Introduction
Atrial fibrillation (AF) is the most common type of
cardiac arrhythmia and a major public health problem. The estimated
number of individuals with AF worldwide was 33.5 million in 2010
(1). The prevalence of AF is
increasing and is estimated to double in the United States by 2050
(2). AF is associated with an
increased risk of heart failure, thromboembolic diseases, such as
cardioembolic stroke, and mortality (3). Although the molecular mechanism
underlying AF is complex and has not been determined definitively,
several risk factors, including age, smoking, obesity,
hypertension, diabetes mellitus, valvular heart disease, coronary
artery disease and heart failure, have been identified clinically
(4). In addition to these
conventional risk factors, recent studies have reported the
importance of genetic factors in the development of AF (5). Genome-wide association studies
(GWASs) and subsequent meta-analyses of such studies have
identified several genes and loci that confer susceptibility to AF
in populations of European ancestry (6–10).
However, susceptibility genes for AF have not been determined
definitively in Japanese populations.
The majority of genetic variants previously reported
to be associated with AF have a minor allele frequency (MAF) of ≥5%
and a small individual effect size. Given that these common
variants explain only a fraction of the heritability of AF, it is
likely that low-frequency (0.5%≤ MAF <5%) or rare (MAF <0.5%)
variants with larger effect sizes contribute to the genetic
architecture of AF (11).
An exome-wide association study (EWAS) with exome
array-based genotyping was performed to identify single nucleotide
polymorphisms (SNPs), especially low-frequency or rare coding
variants with moderate to large effect sizes, that confer
susceptibility to AF in Japanese individuals. Given that GWAS
arrays of previous studies did not include most low-frequency or
rare variants, Illumina human arrays that cover functional SNPs in
entire exons including such variants were used in the current
study.
Materials and methods
Study subjects
A total of 13,166 Japanese individuals (884 patients
with AF and 12,282 controls) were examined. The subjects were
recruited either from individuals who visited outpatient clinics of
or were admitted to participating hospitals (Gifu Prefectural
Tajimi Hospital, Tajimi; Gifu Prefectural General Medical Center,
Gifu; Japanese Red Cross Nagoya First Hospital, Nagoya; Inabe
General Hospital, Inabe; Hirosaki University Hospital and Hirosaki
Stroke and Rehabilitation Center, Hirosaki, Japan) because of
various symptoms or for an annual health checkup between 2002 and
2014; from community-dwelling individuals recruited to a
population-based cohort study in Inabe between 2010 and 2014
(Health Care Center of Inabe General Hospital) or in Tokyo or
Kusatsu between 2011 and 2015 (Tokyo Metropolitan Institute of
Gerontology); or from cases of autopsy performed at the Tokyo
Metropolitan Geriatric Hospital from 1995 to 2012.
Individuals with persistent or paroxysmal AF or with
a history of AF were included in the AF patient group regardless of
medical treatment. Patients with AF who had severe valvular heart
disease, hypertrophic or dilated cardiomyopathy, or congenital
heart disease were excluded from the study. The control individuals
had no history of AF or other significant supraventricular or
ventricular arrhythmias or of taking antiarrhythmic medication.
Autopsy cases were not used as controls.
The study protocol complied with the Declaration of
Helsinki and was approved by the Committees on the Ethics of Human
Research of Mie University Graduate School of Medicine (Tsu,
Japan), Hirosaki University Graduate School of Medicine (Hirosaki,
Japan), Tokyo Metropolitan Institute of Gerontology (Tokyo, Japan)
and participating hospitals. Written informed consent was obtained
from each participant or families of the deceased subjects.
EWAS
Venous blood (5–7 ml) was collected into tubes
containing 50 mmol/l ethylenediaminetetraacetic acid (disodium
salt), peripheral blood leukocytes were isolated, and genomic DNA
was extracted from the cells with the use of a DNA extraction kit
(Genomix supplied by Talent Srl, Trieste, Italy; or SMITEST
EX-R&D supplied by Medical & Biological Laboratories, Co.,
Ltd., Nagoya, Japan) or by standard protocols based on
phenol-chloroform extraction and spin columns. Lysed blood cells
were mixed with an equal volume of a phenol-chloroform mixture.
Following centrifugation at 16,000 × g for 5 min at room
temperature, two distinct phases were formed. The upper aqueous
phase was collected, and genomic DNA was further purified by a spin
column-based DNA extraction method that is based on the fact that
DNA binds to the solid phase of silica under certain conditions. A
buffer solution (Buffer B3; Takara Bio, Inc., Otsu, Japan) was
added to the DNA sample along with 100% ethanol. This solution was
transferred to a spin column, and the column was centrifuged at
11,000 × g for 1 min at room temperature. A wash buffer [Buffer BW
(first wash) or Buffer B5 (second wash); Takara Bio, Inc.] was
added to the column, and the column was centrifuged at 11,000 × g
for 1 min at room temperature again. An elution buffer (Buffer BE;
Takara Bio, Inc.) was added to the column, and the column was
centrifuged at 11,000 × g for 1 min at room temperature again.
Genomic DNA was collected from the bottom of the column. In autopsy
cases, genomic DNA was extracted from kidneys. The EWAS was
performed for 884 subjects with AF and 12,282 control individuals
with the use of a HumanExome-12 v1.1 or v1.2 DNA Analysis BeadChip
or Infinium Exome-24 v1.0 BeadChip (Illumina, Inc., San Diego, CA,
USA). These exome arrays include putative functional exonic
variants selected from >12,000 individual exome or whole-genome
sequences. The exonic content consists of ~244,000 SNPs
representing diverse populations, including European, African,
Chinese and Hispanic individuals (12). SNPs contained in only one of the
exome arrays (~3.6%) were excluded from analysis. Quality control
(13) was performed as follows: i)
Genotyping data with a call rate of <97% were discarded, with
the mean call rate for the remaining data at 99.9%; ii) gender
specification was checked for all samples, and those for which
gender in the clinical records was inconsistent with genetic sex
were discarded; iii) duplicate samples and cryptic relatedness were
checked by calculation of identity by descent, all pairs of DNA
samples with identity by descent of >0.1875 were inspected and
one sample from each pair was excluded; iv) the frequency of SNP
heterozygosity was calculated for all samples, and those with
extremely low or high heterozygosity (>3 standard deviations
from the mean) were discarded; v) SNPs in sex chromosomes or
mitochondrial DNA were excluded from the analysis, as were
nonpolymorphic SNPs or SNPs with a MAF of <0.1%; vi) SNPs whose
genotype distributions deviated significantly (P<0.001) from
Hardy-Weinberg equilibrium in control individuals were discarded;
vii) the genotype data for the EWAS were examined for population
stratification by principal components analysis (14), and population outliers were
excluded from the analysis. A total of 41,243 SNPs passed quality
control and was subjected to analysis.
Statistical analysis
For analysis of characteristics of the study
subjects, quantitative data were compared between patients with AF
and control individuals with the unpaired Student's t test.
Categorical data were compared between the two groups with Fisher's
exact test. Allele frequencies were estimated by the gene counting
method, and Fisher's exact test was applied to identify departure
from Hardy-Weinberg equilibrium. Allele frequencies of SNPs were
compared between subjects with AF and controls with Fisher's exact
test in the EWAS. Multivariable logistic regression analysis was
performed with AF as a dependent variable and independent variables
including age, sex (0, woman; 1, man), the prevalence of
hypertension (0, no history of this condition; 1, positive
history), and genotype of each SNP. Genotypes of SNPs were assessed
according to dominant [0, AA; 1, AB+BB
(A, major allele; B, minor allele)], recessive (0,
AA+AB; 1, BB), and additive genetic models,
and the P-value, odds ratio, and 95% confidence interval
were calculated. Additive models comprised additive 1 (0,
AA; 1, AB; 0, BB) and additive 2 (0,
AA; 0, AB; 1, BB) scenarios, which were
analyzed simultaneously with a single statistical model. The
relation of SNPs to the prevalence of intermediate phenotypes of AF
was compared among genotypes with Fisher's exact test (2×2) or
Pearson's chi-square test (2×3). To compensate for multiple
comparisons of genotypes with AF, we applied Bonferroni's
correction for statistical significance of association. Given that
41,243 SNPs were analyzed, the significance level was set at
P<1.21×10−6 (0.05/41,243) for the EWAS. A
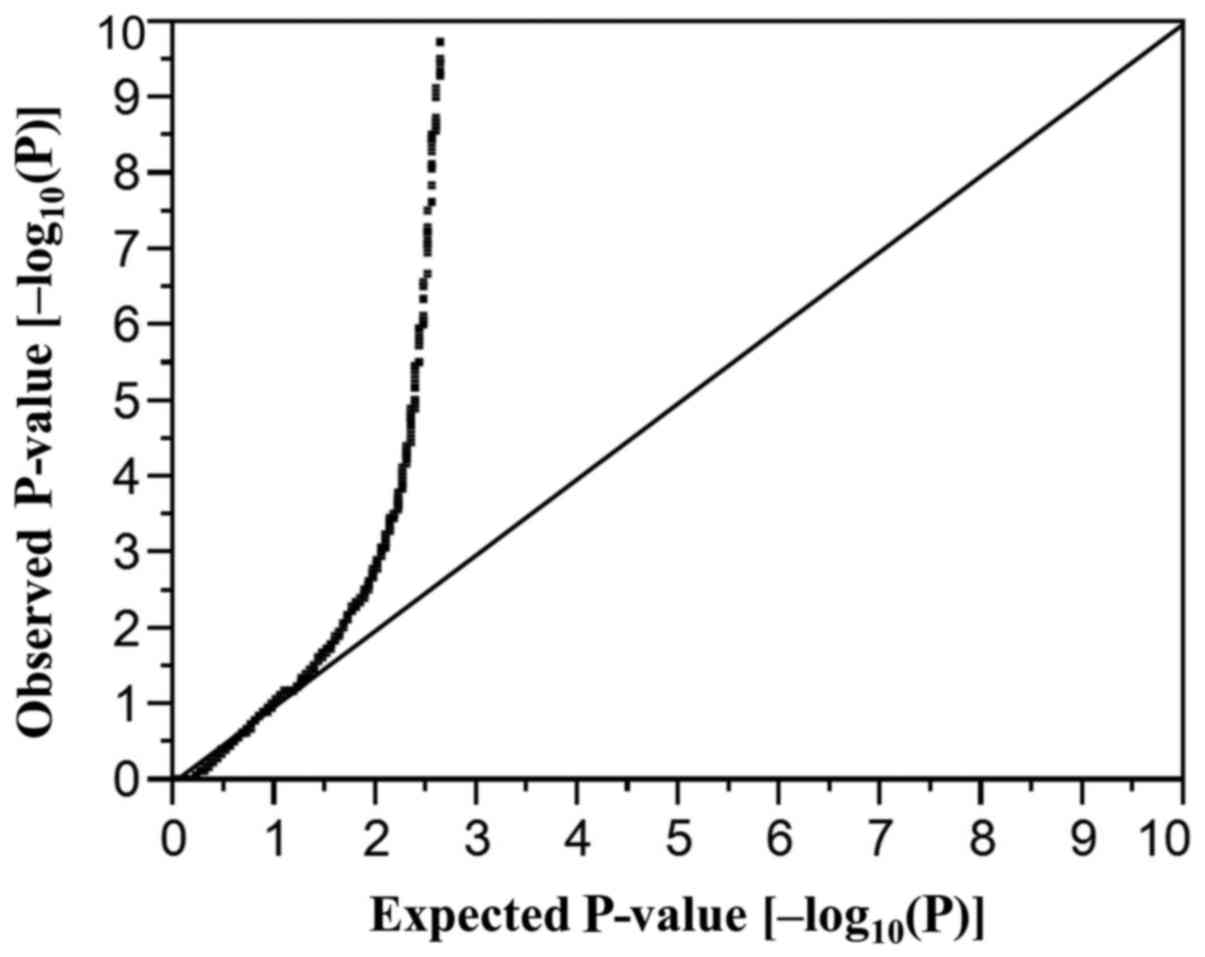
quantile-quantile plot for P-values of allele frequencies in the
EWAS for AF is shown in Fig. 1.
The inflation factor (λ) was 1.52. Bonferroni's correction was also
applied to other statistical comparisons as indicated. Statistical
tests were performed with JMP Genomics version 6.0 software (SAS
Institute, Inc., Cary, NC, USA).
Results
Characteristics of the study
subjects
The characteristics of the subjects enrolled in the
study are presented in Table I.
Age, the number of men, and the prevalence of hypertension,
diabetes mellitus, chronic kidney disease and hyperuricemia were
significantly greater in patients with AF than in control
subjects.
 | Table I.Characteristics of the 13,166 study
subjects. |
Table I.
Characteristics of the 13,166 study
subjects.
| Characteristic | Atrial
fibrillation | Control | P-value |
|---|
| No. of
subjects | 884 | 12,282 |
|
| Age (years) | 75.8±12.3 | 59.4±13.2 | <0.0001 |
| Sex (male/female,
%) | 67.8/32.2 | 56.8/43.2 | <0.0001 |
| Body mass index
(kg/m2) | 23.4±3.7 | 23.3±3.5 | 0.7412 |
| Current or former
smoker (%) | 33.6 | 36.3 | 0.2576 |
| Hypertension
(%) | 83.9 | 51.1 | <0.0001 |
| Diabetes mellitus
(%) | 51 | 23.2 | <0.0001 |
| Dyslipidemia
(%) | 65.3 | 62 | 0.188 |
| Chronic kidney
disease (%) | 40.3 | 23 | <0.0001 |
| Hyperuricemia
(%) | 31.9 | 17 | <0.0001 |
EWAS of AF
The association of AF with allele frequencies of
41,243 SNPs that passed quality control was determined using
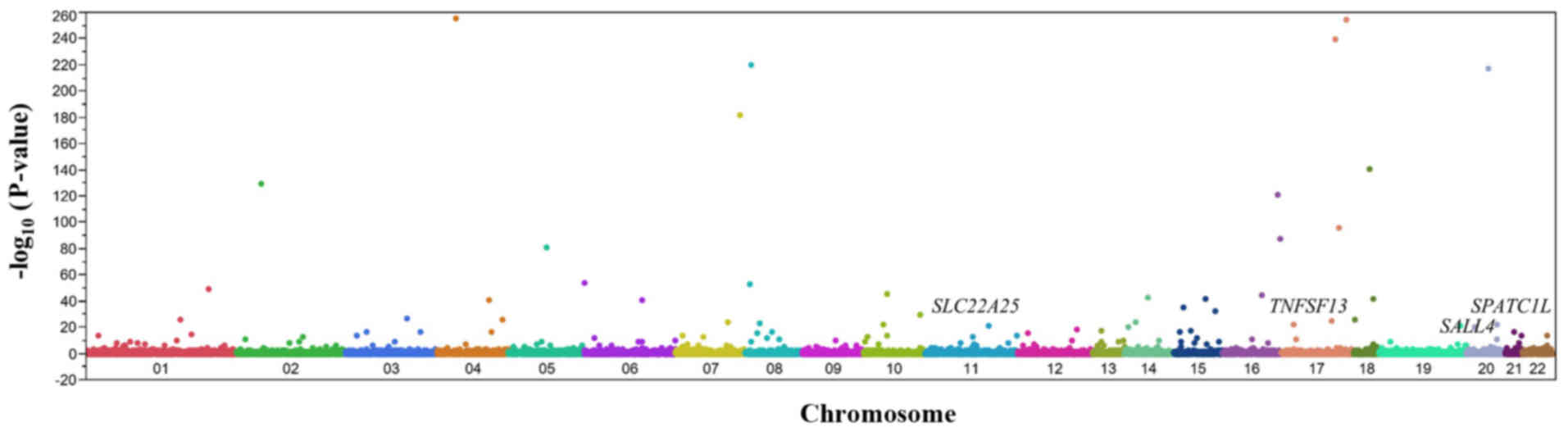
Fisher's exact test. A Manhattan plot for the EWAS of AF is
presented in Fig. 2. Following
Bonferroni's correction, 122 SNPs were significantly
[P<1.21×10−6 (0.05/41,243)] associated with AF
(Table II). The genotype
distributions of these SNPs were in Hardy-Weinberg equilibrium
(P>0.001) both among subjects with AF and among control
individuals (data not shown).
| Table II.The 122 SNPs significantly associated
with atrial fibrillation by exome-wide association analysis. |
Table II.
The 122 SNPs significantly associated
with atrial fibrillation by exome-wide association analysis.
| Gene | SNP | Nucleotide
substitution (amino acid)a | Chromosome:
Position | MAF (%) | P-value
(allele) | Allele OR |
|---|
| N4BP2 | rs61748749 | T/G (S1353R) | 4: 40122170 | 2.3 |
<1.0×10−255 | 0.74 |
| DNAH17 | rs690844 | A/C (I1742M) | 17: 78501838 | 2.5 |
8.94×10−255 | 1.31 |
| HELZ | rs184499441 | C/T (G1288R) | 17: 67114380 | 1.6 |
4.40×10−240 | 1.08 |
|
| rs7828656 | A/C | 8: 11645425 | 44.8 |
1.87×10−220 | 1.1 |
| SLA2 | rs221308 | T/C | 20: 36647995 | 46.7 |
4.11×10−218 | 0.99 |
| SSPO | rs55976638 | G/T | 7: 149832881 | 4.3 |
2.99×10−182 | 1.31 |
| TCEB3B | rs2010834 | A/C (F254C) | 18: 47034504 | 24.5 |
2.60×10−141 | 1.1 |
| FANCL | rs149731356 | T/C (T224A) | 2: 58165745 | 0.7 |
7.35×10−130 | 0.75 |
| PIEZO1 | rs143004911 | G/A (R333C) | 16: 88737957 | 2.7 |
5.07×10−122 | 0.87 |
| TTYH2 | rs9899862 | C/A (D423E) | 17: 74253090 | 6.3 |
1.31×10−96 | 0.86 |
| TUBB3 | rs2302898 | A/G | 16: 89932386 | 23.5 |
1.13×10−87 | 0.87 |
| SLCO6A1 | rs17150488 | T/C (K381R) | 5: 102438751 | 0.6 |
3.30×10−81 | 1.05 |
| GMDS | rs9378305 | C/T | 6: 1703056 | 41.4 |
1.20×10−54 | 0.99 |
| RP1L1 | rs79329877 | T/C | 8: 10622701 | 9.2 |
2.05×10−53 | 1.05 |
| FCMR | rs150080259 | T/G (S61R) | 1: 206902976 | 1.5 |
1.07×10−49 | 1.02 |
| RTKN2 | rs7090884 | A/G | 10: 62202267 | 33.2 |
4.97×10−46 | 1.05 |
| UTP4 | rs193164904 | A/G (I534V) | 16: 69163131 | 0.2 |
1.90×10−45 | 1.58 |
| SNAPC1 | rs74810099 | T/G (M36R) | 14: 61762567 | 2.8 |
1.99×10−43 | 1.02 |
| ALPK2 | rs3809977 | G/T (P1174H) | 18: 58536666 | 17.2 |
3.05×10−42 | 0.93 |
| CSPG4 | rs137981794 | T/C (D1936G) | 15: 75676712 | 0.4 |
4.05×10−42 | ND |
| MDN1 | rs9294445 | A/G (Y3423H) | 6: 89692763 | 7.5 |
1.15×10−41 | 0.99 |
| SETD7 | rs6814310 | C/A | 4: 139534374 | 48.4 |
1.64×10−41 | 0.97 |
| PLA2G4E | rs4924595 | T/C (N400S) | 15: 41995408 | 18.2 |
1.87×10−35 | 1.01 |
| KIF7 | rs117123311 | C/G (S788R) | 15: 89642233 | 3.1 |
1.03×10−32 | 1 |
| CTBP2 | rs3781411 | C/T (R298Q) | 10: 125026867 | 7 |
1.46×10−30 | 0.9 |
| GATA2 | rs78245253 | G/C (A250P) | 3: 128485850 | 4.6 |
1.37×10−27 | 0.79 |
| DLGAP1 | rs3745051 | C/T | 18: 3729175 | 34.4 |
1.64×10−26 | 0.86 |
|
| rs1711393 | T/C | 4: 178074088 | 31 |
1.70×10−26 | 1.14 |
| SLAMF7 | rs117009784 | A/C (R96S) | 1: 160750053 | 5.8 |
3.86×10−26 | 1.07 |
| USP32 | rs8079220 | C/T | 17: 60239372 | 20.5 |
3.56×10−25 | 0.91 |
|
| rs8011192 | T/G | 14: 28319412 | 33.1 |
8.46×10−25 | 0.99 |
| IMPDH1 | rs201001000 | G/A (T369M) | 7: 128395003 | 0.1 |
3.85×10−24 | 0.79 |
| ADRA1A | rs151273238 | G/A (T391M) | 8: 26770378 | 0.2 |
2.12×10−23 | 0.71 |
| TNFSF13 | rs11552708 | G/A (G67R) | 17: 7559238 | 36.6 |
1.32×10−22 | 0.54 |
| SLC18A3 | rs118107581 | A/G (I426V) | 10: 49612016 | 2 |
1.62×10−22 | 1.11 |
| NFATC2 | rs12479626 | T/C (H446R) | 20: 51475656 | 4.7 |
2.79×10−22 | 1.06 |
| TENM4 | rs3812723 | C/T (V396I) | 11: 78863031 | 1.7 |
1.03×10−21 | 0.89 |
| EPN1 | rs200478642 | C/T (P203L) | 19: 55689301 | 0.1 |
1.52×10−21 | 0.35 |
| HNRNPC | rs17197037 | A/G | 14: 21257495 | 29.1 |
3.26×10−21 | 0.99 |
| TMX4 | rs2076015 | T/C (R303G) | 20: 7982394 | 41.1 |
4.49×10−21 | 1.16 |
| FOXN4 | rs140167217 | G/A (S308F) | 12: 109281778 | 0.4 |
4.51×10−19 | 1.12 |
| CEP152 | rs145138194 | G/A (S894F) | 15: 48760148 | 1.2 |
6.92×10−18 | 0.73 |
| FREM2 | rs114864077 | C/T (P128L) | 13: 38687727 | 0.1 |
1.04×10−17 | 1.81 |
| CPA6 | rs4737845 | T/C | 8: 67542707 | 43.3 |
1.87×10−17 | 1.04 |
| KIF15 | rs146292440 | G/A (R1199H) | 3: 44843135 | 0.6 |
3.33×10−17 | 0.57 |
| MFSD1 | rs3765083 | A/G (I230V) | 3: 158819654 | 30.3 |
4.19×10−17 | 1.05 |
| BAHD1 | rs3743143 | A/G (E26G) | 15: 40458541 | 10.5 |
5.85×10−17 | 0.99 |
|
| rs1395821 | A/G | 4: 147126398 | 39.6 |
6.48×10−17 | 1.05 |
| BRWD1 | rs2183573 | G/A (P1511S) | 21: 39202379 | 46 |
1.11×10−16 | 1.06 |
| CD69 | rs199676648 | G/A (R32C) | 12: 9756390 | 0.3 |
2.93×10−16 | 0.9 |
| HR | rs12675375 | C/T (G337D) | 8: 22127432 | 43.4 |
8.47×10−16 | 1.02 |
| SOAT1 | rs143616084 | G/A (R292Q) | 1: 179345008 | 0.1 |
6.19×10−15 | ND |
| JMJD1C | rs149833441 | T/C (K878E) | 10: 63208491 | 1.2 |
9.49×10−15 | 1.44 |
| VWDE | rs848016 | A/G (F142S) | 7: 12389177 | 36.7 |
1.41×10−14 | 1.05 |
| VPS13D | rs143833298 | G/A (R830Q) | 1: 12276077 | 0.8 |
1.59×10−14 | 0.86 |
| SPATC1L | rs113710653 | C/T (E231K) | 21: 46161921 | 1.9 |
1.60×10−14 | 5.36 |
| SNX19 | rs117834100 | C/A (G416C) | 11: 130914694 | 6.9 |
1.83×10−14 | 0.89 |
|
| rs9854207 | A/C | 3: 27572825 | 40.3 |
3.41×10−14 | 1.04 |
| ARHGAP8 | rs5766113 | A/G | 22: 44855543 | 36.8 |
7.18×10−14 | 1.03 |
|
| rs4407763 | G/A | 7: 67553705 | 35.8 |
1.14×10−13 | 1.08 |
|
SLC22A25 | rs11231397 | G/C (R300T) | 11: 63183749 | 43.7 |
1.53×10−13 | 1.55 |
| XIRP2 | rs77219745 | G/A (G1839D) | 2: 167246908 | 7 |
2.89×10−13 | 0.81 |
| MCM10 | rs7905784 | A/T (T541S) | 10: 13192356 | 2.2 |
5.31×10−13 | 1.05 |
|
HIST1H2AC | rs198823 | G/T | 6: 26122705 | 22.9 |
1.60×10−12 | 0.86 |
|
| rs10102598 | G/A | 8: 47178128 | 37.7 |
1.61×10−12 | 0.96 |
| VPS13C | rs77555508 | G/A (S1798F) | 15: 61940726 | 0.6 |
2.47×10−12 | 1.44 |
| ADCY3 | rs7586879 | C/T | 2: 24894108 | 44.7 |
1.18×10−11 | 0.99 |
| CTC1 | rs183966301 | G/A (A1025V) | 17: 8229384 | 0.2 |
1.84×10−11 | 0.56 |
| SALL4 | rs77538589 | C/T (G117R) | 20: 51792134 | 4.3 |
2.24×10−11 | 2.61 |
| ADCY7 | rs201661947 | G/A (A475T) | 16: 50304414 | 0.2 |
3.37×10−11 | ND |
|
TP53INP1 | rs896854 | G/A | 8: 94948283 | 30.7 |
3.76×10−11 | 0.96 |
| TMEM245 | rs2271877 | C/T (A314T) | 9: 109091132 | 11.5 |
8.13×10−11 | 0.98 |
| FCRL1 | rs149740001 | A/T (K103I) | 1: 157803856 | 1 |
8.38×10−11 | 1.22 |
| SCYL2 | rs200554353 | T/C (M256T) | 12: 100312568 | 1.6 |
8.73×10−11 | 0.36 |
| TMCO3 | rs185071949 | C/T (P14L) | 13: 113495622 | 0.4 |
1.97×10−10 | 1.02 |
| WDR27 | rs3734905 | C/T | 6: 169558886 | 19.5 |
3.00×10−10 | 0.99 |
| NGB | rs117207261 | C/G (Q60E) | 14: 77269238 | 0.5 |
3.59×10−10 | 1.24 |
|
| rs6695567 | A/G | 1: 53163913 | 43.6 |
4.89×10−10 | 1.05 |
| FAP | rs151314911 | C/T | 2: 162173786 | 2.9 |
5.26×10−10 | 1.02 |
|
| rs13277113 | A/G | 8: 11491677 | 31.9 |
5.36×10−10 | 1.01 |
| ACER1 | rs72981971 | T/C (M74V) | 19: 6312279 | 36.5 |
5.41×10−10 | 1 |
| FREM2 | rs2496425 | T/C (F1070S) | 13: 38690553 | 38.3 |
7.29×10−10 | 1.01 |
| ASB13 | rs138695721 | A/C (V139G) | 10: 5649071 | 0.6 |
7.67×10−10 | 1.09 |
|
| rs10943716 | T/C | 6: 80543356 | 40.2 |
1.05×10−9 | 1.05 |
| CCDC168 | rs1449707 | A/G (I3015T) | 13: 102741653 | 44.4 |
1.82×10−9 | 0.98 |
| ADGRV1 | rs2366928 | A/G (K3471E) | 5: 90728918 | 17.9 |
2.17×10−9 | 0.97 |
| MDN1 | rs115931523 | G/A (T3130M) | 6: 89695987 | 0.2 |
2.44×10−9 | 1.58 |
| CD96 | rs140727933 | A/G (Y11C) | 3: 111542280 | 0.2 |
2.63×10−9 | 1.16 |
|
| rs4965121 | G/C | 15: 97975562 | 8.5 |
2.69×10−9 | 0.97 |
| KNL1 | rs11858113 | T/C (M598T) | 15: 40621979 | 27.4 |
3.29×10−9 | 0.99 |
| OR4X2 | rs7120775 | C/G (Y27*) | 11: 48245184 | 16.9 |
3.65×10−9 | 1.05 |
| TRPM2 | rs144412484 | A/G (E450G) | 21: 44390934 | 0.4 |
3.68×10−9 | 0.82 |
| MGAT5 | rs66523341 | C/T | 2: 134364804 | 37 |
4.17×10−9 | 0.93 |
| GCOM1 | rs4774980 | G/A | 15: 57691677 | 6 |
5.40×10−9 | 0.98 |
| CSMD2 | rs1874045 | T/C (K2096R) | 1: 33605925 | 45.1 |
7.56×10−9 | 1.08 |
| ADAT1 | rs200524721 | G/C (Q167H) | 16: 75612785 | 0.3 |
8.75×10−9 | 1.12 |
|
| rs4420065 | T/C | 1: 65695778 | 13.1 |
1.52×10−8 | 1.02 |
| NLRX1 | rs149129258 | C/A (P262Q) | 11: 119174034 | 0.5 |
2.31×10−8 | 0.99 |
| DNAAF3 | rs890871 | A/G (L280P) | 19: 55160687 | 25.2 |
3.01×10−8 | 0.98 |
| ZNF25 | rs150582814 | T/C (Y202C) | 10: 37952893 | 0.1 |
3.01×10−8 | ND |
| CMYA5 | rs62621915 | C/T (L1038F) | 5: 79731877 | 49.3 |
5.62×10−8 | 0.93 |
| SYDE2 | rs141587551 | C/A (D173Y) | 1: 85200480 | 6.6 |
6.20×10−8 | 1.03 |
| SLC15A5 | rs3915247 | C/T | 12: 16247679 | 47.6 |
7.01×10−8 | 0.97 |
|
CDC42BPG | rs3741395 | T/C (Q1135R) | 11: 64830034 | 5.6 |
8.31×10−8 | 1.11 |
|
| rs8030485 | G/A | 15: 79116590 | 35.2 |
9.03×10−8 | 0.98 |
|
| rs2564486 | G/T | 18: 59579061 | 12.9 |
9.24×10−8 | 1.05 |
| SLC4A4 | rs1062677 | A/C (I1074L) | 4: 71567828 | 6.1 |
1.20×10−7 | 0.91 |
| STEAP1B | rs17364464 | A/G | 7: 22474434 | 12.2 |
2.03×10−7 | 0.99 |
| KLF17 | rs11210969 | T/A (I35N) | 1: 44129375 | 22.5 |
2.83×10−7 | 1.11 |
|
ADAMTS13 | rs78977446 | C/T (S903L) | 9: 133445796 | 4.5 |
3.10×10−7 | 1.24 |
| ZNF879 | rs17078988 | A/G (T112A) | 5: 179032282 | 33.8 |
4.42×10−7 | 1.01 |
|
| rs1464833 | T/C | 7: 125475608 | 10.2 |
4.58×10−7 | 1.01 |
| PKD1L1 | rs10951936 | A/T | 7: 47882084 | 21 |
7.76×10−7 | 1.01 |
| SNX32 | rs200684568 | G/A (G179R) | 11: 65850787 | 0.5 |
9.00×10−7 | 0.56 |
| NTF3 | rs6332 | G/A | 12: 5494466 | 41.8 |
9.12×10−7 | 1.09 |
| EFHD1 | rs4072149 | T/C | 2: 232643967 | 36.7 |
9.18×10−7 | 1.01 |
| URB2 | rs3811473 | G/T (G778V) | 1: 229636946 | 15.8 |
9.48×10−7 | 1.05 |
| CCDC71 | rs4955419 | A/T (Q317L) | 3: 49163259 | 4.6 |
9.91×10−7 | 1.13 |
|
| rs543588 | T/G | 13: 30096848 | 31.4 |
1.01×10−6 | 0.9 |
| TRIM40 | rs757259 | G/A (E244K) | 6: 30147765 | 16 |
1.06×10−6 | 1.12 |
|
| rs3129264 | T/C | 6: 33133825 | 8.3 |
1.07×10−6 | 0.8 |
| SEMA6A | rs12516652 | G/T (D567E) | 5: 116475552 | 1.9 |
1.14×10−6 | 1.2 |
Multivariable logistic regression
analysis of the relation of SNPs to AF
The association of AF with 122 SNPs identified by
the EWAS was further examined by multivariable logistic regression
analysis with adjustment for age, sex and the prevalence of
hypertension. Eight SNPs were identified to be associated with AF
(P<0.01 in at least one genetic model; Table III). Among these eight SNPs,
rs11552708 [G/A (G67R)] of TNF superfamily member 13
(TNFSF13), rs113710653 [C/T (E231 K)] of spermatogenesis
and centriole associated 1 like (SPATC1L), and
rs11231397 [G/C (R300T)] of solute carrier family 22 member
25 (SLC22A25) were significantly
[P<1.02×10−4 (0.05/488)] associated with AF. The
minor T allele of rs113710653 and the minor C allele
of rs11231397 were risk factors for AF, whereas the minor A
allele of rs11552708 was protective against this condition
(Table III). In addition, the
association of rs77538589 [C/T (G117R)] of spalt like
transcription factor 4 (SALL4) with AF almost achieved
significance, with the minor T allele representing a risk
factor for this condition.
| Table III.Association of SNPs to atrial
fibrillation as determined by multivariable logistic regression
analysis. |
Table III.
Association of SNPs to atrial
fibrillation as determined by multivariable logistic regression
analysis.
|
|
| Dominant | Recessive | Additive 1 | Additive 2 |
|---|
|
|
|
|
|
|
|
|---|
| SNP | Nucleotide
substitution (amino acid) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) | P-value | OR (95% CI) |
|---|
| rs11552708 | G/A (G67R) |
9.36×10−9 | 0.58
(0.49–0.70) |
3.44×10−5 | 0.54
(0.39–0.73) |
3.31×10−6 | 0.63
(0.52–0.77) |
3.66×10−8 | 0.43
(0.30–0.59) |
| rs2076015 | T/C (R303G) | 0.1339 |
| 0.0028 | 1.38
(1.12–1.69) | 0.6253 |
| 0.0038 | 1.41
(1.12–1.78) |
| rs113710653 | C/T (E231K) |
1.09×10−5 | 3.27
(2.00–5.14) | 0.6731 |
|
9.01×10−6 | 3.32
(2.02–5.21) | 0.6802 |
|
| rs11231397 | G/C (R300T) | 0.0016 | 1.40
(1.13–1.73) | 0.0004 | 1.51
(1.20–1.88) | 0.03 | 1.28
(1.02–1.60) |
3.71×10−5 | 1.77
(1.35–2.31) |
| rs77219745 | G/A (G1839D) | 0.2458 |
| 0.0076 | <0.01 (ND) | 0.4205 |
| 0.0072 | <0.01 (ND) |
| rs77538589 | C/T (G117R) | 0.0002 | 1.88
(1.36–2.56) | 0.4697 |
| 0.0002 | 1.90
(1.37–2.58) | 0.4825 |
|
| rs141587551 | C/A (D173Y) | 0.7965 |
| 0.0046 | 3.39
(1.50–7.07) | 0.6564 |
| 0.0048 | 3.37
(1.49–7.02) |
| rs3129264 | T/C | 0.0422 | 0.78
(0.61–0.99) | 0.0016 | <0.01
(0.00–0.36) | 0.1138 |
| 0.0014 | <0.01 (ND) |
Association of SNPs with intermediate
phenotypes of AF
The association of the four SNPs, rs11552708,
rs113710653, rs11231397 and rs77538589, with intermediate
phenotypes of AF was examined. The rs11552708 SNP was significantly
associated with hyperuricemia [P<0.0016 (0.05/32); Table IV].
| Table IV.P-values of the association between
SNPs and intermediate phenotypes of atrial fibrillation. |
Table IV.
P-values of the association between
SNPs and intermediate phenotypes of atrial fibrillation.
| SNP | Nucleotide
substitution (amino acid) | Hypertension | DM | Hyper-TG | Hypo-HDL | Hyper-LDL | CKD | Obesity | HU |
|---|
| rs11552708 | G/A (G67R) | 0.9943 | 0.7439 | 0.8296 | 0.0622 | 0.8902 | 0.0754 | 0.472 | 0.0006 |
| rs113710653 | C/T (E231K) | 0.1278 | 0.5273 | 0.1465 | 0.6862 | 0.2732 | 0.4215 | 0.9986 | 0.1295 |
| rs11231397 | G/C (R300T) | 0.2471 | 0.9445 | 0.12 | 0.1683 | 0.9415 | 0.9571 | 0.1993 | 0.9519 |
| rs77538589 | C/T (G117R) | 0.1179 | 0.8113 | 0.4675 | 0.6656 | 0.5844 | 0.8815 | 0.8741 | 0.6006 |
Association of genes and SNPs
identified in the present study with phenotypes previously examined
in GWASs
The association of the four genes and four SNPs
identified in the present study with phenotypes previously examined
in GWASs available in public databases [GWAS Catalog (http://www.ebi.ac.uk/gwas) and GWAS Central
(http://www.gwascentral.org/browser)]
was determined. None of the genes or SNPs were identified to be
associated with AF in previous studies (data not shown).
Discussion
The development of AF has been reported to be
associated with endothelial dysfunction, including the upregulation
of adhesion molecules, and increased inflammation and oxidative
stress (15). Such endothelial
dysfunction also promotes the electrophysiological remodeling
observed in AF (16) and
accelerates atrial ectopy through electrical excitation of cells
near the pulmonary vein (17). In
addition, coronary atherosclerosis may injure atrial tissue as
result of myocardial ischemia and give rise to histological
changes, including myocyte hypertrophy, necrosis and apoptosis
(18,19). These processes may promote atrial
remodeling with structural, functional, electrical, and metabolic
consequences that underlie the development of AF (18,19).
The current study demonstrated that rs11552708 [G/A
(G67R)] of TNFSF13, rs113710653 [C/T (E231K)] of
SPATC1L and rs11231397 [G/C (R300T)] of SLC22A25 were
significantly associated with AF in Japanese individuals. The minor
T allele of rs113710653 and the minor C allele of
rs11231397 were risk factors for AF, whereas the minor A
allele of rs11552708 was protective against this condition. The
rs77538589 [C/T (G117R)] SNP of SALL4 also showed a tendency
to be associated to AF, although the association did not achieve
statistical significance, with the minor T allele
representing a risk factor for this condition.
The TNFSF13 gene is located at chromosomal
region 17p13.1 (http://www.ncbi.nlm.nih.gov/gene/8741) and is
expressed in various tissues and organs including heart muscle (The
Human Protein Atlas, www.proteinatlas.org/ENSG00000161955-TNFSF13/tissue).
TNFSF13 is a regulator of the development and function of B cells
(20). It has been suggested that
TNFSF13 has important roles in normal immune responses, and also in
the development of autoimmune and inflammatory diseases. Plasma
levels of TNFSF13 were reported to be increased in patients with
multiple sclerosis (21),
Sjögren's syndrome (22), systemic
lupus erythematosus (23) and
rheumatoid arthritis (24). The
levels of TNFSF13 were also demonstrated to be significantly higher
in the synovial fluid of patients with rheumatoid arthritis than in
the serum, suggesting that this protein may be produced locally at
the site of inflammation (25).
Previous GWASs identified TNFSF13 as a susceptibility locus
for immunoglobulin A nephropathy (26). Chronic vascular inflammation is an
important feature of atherosclerosis (27), and atherosclerosis was previously
reported to be associated with increased expression of TNFSF13. The
plasma levels of TNFSF13 were demonstrated to be greater in
patients with coronary artery disease than in healthy controls, and
TNFSF13 immunoreactivity was detected in aggregated platelets
within the ruptured plaque of patients with myocardial infarction
(28). Pronounced TNFSF13
immunostaining was also observed in macrophages from carotid
plaques (28). The current study
demonstrated that rs11552708 [G/A (G67R)] of TNFSF13 was
significantly associated with AF, with the minor A allele
being protective against this condition. Chronic vascular
inflammation, atherosclerosis, endothelial dysfunction and
subsequent atrial remodeling are important in the pathogenesis of
AF. Given that TNFSF13 is implicated in vascular inflammation and
atherosclerosis (28), the
association of TNFSF13 rs11552708 [G/A (G67R)] with AF may
be attributable to the effect of this gene on atrial
remodeling.
The SPATC1L gene is located at chromosomal
region 21q22.3 (http://www.ncbi.nlm.nih.gov/gene/84221) and is
expressed in various tissues and organs including heart muscle (The
Human Protein Atlas, www.proteinatlas.org/ENSG00000160284-SPATC1L/tissue).
SPATC1L is distributed in the cytoplasm, nucleus and perinuclear
region of cells, and it translocates to sites of cell-cell
junctions in response to cell stimulation with neurokinin A
(29). Expression of
SPATC1L modulates the response of human cells to
N-methyl-N'-nitro-N-nitrosoguanidine,
suggesting that the encoded protein may have a role in protecting
cells from cell death induced by this DNA-damaging agent (30). The current study demonstrated that
rs113710653 [C/T (E231K)] of SPATC1L was significantly
associated with AF, with the minor T allele representing a
risk factor for this condition, although the underlying molecular
mechanism of this association remains unclear.
The SLC22A25 gene is located at chromosomal
region 11q12.3 (http://www.ncbi.nlm.nih.gov/gene/387601) and is
abundantly expressed in the liver (The Human Protein Atlas,
www.proteinatlas.org/ENSG00000196600-SLC22A25/tissue).
SLC22 is a large family of transmembrane organic cation and anion
transporter proteins expressed predominantly in kidney and liver
that mediate the uptake and excretion of environmental toxins,
endogenous substances and drugs in the body (31). The results of the present study
indicated that rs11231397 [G/C (R300T)] of SLC22A25 was
significantly associated with AF, with the minor C allele
representing a risk factor for this condition, although the
functional relevance of this association remains unknown.
The SALL4 gene is located at chromosomal
region 20q13.2 (http://www.ncbi.nlm.nih.gov/gene/57167) and is
expressed in various tissues and organs including heart muscle (The
Human Protein Atlas, www.proteinatlas.org/ENSG00000101115-SALL4/tissue).
Mutations in SALL4 have been identified as a cause of
Okihiro syndrome, which is characterized by developmental defects
in limbs and multiple organs including the heart (32,33).
Expression of SALL4 was detected in the left ventricular
myocardium and interventricular septum of the mouse heart at
embryonic day 10.5 (34). The
current study demonstrated that rs77538589 [C/T (G117R)] of
SALL4 tended to be associated with AF, with the minor
T allele representing a risk factor for this condition.
Given that SALL4 may have an important role in heart development,
the association of rs77538589 of SALL4 with AF may reflect
an effect of this gene on atrial structure or electrical
function.
A meta-analysis of GWASs for AF (10) identified 10 SNPs (rs6666258 of
potassium calcium-activated channel subfamily N member 3,
rs3903239 of paired related homeobox 1, rs6817105 of
paired like homeodomain 2, rs2040862 of Wnt family member
8A, rs3807989 of caveolin 1, rs10821415 of chromosome
9 open reading frame 3, rs10824026 of synaptopodin 2
like, rs1152591 of spectrin repeat containing nuclear
envelope protein 2, rs7164883 of hyperpolarization activated
cyclic nucleotide gated potassium channel 4 and rs2106261 of
zinc finger homeobox 3) as susceptibility loci for this
condition. The MAF of these SNPs ranged from 13.1–44.7% and the
odds ratio from 0.87–1.64 (10).
In the current study, four novel loci that may confer
susceptibility to AF were identified, with the allele odds ratio
(MAF, %) of rs11552708 of TNFSF13, rs113710653 of
SPATC1L, rs11231397 of SLC22A25 and rs77538589 of
SALL4 being 0.54 (36.6%), 5.36 (1.9%), 1.55 (43.7%) and 2.61
(4.3%), respectively. Both rs11552708 of TNFSF13 and
rs11231397 of SLC22A25 were thus common variants with a low
effect size, whereas rs113710653 of SPATC1L and rs77538589
of SALL4 were low-frequency variants with a moderate to high
effect size.
There are several limitations to the present study:
i) Given that the results of the study were not replicated, the
findings will require validation in other independent subject
panels or other ethnic groups; ii) it is possible that rs11552708
of TNFSF13, rs113710653 of SPATC1L, rs11231397 of
SLC22A25 or rs77538589 of SALL4 is in linkage
disequilibrium with other polymorphisms in the same gene or other
nearby genes that are actually responsible for the development of
AF; and iii) the functional relevance of these SNPs to the
pathogenesis of AF remains to be elucidated.
In conclusion, rs11552708 of TNFSF13,
rs113710653 of SPATC1L, rs11231397 of SLC22A25, and
rs77538589 of SALL4 may be susceptibility loci for AF in
Japanese individuals. Determination of genotypes of these SNPs may
prove informative for assessment of the genetic risk for AF in
Japanese individuals.
Acknowledgements
This work was supported by CREST (grant no.
JPMJCR1302), Japan Science and Technology Agency (to Y.Y, J.S. and
I.T.) and by Japan Society for the Promotion of Science KAKENHI
grants no. JP15H04772 (to Y.Y.), no. JP25242062 (to M.T.) and no.
JP16H01872 (to M.T.).
References
|
1
|
Chugh SS, Havmoeller R, Narayanan K, Singh
D, Rienstra M, Benjamin EJ, Gillum RF, Kim YH, McAnulty JH Jr,
Zheng ZJ, et al: Worldwide epidemiology of atrial fibrillation: A
global burden of disease 2010 study. Circulation. 129:837–847.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Go AS, Hylek EM, Phillips KA, Chang Y,
Henault LE, Selby JV and Singer DE: Prevalence of diagnosed atrial
fibrillation in adults: National implications for rhythm management
and stroke prevention: The An ticoagulation and risk factors in
atrial fibrillation (ATRIA) study. JAMA. 285:2370–2375. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lip GY, Tse HF and Lane DA: Atrial
fibrillation. Lancet. 379:648–661. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schnabel RB, Sullivan LM, Levy D, Pencina
MJ, Massaro JM, D'Agostion RB Sr, Newton-Cheh C, Yamamoto JF,
Magnani JW, Tadros TM, et al: Development of a risk score for
atrial fibrillation (Framingham Heart Study): A community-based
cohort study. Lancet. 373:739–745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lubitz SA, Ozcan C, Magnani JW, Kääb S,
Benjamin EJ and Ellinor PT: Genetics of atrial fibrillation:
Implications for future research directions and personalized
medicine. Circ Arrhythm Electrophysiol. 3:291–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gudbjartsson DF, Arnar DO, Helgadottir A,
Gretarsdottir S, Holm H, Sigurdsson A, Jonasdottir A, Baker A,
Thorleifsson G, Kristjansson K, et al: Variants conferring risk of
atrial fibrillation on chromosome 4q25. Nature. 448:353–357. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gudbjartsson DF, Holm H, Gretarsdottir S,
Thorleifsson G, Walters GB, Thorgeirsson G, Gulcher J, Mathiesen
EB, Njølstad I, Nyrnes A, et al: A sequence variant in ZFHX3 on
16q22 associates with atrial fibrillation and ischemic stroke. Nat
Genet. 41:876–878. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Benjamin EJ, Rice KM, Arking DE, Pfeufer
A, van Noord C, Smith AV, Schnabel RB, Bis JC, Boerwinkle E, Sinner
MF, et al: Variants in ZFHX3 are associated with atrial
fibrillation in individuals of European ancestry. Nat Genet.
41:879–881. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ellinor PT, Lunetta KL, Glazer NL, Pfeufer
A, Alonso A, Chung MK, Sinner MF, de Bakker PI, Mueller M, Lubitz
SA, et al: Common variants in KCNN3 are associated with lone atrial
fibrillation. Nat Genet. 42:240–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellinor PT, Lunetta KL, Albert CM, Glazer
NL, Ritchie MD, Smith AV, Arking DE, Müller-Nurasyid M, Krijthe BP,
Lubitz SA, et al: Meta-analysis identifies six new susceptibility
loci for atrial fibrillation. Nat Genet. 44:670–675. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Manolio TA, Collins FS, Cox NJ, Goldstein
DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR,
Chakravarti A, et al: Finding the missing heritability of complex
diseases. Nature. 461:747–753. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Grove ML, Yu B, Cochran BJ, Haritunians T,
Bis JC, Taylor KD, Hansen M, Borecki IB, Cupples LA, Fornage M, et
al: Best practices and joint calling of the HumanExome BeadChip:
The CHARGE consortium. PLoS One. 8:e680952013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Anderson CA, Pettersson FH, Clarke GM,
Cardon LR, Morris AP and Zondervan KT: Data quality control in
genetic case-control association studies. Nat Protoc. 5:1564–1573.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Price AL, Patterson NJ, Plenge RM,
Weinblatt ME, Shadick NA and Reich D: Principal components analysis
corrects for stratification in genome-wide association studies. Nat
Genet. 38:904–909. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
O'Neal WT, Efird JT, Yeboah J, Nazarian S,
Alonso A, Heckbert SR and Soliman EZ: Brachial flow-mediated
dilation and incident atrial fibrillation: The multi-ethnic study
of atherosclerosis. Arterioscler Thromb Vasc Biol. 34:2717–2720.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim YM, Guzik TJ, Zhang YH, Zhang MH,
Kattach H, Ratnatunga C, Pillai R, Channon KM and Casadei B: A
myocardial Nox2 containing NAD (P)H oxidase contributes to
oxidative stress in human atrial fibrillation. Circ Res.
97:629–636. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haïssaguerre M, Jaïs P, Shah DC, Takahashi
A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Métayer P and
Clémenty J: Spontaneous initiation of atrial fibrillation by
ectopic beats origination in the pulmonary veins. N Engl J Med.
339:659–666. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Herringa J, Van Der Kuip DA, Hofman A,
Kors JA, van Rooij FJ, Lip GY and Witteman JC: Subclinical
atherosclerosis and risk of atrial fibrillation: The rotterdam
study. Arch Intern Med. 167:382–387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Casaclang-Verzosa G, Gresh BJ and Tsang
TS: Structural and functional remodeling of the left atrium:
Clinical and therapeutic implications for atrial fibrillation. J Am
Coll Cardiol. 51:1–11. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dillon SR, Gross JA, Ansell SM and Novak
AJ: An APRIL to remember: Novel TNF ligands as therapeutic targets.
Nat Rev Drug Discov. 5:235–246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Thangarajh M, Masterman T, Rot U, Duvefelt
K, Brynedal B, Karrenbauer VD and Hillert J: Increased levels of
APRIL (a proliferation-inducing ligand) mRNA in multiple sclerosis.
J Neuroimmunol. 167:210–214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jonsson MV, Szodoray P, Jellestad S,
Jonsson R and Skarstein K: Association between circulating levels
of the novel TNF family members APRIL and BAFF and lymphoid
organization in primary Sjögren's syndrome. J Clin Immunol.
25:189–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Koyama T, Tsukamoto H, Miyagi Y, Himeji D,
Otsuka J, Miyagawa H, Harada M and Horiuchi T: Raised serum APRIL
levels in patients with systemic lupus erythematosus. Ann Rheum
Dis. 64:1065–1067. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cheema GS, Roschke V, Hilbert DM and Stohl
W: Elevated serum B lymphocyte stimulator levels in patients with
systemic immune-based rheumatic diseases. Arthritis Rheum.
44:1313–1319. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan SM, Xu D, Roschke V, Perry JW, Arkfeld
DG, Ehresmann GR, Migone TS, Hilbert DM and Stohl W: Local
production of B lymphocyte stimulator protein and APRIL in
arthritic joints of patients with inflammatory arthritis. Arthritis
Rheum. 48:982–992. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi
S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, et
al: Discovery of new risk loci for IgA nephropathy implicates genes
involved in immunity against intestinal pathogens. Nat Genet.
46:1187–1196. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sandberg WJ, Otterdal K, Gullestad L,
Halvorsen B, Ragnarsson A, Frøland SS, Damås JK, Oie E, Aukrust P,
Hansson GK and Yndestad A: The tumour necrosis factor superfamily
ligand APRIL (TNFSF13) is released upon platelet activation and
expressed in atherosclerosis. Thromb Haemost. 102:704–710.
2009.PubMed/NCBI
|
|
29
|
Lecat S, Matthes HW, Pepperkok R, Simpson
JC and Galzi JL: A fluorescent live imaging screening assay based
on translocation criteria identifies novel cytoplasmic proteins
implicated in G protein-coupled receptor signaling pathways. Mol
Cell Proteomics. 14:1385–1399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fry RC, Svensson JP, Valiathan C, Wang E,
Hogan BJ, Bhattacharya S, Bugni JM, Whittaker CA and Samson LD:
Genomic predictors of interindividual differences in response to
DNA damaging agents. Genes Dev. 22:2621–2626. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jacobsson JA, Haitina T, Lindblom J and
Fredriksson R: Identification of six putative human transporters
with structural similarity to the drug transporter SLC22 family.
Genomics. 90:595–609. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kohlhase J, Heinrich M, Schubert L,
Liebers M, Kispert A, Laccone F, Turnpenny P, Winter RM and Reardon
W: Okihiro syndrome is caused by SALL4 mutations. Hum Mol Genet.
11:2979–2987. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sakaki-Yumoto M, Kobayashi C, Sato A,
Fujimura S, Matsumoto Y, Takasato M, Kodama T, Aburatani H,
Asashima M, Yoshida N and Nishinakamura R: The murine homolog of
SALL4, a causative gene in Okihiro syndrome, is essential for
embryonic stem cell proliferation, and cooperates with Sall1 in
anorectal, heart, brain and kidney development. Development.
133:3005–3013. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koshiba-Takeuchi K, Takeuchi JK, Arruda
EP, Kathiriya IS, Mo R, Hui CC, Srivastava D and Bruneau BG:
Cooperative and antagonistic interactions between Sall4 and Tbx5
pattern the mouse limb and heart. Nat Genet. 38:175–183. 2006.
View Article : Google Scholar : PubMed/NCBI
|