodium hydrosulfide (NaHS) treatment
exerted a positive effect on Akt phosphorylation and such a
positive effect depends on treatment dose and duration, indicating
that Akt phosphorylation was induced by H2S (5). Another study demonstrates that
H2S exhibits a stimulatory role in endothelial nitric
oxide synthases (eNOS) phosphorylation via an Akt-dependent and p38
mitogen-activated protein kinases (MAPK) mechanism, indicating that
increased generation of NO eventually triggers the corresponding
stimulatory effects of H2S on angiogenesis (6). Thus, it is hypothesized that the
Akt/eNOS signaling pathway may be investigated to elucidate whether
the underlying Ang II-induced endothelial cell injury may be
restored under certain circumstances.
Therefore, the aim of the present study was to
evaluate whether H2S stimulates the release of NO for
protecting human umbilical vein endothelial cells (HUVECs) against
Ang II-induced injury and whether the underlying mechanism involves
the Akt/eNOS signaling pathway.
Materials and methods
Culture and treatment of HUVECs
HUVECs [American Type Culture Collection (ATCC)
CRL-1730; ATCC, Manassas, VA, USA) were seeded into six-well plates
at a density of 105/ml. The cells were cultivated with
Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), at 37°C for 24 h under an atmosphere of 5% CO2
(4). The HUVECs (1×105)
were divided into five treatment groups as follows: Normal control,
Ang II, Ang II + NaHS (H2S donor), Ang II + Akt
inhibitors + NaHS, and Ang II + eNOS inhibitors + NaHS. HUVECs were
treated with Sigma-Aldrich Ang II (1 µM; Merck KGaA, Darmstadt,
Germany) for 48 h to induce injury, while the treatment with 100 µM
NaHS (H2S donor) was for 30 min. Incubation of HUVECs
with the Akt inhibitor, LY 294002 (10 µM) and the eNOS inhibitor,
L-NAME (200 µM; Sigma-Aldrich; Merck KGaA), were for 1 h, as
previously described (6).
Cell viability detection by
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide
(MTT)
Cells were incubated at 37°C for 3 h with fresh
medium containing 1 g/l MTT. The unbounded MTT was washed with PBS
and the formazan product in the form of blue-violet crystalline was
dissolved with dimethyl sulfoxide. Finally, cell viability was
indirectly measured by enzyme-linked immunosorbent assay at a wave
length of 570 nm, after removing the background absorbance at a
wave length of 690 nm, as previously described (8).
Cell apoptosis detected by flow
cytometry
HUVECs were digested with trypsin, separated by
centrifugation (1,000 rpm for 5 min) and washed gently with PBS 3
times. Apoptosis was detected in accordance with the procedures of
an Annexin V-allophycocyanin/propidium iodide (PI) apoptosis
detection kit (Beyotime Institute of Biotechnology, Haimen, China).
The cells (5×105) were re-suspended in Annexin V binding
solution (195 µl) and incubated with Annexin V-fluorescein
isothiocyanate (5 µl) and PI (10 µl) for 20 min in the dark at room
temperature. Finally, the detection of apoptosis was performed
using flow cytometry (Bio-Rad Laboratories, Inc.- Hercules, CA,
USA), as previously described (4).
Cell migration detected by wound
healing method
Hydroxyurea (5 mmol/l) was added to the cell culture
medium to inhibit the proliferation of HUVECs. Scratches were made
using a yellow pipette tip, and the cells were then washed twice
using growth medium. Images were obtained before and 24 h after
formation of the scratches, and the cell migration rate was
calculated by measuring the loss area, as previously described
(5).
Cell proliferation detected using the
5-bromo-2′-deoxyuridine (BrdU) incorporation method
Cells were counted using a Nikon Eclipse TS100
optical microscope (Nikon Corporation, Tokyo, Japan) and seeded in
96-well plates (1×104 cells/well) for 24 h. The
serum-free medium was replaced and the cells were incubated
overnight at 37°C. Cell proliferation was then detected by BrdU
immunostaining (Beyotime Institute of Biotechnology) (6).
Cell adhesion measured by hematoxylin
staining
The serum-starved endothelial cells (density,
5×104 cells/well) were seeded into 12-well plates that
were coated with type I collagen (2 mg/ml) at 37°C for 30 min. The
medium was aspirated and washed gently twice with PBS.
Subsequently, 4% paraformaldehyde was used to fix the adherent
cells. The cells were stained with hematoxylin and observed using a
Nikon Eclipse TS100 optical microscope (Nikon Corporation), as
previously described (5).
Observation of tubular structures with
an artificial basement membrane
The 24-well plates were coated with matrigel (300
µl) at 37°C for 30 min. Then the treated cells (density,
5×104 cells/well) were seeded in 24-well plates at 37°C
for 16 h. Microscopic images were obtained using a Nikon Eclipse
TS100 optical microscope (Nikon Corporation), and the length of the
capillary was measured using Image J software version 1.46
(National Institutes of Health, Bethesda, MD, USA) to evaluate the
formation of tubular structures (5).
NO production detected using the
Griess method
Cells were incubated with
4-amino-5-methylamino-2′,7′-difluorofluorescein (5 µM) at 37°C for
30 min. Subsequently, the unbound dye was washed in DMEM medium and
replaced with fresh medium. The results were observed by
fluorescence microscopy (6).
Western blot analysis
HUVECs were washed with pre-cooled PBS buffer 3
times and then lysed with lysis solution [0.5 M EDTA and 1 M
Tris-Cl (pH 7.4)], sucrose (0.3 M) and protease inhibitors
(Sigma-Aldrich; Merck KGaA). The mixture underwent ultrasound
processing on ice 3 times, for 5–10 sec each time. Subsequently,
the lysate was centrifuged at 13,400 × g at 4°C for 5 min, after
which the supernatant was collected. Electrophoresis with 10%
sodium dodecyl sulfate polyacrylamide gel electrophoresis was
performed, using 50 µg protein, at 4°C for 1–2 h. After completion
of electrophoresis, proteins were transferred to polyvinylidene
fluoride membranes at 80 V for 1 h and incubated with primary
antibodies at 4°C overnight. The antibodies were as follows:
Phosphorylated eNOS (Ser1177; catalog no. 9570; 1:1,000), eNOS)
catalog no. 5880; 1:1,000), phosphorylated Akt (S473; catalog no.
11962; 1:1,000) and Akt (catalog no. 2920; 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-CSE (catalog no.
22219–1-AP; 1:5,000; ProteinTech Group, Inc., Chicago, IL, USA) and
anti-β-actin (catalog no. A5441; 1:10,000; Sigma-Aldrich; Merck
KGaA). Finally, the proteins were incubated with a secondary
antibody conjugated to horse radish peroxidase (catalog no. A7289;
1:2,000; Sigma-Aldrich, Merck KGaA), and were then exposed to
enhanced chemiluminescence (ECL) liquid. Data analysis was
conducted following X-ray tableting, development and fixing
(6).
Statistical analysis
The data were processed using GraphPad Prism version
5.0 software (GraphPad Software, Inc., La Jolla, CA, USA) for
statistical analysis and were presented as the mean ± standard
deviation. When data conformed to normal distribution,
between-group comparisons of measurement data were performed using
a two-tailed t-test; otherwise, the non-parametric Mann-Whitney
test was applied. Analysis of variance and the non-parametric
Kruskal-Wallis test were performed for comparisons between the 5
groups. Regarding enumeration data, the χ2 test was
implemented to compare the groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
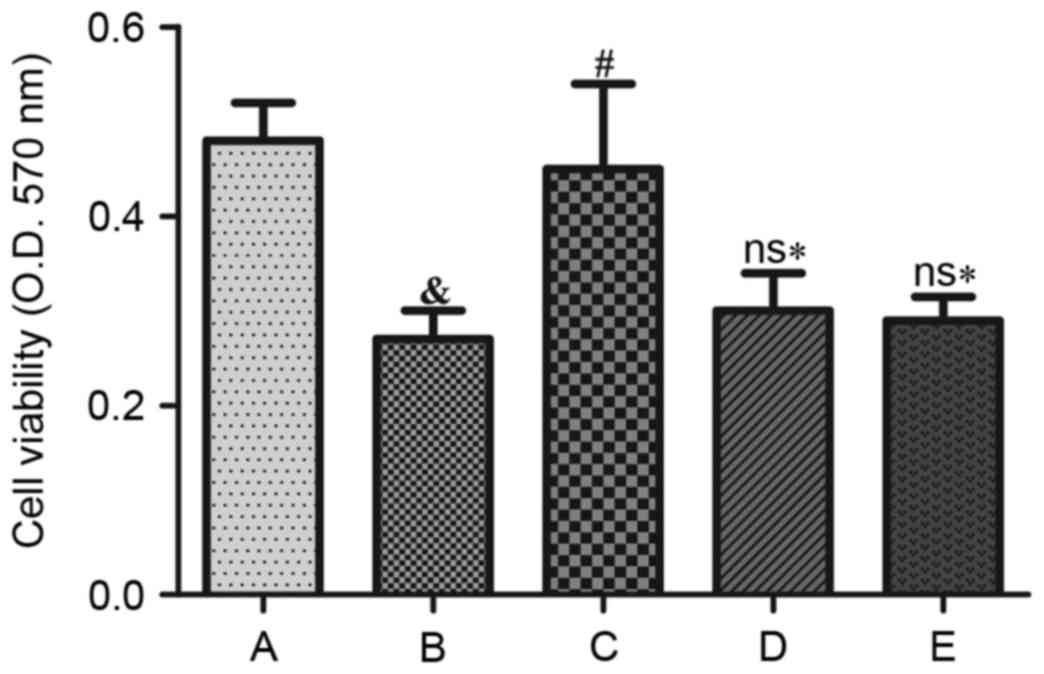
Effects of Ang II and H2S on cell
viability of HUVECs
Compared with the control group, cell viability
decreased following treatment with Ang II for 48 h (P<0.05).
Additionally, the cell survival rate was significantly improved by
treatment with NaHS (P<0.05). However, cell survival did not
significantly increase when HUVECs were exposed to LY 294002 (Akt
inhibitor) or L-NAME (eNOS inhibitor) prior to NaHS treatment,
indicating that cell viability was significantly reduced
(P<0.05) when compared with NaHS treatment (Fig. 1).
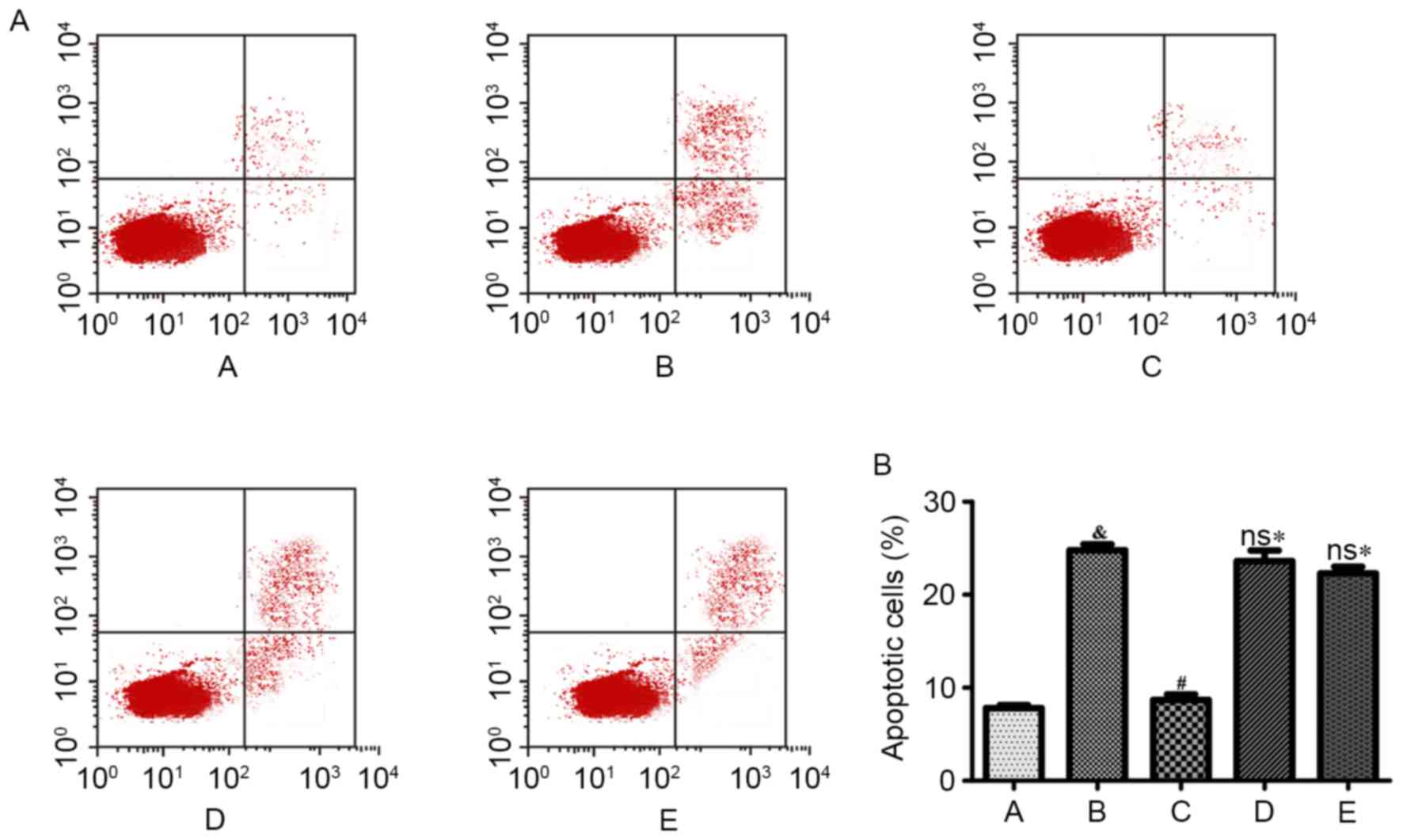
Effects of Ang II and H2S on cell
apoptosis of HUVECs
Compared with the control group, cell apoptosis was
significantly increased following Ang II treatment for 48 h
(P<0.001). NaHS treatment significantly reduced the percentage
of apoptotic cells induced by Ang II (P<0.001). However, NaHS
treatment was ineffective following simultaneous treatment with LY
294002 (Akt inhibitor) or L-NAME (eNOS inhibitor; Fig. 2).
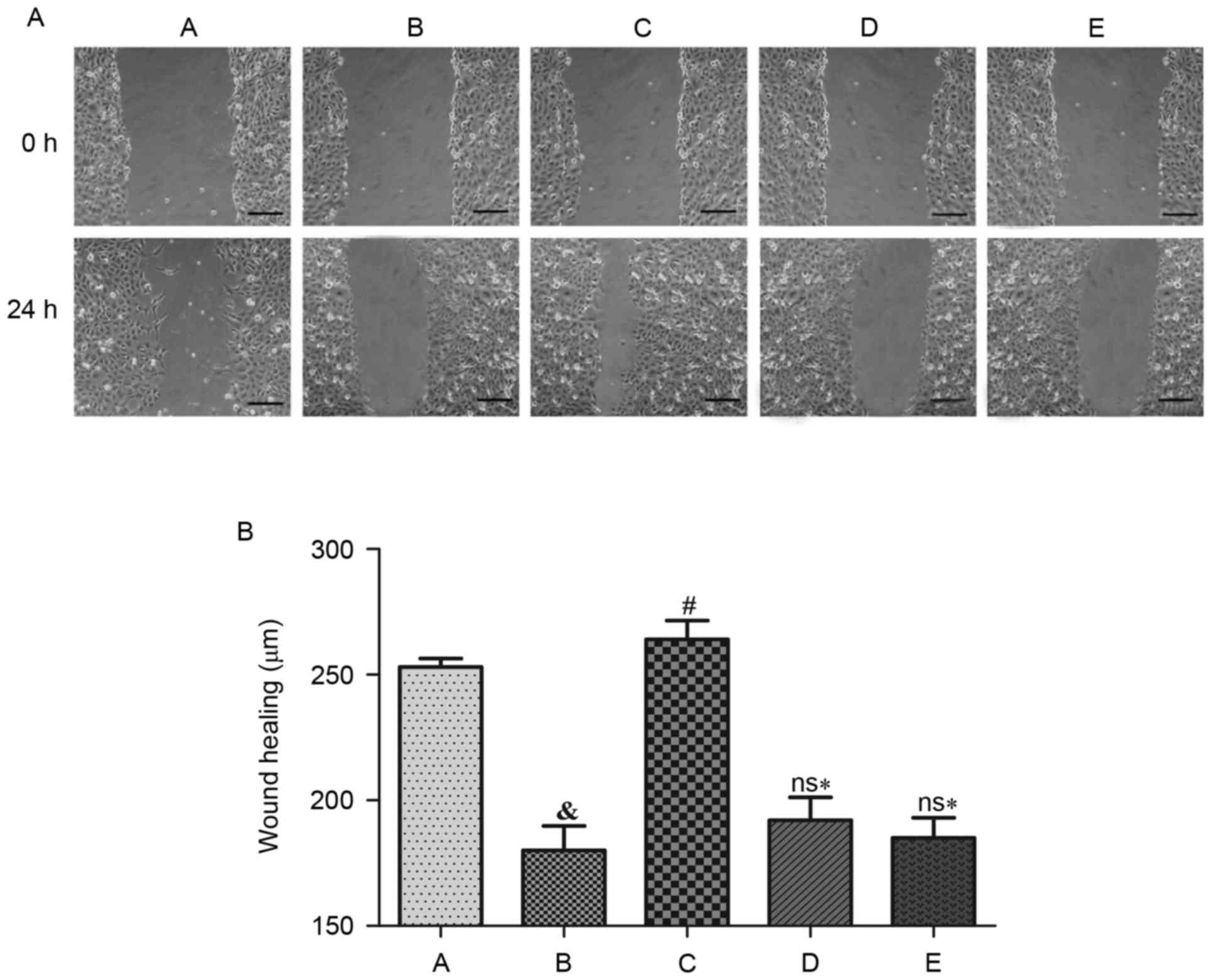
Effects of Ang II and H2S on cell
migration rate of HUVECs
The cell migration rate dropped significantly
following treatment with Ang II when compared with the control
group (P<0.05). NaHS treatment significantly improved the cell
migration rate (P<0.05), although it did not enhance the cell
migration rate when HUVECs were concurrently treated with LY 294002
(Akt inhibitor) or L-NAME (eNOS inhibitor). Thus, the cell
migration rates were significantly declined compared with NaHS
treatment alone (P<0.05; Fig.
3).
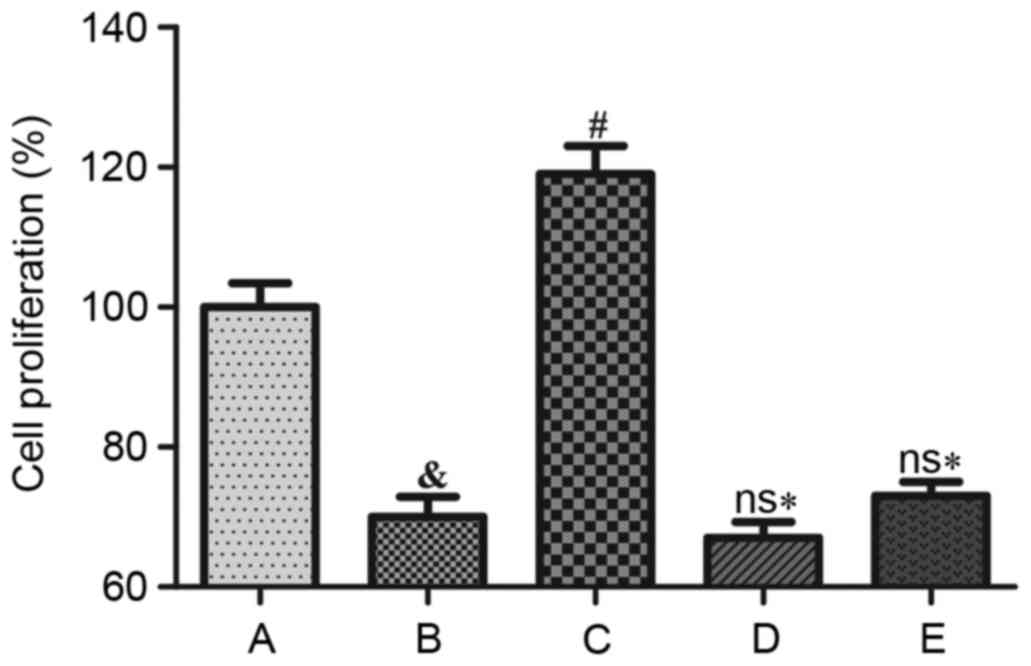
Effects of Ang II and H2S on cell
proliferation of HUVECs
Cell proliferation decreased significantly
subsequent to Ang II treatment when compared with the control group
(P<0.05). NaHS treatment significantly increased cell
proliferation (P<0.001); however, NaHS did not significantly
increase cell proliferation following pretreatment with LY 294002
(Akt inhibitor) or L-NAME (eNOS inhibitor; Fig. 4).
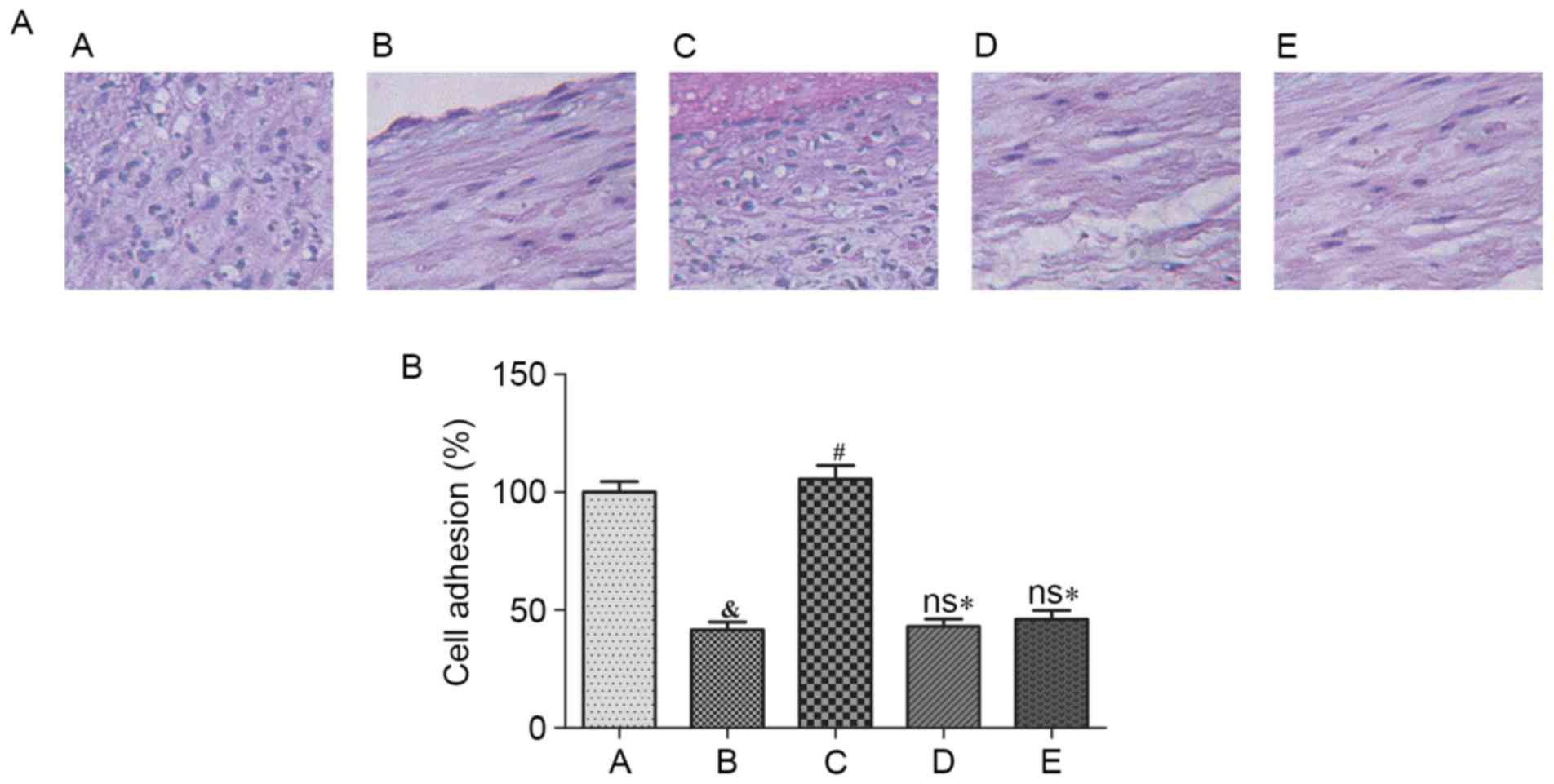
Cell adhesion ability of HUVECs
The hematoxylin staining results demonstrated that
cell adhesion ability decreased significantly following Ang II
treatment (P<0.05), and NaHS exposure significantly improved
cell adhesion ability (P<0.05). NaHS was, however, unable to
markedly increase cell adhesion ability following pretreatment with
LY 294002 (Akt inhibitor) or L-NAME (eNOS inhibitor; Fig. 5).
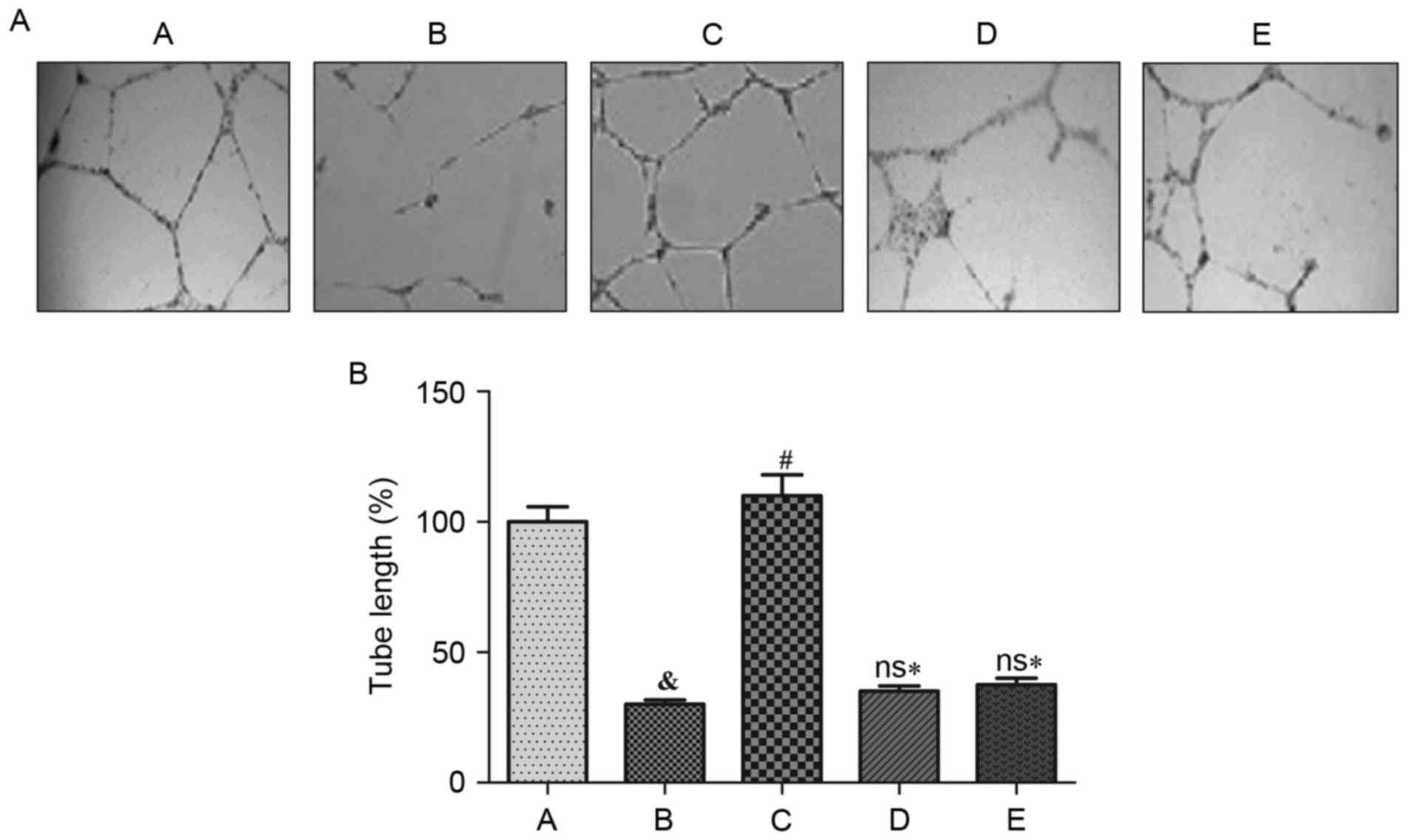
Cell tube formation assay of
HUVECs
Cell tube formation ability decreased significantly
subsequent to treatment with Ang II, compared with the control
group (P<0.05). NaHS treatment significantly increased the
length of capillaries, implying that the tube formation of HUVECs
was stimulated by H2S (P<0.05). Conversely, NaHS
failed to significantly promote HUVECs to form tubular structures
when exposed to LY 294002 (Akt inhibitor) or L-NAME (eNOS
inhibitor). Consequently, their cell tube formation ability
decreased significantly when compared with treatment with NaHS
alone (P<0.05; Fig. 6).
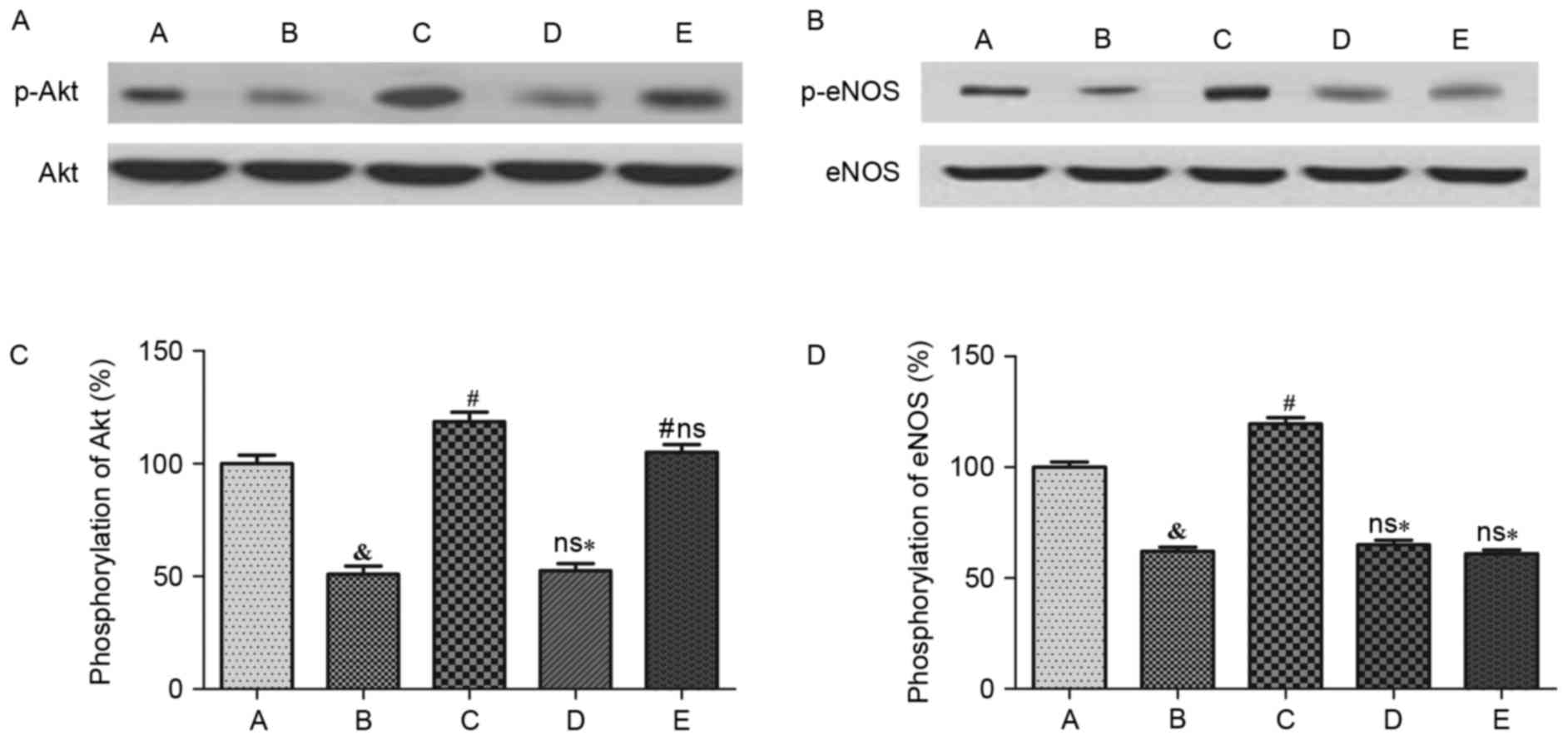
Effects of H2S on Akt and eNOS
phosphorylation in HUVECs, and NO production
Compared with the control group, intracellular
phosphorylation of Akt was significantly reduced following Ang II
treatment (P<0.05; Fig. 7).
NaHS treatment may significantly reverse this effect (P<0.05);
however, treatment with NaHS + LY 294002 did not promote Akt
phosphorylation. Furthermore, Akt phosphorylation was significantly
reduced when compared with NaHS treatment (P<0.05), although it
was not inhibited following treatment with L-NAME + NaHS
(P<0.05). With respect to NaHS treatment, Akt phosphorylation in
HUVECs was not statistically significant (Fig. 7A and C). In addition, the
intracellular eNOS phosphorylation level was significantly lower
than the control group following Ang II treatment (P<0.05) and
NaHS treatment significantly reversed this effect (P<0.05). NaHS
cannot, however, stimulate eNOS phosphorylation following LY 294002
or L-NAME pretreatment. Furthermore, eNOS phosphorylation decreased
significantly (P<0.05) compared with the NaHS alone treatment
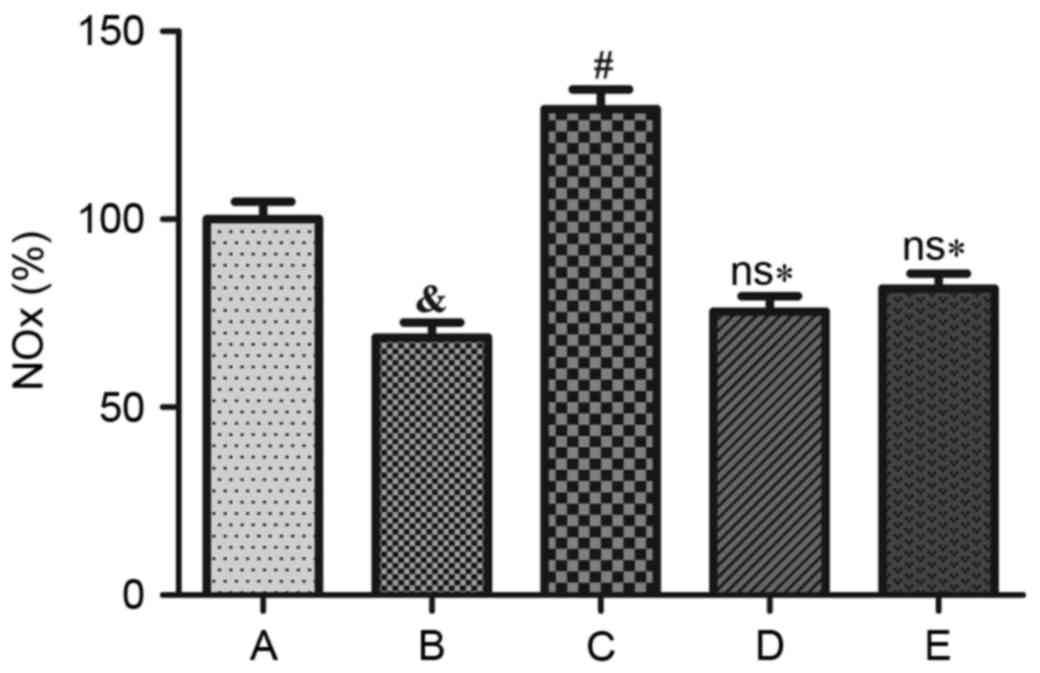
group (Fig. 7B and D). Compared
with the control group, the intracellular NO release was
significantly reduced following Ang II treatment (P<0.05) and
NaHS treatment significantly reversed this effect (P<0.05). NaHS
treatment did not promote NO production when LY29400 or L-NAME was
added and NO generation was significantly reduced compared with the
NaHS alone treatment group (P<0.05; Fig. 8).
Discussion
The endothelium that lines the inner surface of the
entire vascular tree is responsible for cardiovascular homeostasis
(9,10). Endothelial dysfunction may be
detected in various cardiovascular diseases, and reflects
pathophysiological changes in the phenotype and functions of ECs
(9). Four major findings have been
described in the current study, following treatment of a culture of
HUVECs. Firstly, Ang II (1 µM) significantly decreased cell
viability, migration, proliferation, adhesion and tube-like
structure formation in HUVECs, whilst it significantly increased
cell apoptosis. Secondly, Ang II treatment significantly reduced
the phosphorylation levels of Akt and eNOS, and NO metabolite
concentrations in the culture media. Furthermore, all of the Ang
II-induced responses were prevented by NaHS, an external
H2S donor. Finally, all protective effects caused by
NaHS were sensitive to MLY 294002 and L-NAME, selective antagonists
of Akt and eNOS, respectively.
The primary function of Ang II lies in the adaptive
changes in blood pressure and vascular homeostasis via regulation
of vascular tone, sympathetic nervous excitability and
electrolyte/water balance (11).
Ang II, originating in either blood circulation or locally secreted
vascular walls, activates multiple intracellular signaling pathways
via Ang II type 1 (AT1) and AT2 receptors, of which are widely
expressed in ECs. Specifically, Ang II activates NAD (P) H oxidase
with a consequent increment in the production of reactive oxygen
species (ROS) (12). Hence,
excessive Ang II triggers the imbalance of ROS metabolism, and
thereby causes DNA damage, mitochondrial dysfunction, as well as
endothelium injury within HUVECs (13). The viability of HUVECs will be
reduced and the corresponding apoptosis ratio will rise once the
above-mentioned cellular damage occurs (13). As the migratory capability of ECs
is pivotal in the maintenance of vessel wall integrity, and the
precondition of angiogenesis is a normal endothelial function
(14), it is hypothesized that Ang
II treatment may significantly reduce cell proliferation, adhesion
and tube-like structure formation in HUVECs.
H2S, a gasotransmitter produced in ECs,
may deter the progression of various cardiovascular diseases
through molecular inhibition of vascular inflammation and reduction
of ROS levels within ECs (10,15,16).
When synthesis of H2S in HUVECs is suppressed, cell
apoptosis is caused and thereby a series of endocellular reactions
are depressed, including cell migration, proliferation and
tube-like structure formation (16,17).
Thus, certain Ang II-induced types of damage are, to a certain
extent, prevented by NaHS, and the present experimental results
confirm the findings.
NO, a vital endogenous vasodilator, is produced in
ECs by eNOS, but its availability is impaired in various
cardiovascular diseases (9). In
addition, increasing evidence demosntrates that H2S may
exert protective effects on the cardiovascular systems by adjusting
NO synthesis and/or its functions (6,9,18,19).
The production of NO in ECs is based on activation of a cascade of
phosphorylation events, for example, actions relevant to p38 MAPK,
Akt and eNOS (6,20). Previous studies have demonstrated
that Ang II significantly inhibited NO synthesis in HUVECs by
increasing the generation of ROS and sequentially inducing injury
of ECs (12,21).
There were certain limitations of the present study.
The experimental results were obtained in vitro, therefore
in vivo experiments are required for further consolidation.
Furthermore, whether there are time- or dosage-dependent effects of
Ang II and NaHS treatment remains unknown, as only a single dose
and intervention time was used. Ang II induces dysfunctions of
HUVECs via reducing NO generation and H2S weakens this
malfunction via promoting the Akt/eNOS signaling pathway.
Initially, Ang II and H2S were linked via their
association with NO in the present study, and the results may
provide a novel research approach for treating endothelial
dysfunctions.
In conclusion, the present study demonstrates that
H2S promotes NO synthesis, which may counteract the
damage caused by Ang II. Furthermore, via a systematic experimental
design, the present study indicates that H2S weakens Ang
II-induced EC dysfunction via regulation of the Akt/eNOS signaling
pathway.
References
|
1
|
Li B, Zani A, Martin Z, Lee C,
Zani-Ruttenstock E, Eaton S and Pierro A: Intestinal epithelial
cell injury is rescued by hydrogen sulfide. J Pediatr Surg.
51:775–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakashima H, Suzuki H, Ohtsu H, Chao JY,
Utsunomiya H, Frank GD and Eguchi S: Angiotensin II regulates
vascular and endothelial dysfunction: Recent topics of Angiotensin
II type-1 receptor signaling in the vasculature. Curr Vasc
Pharmacol. 4:67–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhi Z, Pengfei Z, Xiaoyi T and Genshan M:
Adiponectin ameliorates angiotensin II-induced vascular endothelial
damage. Cell Stress Chaperones. 19:705–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luo P, Zhang WF, Qian ZX, Xiao LF, Wang H,
Zhu TT, Li F, Hu CP and Zhang Z: MiR-590-5p-meidated LOX-1
upregulation promotes Angiotensin II-induced endothelial cell
apoptosis. Biochem Biophys Res Commun. 471:402–408. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T
and Zhu YC: The novel proangiogenic effect of hydrogen sulfide is
dependent on Akt phosphorylation. Cardiovasc Res. 76:29–40. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Altaany Z, Yang G and Wang R: Crosstalk
between hydrogen sulfide and nitric oxide in endothelial cells. J
Cell Mol Med. 17:879–888. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Osipov RM, Robich MP, Feng J, Liu Y,
Clements RT, Glazer HP, Sodha NR, Szabo C, Bianchi C and Sellke FW:
Effect of hydrogen sulfide in a porcine model of myocardial
ischemia-reperfusion: Comparison of different administration
regimens and characterization of the cellular mechanisms of
protection. J Cardiovasc Pharmacol. 54:287–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marampon F, Gravina GL, Scarsella L,
Festuccia C, Lovat F, Ciccarelli C, Zani BM, Polidoro L, Grassi D,
Desideri G, et al: Angiotensin-converting-enzyme inhibition
counteracts angiotensin II-mediated endothelial cell dysfunction by
modulating the p38/SirT1 axis. J Hypertens. 31:1972–1983. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Altaany Z, Moccia F, Munaron L, Mancardi D
and Wang R: Hydrogen sulfide and endothelial dysfunction:
Relationship with nitric oxide. Curr Med Chem. 21:3646–3661. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang R, Szabo C, Ichinose F, Ahmed A,
Whiteman M and Papapetropoulos A: The role of H2S bioavailability
in endothelial dysfunction. Trends Pharmacol Sci. 36:568–578. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kanaide H, Ichiki T, Nishimura J and
Hirano K: Cellular mechanism of vasoconstriction induced by
angiotensin II: It remains to be determined. Circ Res.
93:1015–1017. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Desideri G, Bravi MC, Tucci M, Croce G,
Marinucci MC, Santucci A, Alesse E and Ferri C: Angiotensin II
inhibits endothelial cell motility through an AT1-dependent
oxidant-sensitive decrement of nitric oxide availability.
Arterioscler Thromb Vasc Biol. 23:1218–1223. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lu Y, Wang RH, Guo BB and Jia YP:
Quercetin inhibits angiotensin II induced apoptosis via
mitochondrial pathway in human umbilical vein endothelial cells.
Eur Rev Med Pharmacol Sci. 20:1609–1616. 2016.PubMed/NCBI
|
|
14
|
Jang H, Oh MY, Kim YJ, Choi IY, Yang HS,
Ryu WS, Lee SH and Yoon BW: Hydrogen sulfide treatment induces
angiogenesis after cerebral ischemia. J Neurosci Res. 92:1520–1528.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang G and Wang R: H2S and blood vessels:
An overview. Handb Exp Pharmacol. 230:85–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang MJ, Cai WJ and Zhu YC: Mechanisms of
angiogenesis: Role of hydrogen sulphide. Clin Exp Pharmacol
Physiol. 37:764–771. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen Y, Guo W, Wang Z, Zhang Y, Zhong L
and Zhu Y: Protective effects of hydrogen sulfide in hypoxic human
umbilical vein endothelial cells: A possible mitochondria-dependent
pathway. Int J Mol Sci. 14:13093–13108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coletta C, Papapetropoulos A, Erdelyi K,
Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I,
Martin E and Szabo C: Hydrogen sulfide and nitric oxide are
mutually dependent in the regulation of angiogenesis and
endothelium-dependent vasorelaxation. Proc Natl Acad Sci USA.
109:9161–9166. 2012; View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Go YM, Lee HR and Park H: H(2)S inhibits
oscillatory shear stress-induced monocyte binding to endothelial
cells via nitric oxide production. Mol Cells. 34:449–455. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Predmore BL, Julian D and Cardounel AJ:
Hydrogen sulfide increases nitric oxide production from endothelial
cells by an akt-dependent mechanism. Front Physiol. 2:1042011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu H, Chen T, Li N, Wang S and Bu P: Role
of SIRT3 in angiotensin II-induced human umbilical vein endothelial
cells dysfunction. BMC Cardiovasc Disord. 15:812015. View Article : Google Scholar : PubMed/NCBI
|