Introduction
The risk of death by cardiovascular disease (CVD) is
greatly increased in chronic kidney disease (CKD). Besides the
traditional risk factors, uremic toxins may be directly responsible
for the pathogenesis of CVD in CKD. Asymmetric dimethylarginine
(ADMA) is one of the most important uremic toxins that promote the
progression of CKD (1). There is
evidence of a strong association among atherosclerosis risk factors
and ADMA levels (2–6). In previous reports, ADMA induced
endothelial cell apoptosis (7,8), but
the molecular biological mechanisms are not clear, yet. Endothelial
cell injury and apoptosis can increase vascular permeability and
enhance platelet activation and aggregation, thus promoting
atherosclerosis and CVD.
Several recent studies proposed a role for
endoplasmic reticulum (ER) stress in endothelial cell apoptosis and
atherosclerosis. The ER facilitates protein folding and secretion.
Impairment of normal ER function leads to an imbalance in protein
homeostasis, accompanied by accumulation of misfolded proteins in
the ER lumen, a condition referred to as ER stress. Activation of
ER stress can ultimately return the ER to its normal physiological
state. Three pathways activated during ER stress have been
identified so far: Protein kinase RNA-like ER kinase (PERK),
inositol requiring enzyme-1 (IRE1) and activating transcription
factor 6 (ATF6). After activation of ER stress, improperly folded
proteins undergo degradation, leading to expression of target genes
including the ER chaperone, 78-kDa glucose-regulated protein
(GRP78/BiP), and factors involved in degradation of unfolded
proteins. The hypothesized purpose of ER stress is to promote cell
survival by restoring ER function. However, prolonged activation of
ER stress, an indication that the ER stress cannot be mitigated and
homeostasis cannot be reestablished, correlates with cell death.
Recently, PERK and IRE1 have been identified as factors involved in
cell death (9,10).
The ER stress pathway can be evoked by abnormalities
in calcium homeostasis. Calcium actively accumulates by
sarco/endoplasmic reticulum calcium-ATPases (SERCAs) in the ER
(11,12). Because the SERCA-dependent calcium
transport is the only calcium uptake system in this organelle,
regulation of SERCA function by the cell constitutes a key
regulatory mechanism of calcium homeostasis in the ER. Impaired
SERCA function is regarded as a major contributor to ER stress
(13,14). The SERCA pump is encoded by a
family of three genes, SERCA1, 2 and 3, which
are highly conserved and located on different chromosomes (15). Expression of SERCA isoforms is
cell-specific and developmentally regulated. SERCA3 has been
consistently shown to be co-expressed with SERCA2b in
differentiated human umbilical vein endothelial cells (HUVECs)
(16,17).
In this study, we investigated whether ER stress is
involved in ADMA-induced HUVEC apoptosis. We also explored whether
the SERCA pathway is responsible for the ADMA-induced ER
stress.
Materials and methods
Chemical reagents
ADMA was from Sigma-Aldrich (St. Louis, MO, USA).
GRP78/BiP, IRE1 and cleaved caspase-3 antibodies were obtained from
Cell Signaling Technology (Beverly, MA, USA). Bcl2, p-PERK, GAPDH
and β-actin antibodies were purchased from Santa Cruz Biotechnology
Inc. (Santa Cruz, CA, USA). SERCA2b and SERCA3 antibodies were
purchased from Abcam Inc. (Cambridge, MA, USA). All other reagents,
unless specially stated, were purchased from Sigma-Aldrich.
Cell culture
Primary HUVECs were from ScienCell Research
Laboratories Inc. (Carlsbad, CA, USA). HUVECs were cultured in
fibronectin-coated flasks using endothelial cell medium (ECM,
Sciencell Inc.) with bullet kit additives (Sciencell Inc.) and 10%
(v/v) fetal bovine serum (Sciencell Inc.). HUVECs used in the
experiments were at passages 3–4.
Western blot analysis
Total protein was extracted from HUVECs with lysis
buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1%
sodium deoxycholate, 0.1% SDS and 1% PMSF). Protein concentration
was determined using a BCA protein assay kit (Thermo Fisher
Scientific, Waltham, MA, USA). Samples were mixed with an equal
volume of 5´SDS loading buffer (125 mM Tris-HCl, 4% SDS, 100 mM
DTT, 20% glycerol and 0.2% bromophenol blue) and heated at 99°C for
10 min. Subsequently, sample aliquots (20 µg protein each) were
resolved on an 8–12% SDS-PAGE gel and the protein bands were
electrophoretically transferred onto a nitrocellulose membrane
(Amersham International plc., Cardiff, UK). Membranes were blocked
with 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1%
Tween 20 (TBST) for 2.5 h at room temperature, followed by an
overnight incubation with primary antibodies at 4°C. The membranes
were then incubated with horseradish peroxidase-conjugated
secondary antibodies for 1 h at room temperature, exposed to
enhanced chemiluminescence (ECL) kit (Millipore Co., Bedford, MA,
USA) and then to Kodak X-OMAT film (Eastman Kodak Inc., Rochester,
NY, USA). Primary antibodies and dilutions were: anti-Bcl2
(1:1,000), anti-cleaved caspase-3 (1:1,000), anti-IRE1 (1:1,000),
anti-p-PERK (1:1,000), anti-GPR78/BiP (1:1,000), anti-SERCA2b
(1:500), anti-SERCA3 (1:500), anti-GAPDH (1:3,000) and anti-β-actin
(1:5,000).
Terminal deoxynucleotidyl transferase
(TdT)-mediated-digoxigenin-11-dUTP nick end labeling (TUNEL)
assay
Apoptotic cells were evaluated by the TUNEL assay,
using the In Situ Cell Death Detection kit (Roche
Diagnostics, Mannheim, Germany) according to the manufacturer's
recommendations. Briefly, cells were fixed with 4% paraformaldehyde
for 60 min at room temperature. After washing with
phosphate-buffered saline (PBS), the cells were permeabilized with
0.1% Triton X-100 for 2 min at 4°C, and then each sample was
incubated with 45 µl of labeling solution plus 5 µl of enzyme
solution at 37°C for 1 h. The cells were then washed three times
with PBS. Cell nuclei were stained with
4′,6-diamidino-2-phenylindole (DAPI) for 15 min, and then washed
with PBS for 5 min at room temperature. Finally, the cells were
mounted onto coverslips. Cell images were acquired by fluorescence
microscopy (Leica DM 4000B, Germany).
Measurement of SERCA activity
SERCA activity was measured based on
Ca2+-dependent ATP hydrolysis and phosphate (Pi)
release. HUVECs were incubated with various concentrations of ADMA
for 24 h. They were then washed once with cold saline (0.9% NaCl in
distilled water) and resuspended in saline. After sonication of
samples for 5s, SERCA activity was measured with a Super Microscale
Ca2+-ATPase Detection Kit (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) according to the
manufacturer's protocol.
Cytosolic
Ca2+([Ca2+]cyt) measurements
Cytosolic calcium concentrations were measured using
a LEICA TCS SP5 confocal microscope with the calcium-sensitive
indicator Fluo-3 AM. Cells were incubated with loading buffer (10
mM HEPES, 0.5 mM Na2HPO4, 137 mM NaCl, 5 mM
KCl, 1.3 mM CaCl2, 0.7 mM MgCl2 and 5 mM
glucose, pH 7.4) containing 5 µM Fluo-3 AM (Molecular Probes,
Eugene, OR, USA) and 0.02% pluronic acid (Molecular Probes) at 37°C
for 45 min. The cells were then washed twice with loading buffer.
Before measuring fluorescent signals, cells were washed in
calcium-free medium to remove any dye nonspecifically associated
with the cell surface, and then incubated for another 30 min to
allow complete de-esterification of the intracellular AM esters.
Changes in intracellular Ca2+ were then monitored by
confocal fluorescence microscopy with emission and excitation at
520 and 485 nm, respectively.
Statistical analysis
Data are expressed as mean ± SE. Comparisons among
experimental groups were made by one-way ANOVA. Differences in mean
values were considered significant at P<0.05.
Results
ADMA treatment of HUVECs induces
apoptosis
TUNEL staining was used to confirm apoptosis. As
shown by TUNEL analysis (Fig. 1),
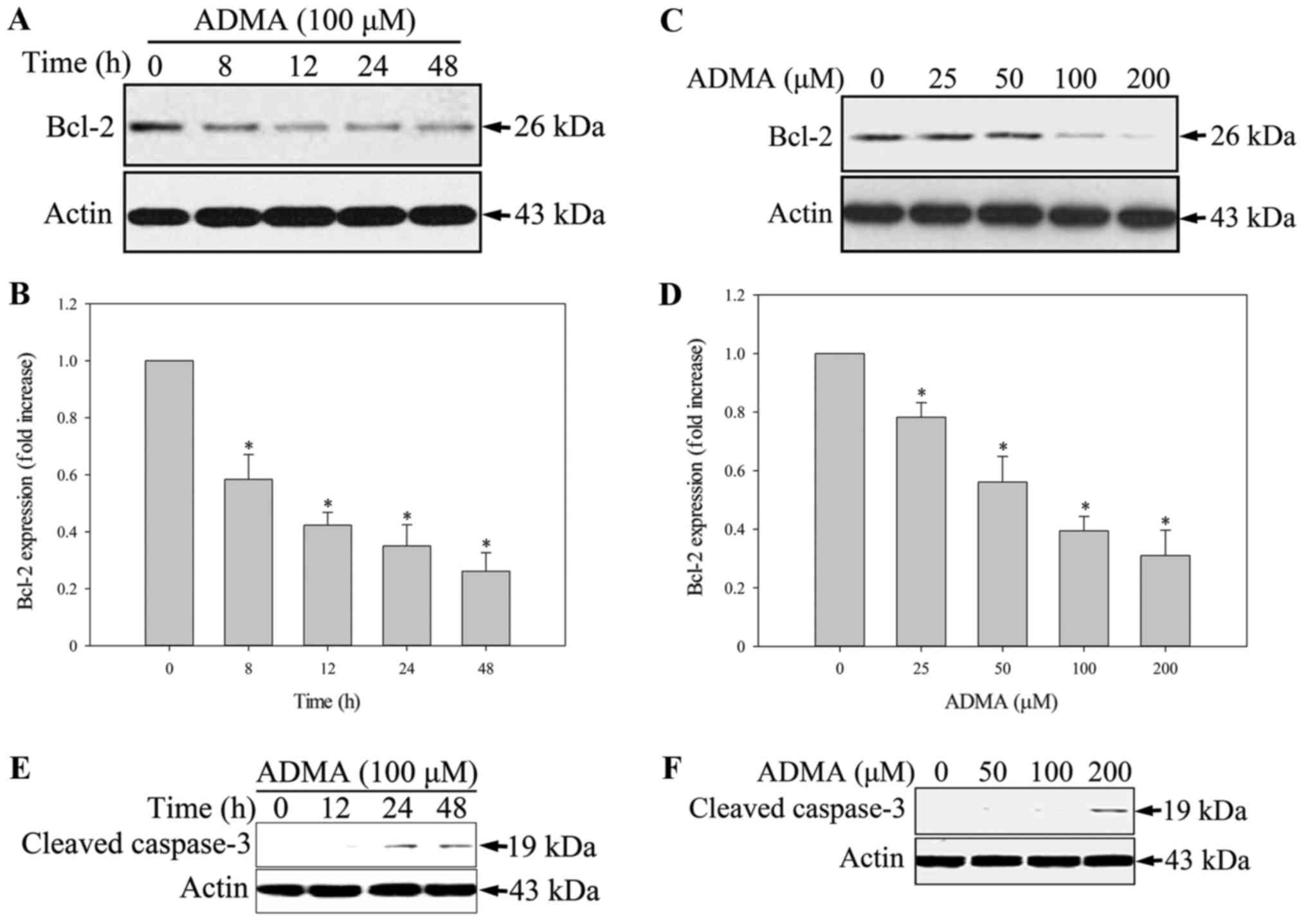
HUVECs treated with 100 µM ADMA had a higher proportion of
apoptotic cells than the control HUVECs. Bcl2, an apoptosis
inhibitor, is a central modulator of intrinsic apoptosis. In HUVECs
incubated with 100 µM ADMA for 0–48 h, Bcl2 was significantly
downregulated (Fig. 2A and B).
When HUVECs were incubated with various concentrations of ADMA
(0–200 µM) for 24 h, the Bcl2 levels decreased in a dose-dependent
manner (Fig. 2C and D).
Furthermore, we analyzed the expression of cleaved caspase-3, a
critical executioner of apoptosis. We found that ADMA increased
cleaved caspase-3 expression in a time- and dose-dependent manner
(Fig. 2E and F).
ER stress is involved in ADMA-induced
HUVEC apoptosis
The ER stress pathway plays a role in apoptosis
induction. Therefore, we investigated whether the ER stress pathway
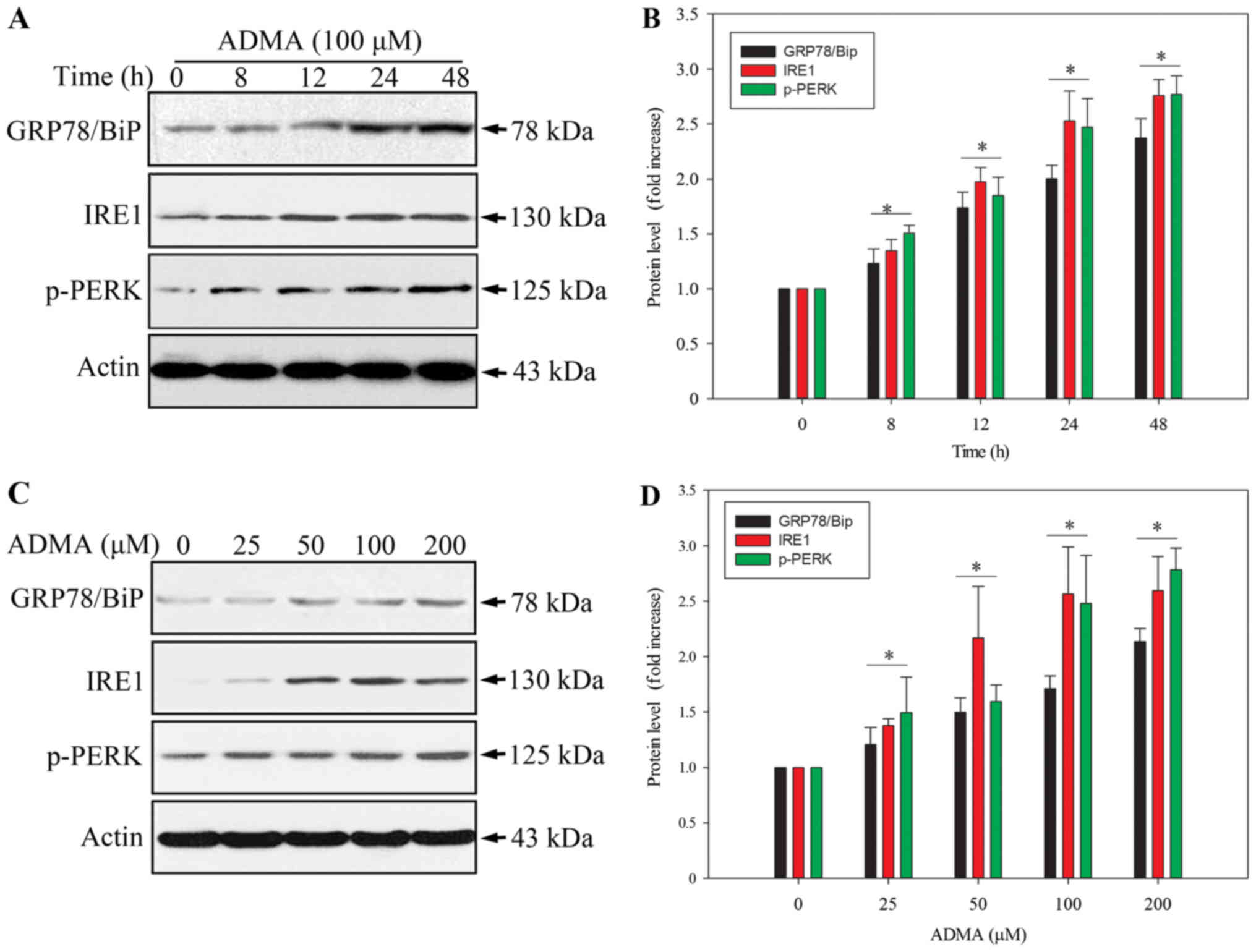
is involved in the ADMA-induced apoptosis in HUVECs. The levels of
the ER stress-related protein, GPR78/BiP, and the apoptosis-related
ER stress proteins, PERK and IRE1 (9,10),
were analysed by western blot. As PERK activation requires
phosphorylation, we examined the level of phosphorylated PERK
(p-PERK). As expected, ADMA induced PERK phosphorylation, and
increased GPR78/BiP and IRE1 expression in HUVECs in a
time-dependent manner (Fig. 3A and
B). Moreover, ADMA induced p-PERK, GPR78/BiP and IRE1
expression in a dose-dependent manner (Fig. 3C and D). These results indicated
that ER stress was involved in ADMA-induced HUVEC apoptosis.
ADMA suppresses SERCA3 expression in
HUVECs
A recent study supported an important role for ER
calcium homeostasis in the development of apoptosis. Because SERCA
is the only calcium pump in the ER, we further hypothesized that
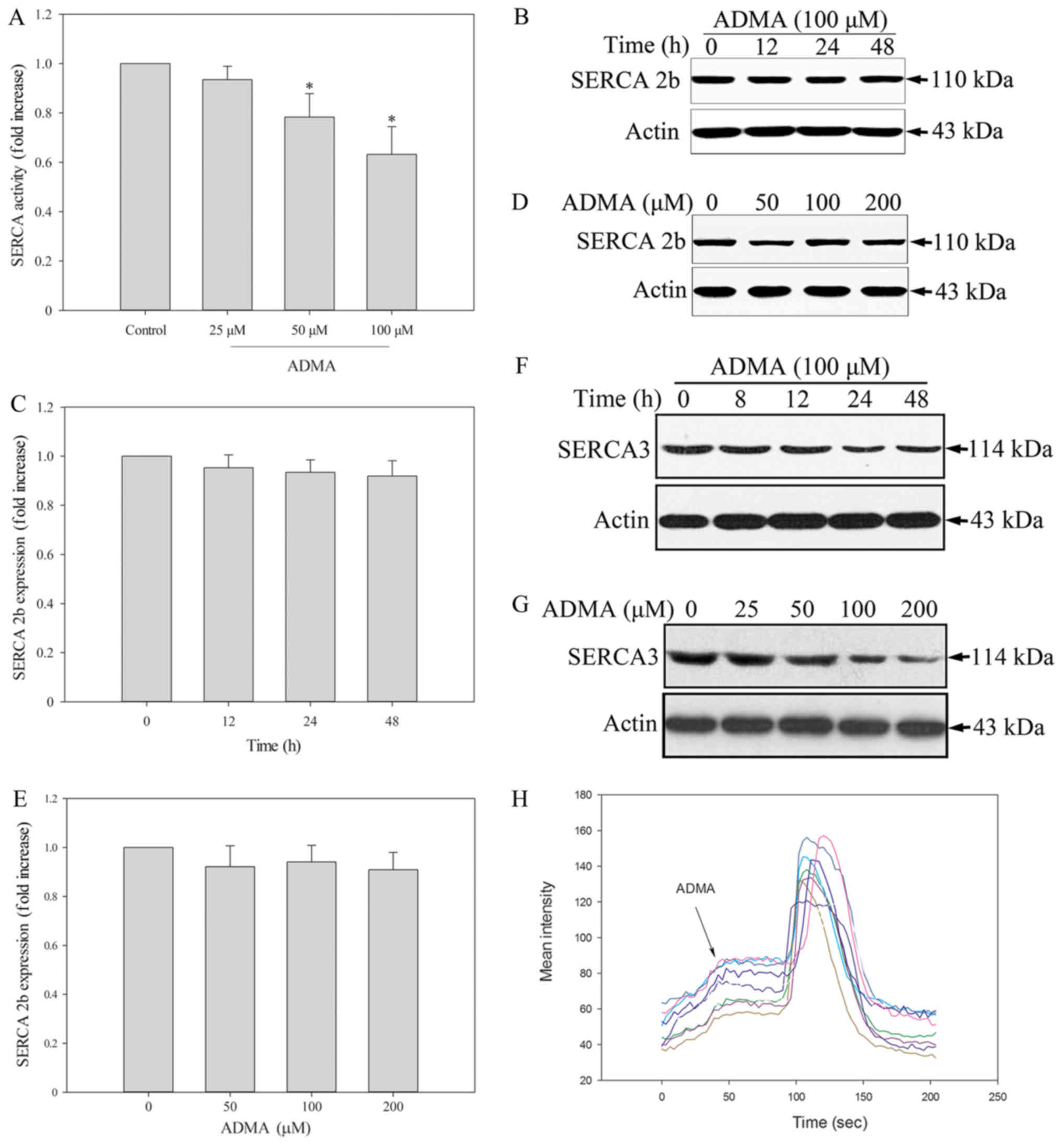
its inhibition may evoke ER stress. Thus, we tested whether SERCA
is modulated by ADMA by measuring the Ca2+-ATPase
activity. The results showed that ADMA (50–100 µM) significantly
inhibited SERCA activity in HUVECs (Fig. 4A). Endothelial cells express two
distinct isoforms, SERCA2b and SERCA3. It has been reported that in
freshly isolated and early passage (P0-P1) HUVECs, SERCA3 was the
dominant isoform. After further cell proliferation (P2-P4), there
was a reduction in the expression of the SERCA3 isoform in HUVECs
and a corresponding increase in SERCA2b (18). Therefore, we measured the levels of
both HUVEC-associated SERCA isoforms by western blotting. We found
that ADMA induced a slight, but not significant, decrease in
SERCA2b protein expression (Fig.
4B-E). In contrast, ADMA induced a statistically significant
downregulation of SERCA3 (Fig. 4F and
G). This may be the mechanism of ADMA-induced ER stress and
apoptosis in HUVECs. A decrease in SERCA expression leads to
depletion of ER Ca2+ and increased cytosolic
Ca2+ levels ([Ca2+]cyt). We
therefore examined the [Ca2+]cyt levels in
HUVECs. The change in the [Ca2+]cyt level was
measured by confocal imagining using Fluo-3 dye in the presence of
extracellular Ca2+. The change in fluorescence intensity
after ADMA treatment was recorded, indicating the level of
extracellular Ca2+. Fluorescence intensity measurements
were made in every 3 sec. We found that ADMA treatment led to
increased fluorescence intensity, which means that
[Ca2+]cyt increased (Fig. 4H). Coloured lines represent every
single cells. This may be the mechanism of ADMA-induced ER stress
and apoptosis in HUVECs.
Discussion
CVD is the leading cause of death among CKD
patients. There is an urgent need to characterize novel pathways
and identify new therapeutic targets to control CVD progression.
ADMA is a significant uremic toxin and its levels are elevated in
CKD, even at the early stages. In a large cohort of subjects, those
with higher ADMA levels had an increased risk of death attributed
to cardiovascular disorders (19).
Consistently, circulating ADMA levels have been shown to be
independently associated with cardiovascular risk, death and
incidence of CVD outcomes (5,6,20,21).
As an endogenous inhibitor of nitric oxide (NO)
synthase, ADMA can reduce NO production. NO is a key factor
maintaining vascular homeostasis. By reducing NO generation, ADMA
can influence endothelial dysfunction (1). However, ADMA acts through mechanisms
beyond its NO synthase inhibition. In previous reports, ADMA
induced endothelial cell apoptosis (7,8). In
our study, we also found that ADMA induced cleaved caspase-3
expression and decreased Bcl2 expression. These results
demonstrated that endothelial cell apoptosis may contribute to
ADMA-induced vascular injury.
ER stress results from the accumulation of misfolded
proteins in the ER lumen. The likely purpose of ER stress is to
help remove misfolded proteins and promote cell survival. However,
prolonged ER stress triggers apoptosis. Among the three known ER
stress sensors, PERK and IRE1 can trigger the cellular apoptosis
program. Under normal conditions, BiP/GRP78 associates with and
inactivates ER stress sensor proteins. Under stress conditions,
GRP78/BiP dissociates from the ER sensor proteins, activating
downstream signal transduction to rescue ER homeostasis. Increased
GRP78/BiP levels in the ER compartment have been regarded as
evidence for induction of ER stress. Several studies have strongly
supported a central role for the ER stress response in the
pathogenesis of endothelial dysfunction and CVD. Alleviating ER
stress may thus represent a promising therapeutic approach for
preventing or improving CVD. These studies led us to investigate
whether ADMA induces ER stress in HUVECs. We confirmed that ADMA
induced p-PERK, IRE1 and GRP78/BiP expression in a time- and
dose-dependent manner, indicating activation of the ER stress
response. We further explored the possible mechanisms involved in
ADMA-induced ER stress.
High concentrations of Ca2+ in the ER
lumen ([Ca2+]ER) are essential for normal ER
folding capacity (13). SERCA,
localized in the ER membrane, lowers
[Ca2+]cyt by pumping Ca2+ from the
cytosol into the ER. Inhibition of SERCA activity impaired ER
Ca2+ homeostasis and caused prolonged ER stress
(13,14). We therefore hypothesized that a
disturbance in SERCA activity is the major cause of ADMA-induced ER
stress in HUVECs. We found that ADMA inhibited the SERCA activity
in HUVECs. The SERCA pump is coded by a family of three genes,
SERCA1, 2 and 3, encoding the SERCA1, SERCA2
and SERCA3 isoforms, respectively. SERCA isoform expression is
specific to cell type and developmental stages (15). While SERCA2b is a ubiquitous
isoform expressed in muscle and non-muscle cells, the SERCA3
isoform is primarily expressed in non-muscle, especially
endothelial, cells (16,17). Freshly isolated HUVECs contain only
SERCA3 and no SERCA2b mRNA. These SERCA
expression patterns change with increased cell proliferation.
SERCA3 mRNA decreased, while that for SERCA2b
increased, with increasing passages in culture. Because we used
HUVECs at passages 3 and 4, we examined both SERCA2b and SERCA3
protein expression after ADMA stimulation. We found that ADMA
slightly decreased SERCA2b expression, but more significantly
reduced SERCA3 expression. This indicated that the ADMA-induced
effects on SERCA were isoform-dependent. We also examined the
changes in [Ca2+]cyt in HUVECs in response to
ADMA. We used Fluo-3 to detect the calcium levels in the cytoplasm.
We found that the fluorescence intensity greatly increased after
addition of ADMA to the culture medium. This result indicated that
the [Ca2+]cyt levels increased in HUVECs
after ADMA stimulation.
Our results are consistent with other studies
reporting that ADMA induced the ER stress response in many
different cell types, such as mesangial cells, adipocytes,
macrophages, glomerular endothelial cells and lung epithelial cells
(22–27). These results implicated ER stress
as the common pathway through which ADMA contributes to a variety
of human diseases, including CKD, diabetes mellitus and liver
disease. Thus, inhibition of ER stress and related pathways may be
effective as a wide range treatment. However, there are a few
limitations in our study. First, the SERCA isoforms expressed in
HUVECs are influenced by the number of passages. Additionally,
after 5 or 6 passages the cell phenotype changes to another
phenotype, such as fibroblasts. Therefore, in this study we used
cells from passages 3 and 4, which may be the main reason behind
the finding that ADMA only obviously inhibited SERCA3, as it is the
main isoform in the these passages. Hence, this in vitro
expreiement is limited by the cell culture. Thus, futher in
vivo experiments are required, to elucidate which isoforms are
the main ones responsible for ADMA-induced ER stress in vascular
cells. Moreover, in the normal population, the plasma level of ADMA
is 0.92–0.11 µM (28,29). However, the plasma levels of ADMA
are increased in some diseases, such as CKD, diabetes,
cardiovascular disease and hypertension (30–32).
Hemodialysis patients with atherosclerosis show significantly
higher ADMA levels of up to 7.31 µM (33). However, when we performed
experiments to study the toxicity mechanisms of ADMA in
vitro, much larger doses (0–200 µM) than the pathological
concentrations in clinical patients were required (22,25,34).
Therefore, wheher ADMA inhibits SERCA3 expression and induces ER
stress during long-term ADMA accumulation in the human body still
need to be explored in future in vivo studies.
In conclusion, we think that ADMA-induced ER stress
and apoptosis are closely related to the decrease in SERCA3
expression.
In this study, we found that ADMA induced ER stress,
decreased Bcl2 expression and ultimately induced apoptosis in
HUVECs. In addition, ADMA decreased SERCA3 activity and expression.
Together, our results indicated that ADMA induced growth
suppression and apoptosis in HUVECs, effects that are related to
activation of ER stress and inhibition of SERCA3 expression.
Developing therapies that directly target the defective endogenous
SERCA enzyme to correct Ca2+ imbalances in the ER may
constitute a novel approach for treating ADMA-induced vascular
injury.
Acknowledgements
The authors would like to thank the BeiJing Talents
Fund (no. 2016000021469G223) for financial support.
References
|
1
|
Vallance P, Leone A, Calver A, Collier J
and Moncada S: Accumulation of an endogenous inhibitor of nitric
oxide synthesis in chronic renal failure. Lancet. 339:572–575.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Konya H, Miuchi M, Satani K, Matsutani S,
Yano Y, Tsunoda T, Ikawa T, Matsuo T, Ochi F, Kusunoki Y, et al:
Asymmetric dimethylarginine, a biomarker of cardiovascular
complications in diabetes mellitus. World J Exp Med. 5:110–119.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Böger RH, Maas R, Schulze F and Schwedhelm
E: Asymmetric dimethylarginine (ADMA) as a prospective marker of
cardiovascular disease and mortality-an update on patient
populations with a wide range of cardiovascular risk. Pharmacol
Res. 60:481–487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Landim MB, Filho A Casella and Chagas AC:
Asymmetric dimethylarginine (ADMA) and endothelial dysfunction:
Implications for atherogenesis. Clinics. 64:471–478. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schnabel R, Blankenberg S, Lubos E,
Lackner KJ, Rupprecht HJ, Espinola-Klein C, Jachmann N, Post F,
Peetz D, Bickel C, et al: Asymmetric dimethylarginine and the risk
of cardiovascular events and death in patients with coronary artery
disease: Results from the AtheroGene Study. Circ Res. 97:e53–e59.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu TM, Ding YA, Charng MJ and Lin SJ:
Asymmetrical dimethylarginine: A novel risk factor for coronary
artery disease. Clin Cardiol. 26:458–464. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Zhang Z, Lv L, Qiao H, Chen X and
Zou C: (−)-epigallocatechin gallate inhibits asymmetric
dimethylarginine-induced injury in human brain microvascular
endothelial cells. Neurochem Res. 41:1868–1876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma J, Zhao S, Gao G, Chang H, Ma P and Jin
B: probucol protects against asymmetric dimethylarginine-induced
apoptosis in the cultured human brain microvascular endothelial
cells. J Mol Neurosci. 57:546–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Woehlbier U and Hetz C: Modulating stress
responses by the UPRosome: a matter of life and death. Trends in
Biochem Sci. 36:329–337. 2011. View Article : Google Scholar
|
|
11
|
Zarain-Herzberg A, Garcia-Rivas G and
Estrada-Aviles R: Regulation of SERCA pumps expression in diabetes.
Cell Calcium. 56:302–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Brini M, Cali T, Ottolini D and Carafoli
E: The plasma membrane calcium pump in health and disease. FEBS J.
280:5385–5397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fu S, Yang L, Li P, Hofmann O, Dicker L,
Hide W, Lin X, Watkins SM, Ivanov AR and Hotamisligil GS: Aberrant
lipid metabolism disrupts calcium homeostasis causing liver
endoplasmic reticulum stress in obesity. Nature. 473:528–531. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li W, Ouyang Z, Zhang Q, Wang L, Shen Y,
Wu X, Gu Y, Shu Y, Yu B, Wu X, et al: SBF-1 exerts strong
anticervical cancer effect through inducing endoplasmic reticulum
stress-associated cell death via targeting sarco/endoplasmic
reticulum Ca(2+)-ATPase 2. Cell Death Dis. 5:e15812014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Periasamy M and Kalyanasundaram A: SERCA
pump isoforms: Their role in calcium transport and disease. Muscle
Nerve. 35:430–442. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lipskaia L, Keuylian Z, Blirando K,
Mougenot N, Jacquet A, Rouxel C, Sghairi H, Elaib Z, Blaise R,
Adnot S, et al: Expression of sarco (endo) plasmic reticulum
calcium ATPase (SERCA) system in normal mouse cardiovascular
tissues, heart failure and atherosclerosis. Biochim Biophys Acta.
1843:2705–2718. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mountian I, Manolopoulos VG, De SH, Parys
JB, Missiaen L and Wuytack F: Expression patterns of
sarco/endoplasmic reticulum Ca(2+)-ATPase and inositol
1,4,5-trisphosphate receptor isoforms in vascular endothelial
cells. Cell Calcium. 25:371–380. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Anger M, Samuel JL, Marotte F, Wuytack F,
Rappaport L and Lompré AM: In situ mRNA distribution of sarco
(endo)plasmic reticulum Ca (2+)-ATPase isoforms during ontogeny in
the rat. J Mol Cell Cardiol. 26:1–550. 1994. View Article : Google Scholar
|
|
19
|
Valkonen VP, Päivä H, Salonen JT, Lakka
TA, Lehtimäki T, Laakso J and Laaksonen R: Risk of acute coronary
events and serum concentration of asymmetrical dimethylarginine.
Lancet. 358:2127–2128. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nicholls SJ, Wang Z, Koeth R, Levison B,
DelFraino B, Dzavik V, Griffith OW, Hathaway D, Panza JA, Nissen
SE, et al: Metabolic profiling of arginine and nitric oxide
pathways predicts hemodynamic abnormalities and mortality in
patients with cardiogenic shock after acute myocardial infarction.
Circulation. 116:2315–2324. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tousoulis D, Bouras G, Antoniades C,
Marinou K, Papageorgiou N, Miliou A, Hatzis G, Stefanadi E,
Tsioufis C and Stefanadis C: Methionine-induced homocysteinemia
impairs endothelial function in hypertensives: The role of
asymmetrical dimethylarginine and antioxidant vitamins. Am J
Hypertens. 24:936–942. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park MJ, Oh KS, Nho JH, Kim GY and Kim DI:
Asymmetric dimethylarginine (ADMA) treatment induces apoptosis in
cultured rat mesangial cells via endoplasmic reticulum stress
activation. Cell Biol Int. 40:662–670. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong D, Gao HC, Wang X, Li LF, Li CC, Luo
Y, Wang KK, Bai YP and Zhang GG: Asymmetric dimethylarginine
triggers macrophage apoptosis via the endoplasmic reticulum stress
pathway. Mol Cell Biochem. 398:31–38. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leng YP, Qiu N, Fang WJ, Zhang M, He ZM
and Xiong Y: Involvement of increased endogenous asymmetric
dimethylarginine in the hepatic endoplasmic reticulum stress of
type 2 diabetic rats. PLoS One. 9:e971252014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guo W, Ding J, Zhang A, Dai W, Liu S, Diao
Z, Wang L, Han X and Liu W: The inhibitory effect of quercetin on
asymmetric dimethylarginine-induced apoptosis is mediated by the
endoplasmic reticulum stress pathway in glomerular endothelial
cells. Int J Mol Sci. 15:484–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou QG, Zhou M, Hou FF and Peng X:
Asymmetrical dimethylarginine triggers lipolysis and inflammatory
response via induction of endoplasmic reticulum stress in cultured
adipocytes. Am J Physiol Endocrinol Metab. 296:E869–E878. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lim SK, Choi H, Park MJ, Kim DI, Kim JC,
Kim GY, Jeong SY, Rodionov RN, Han HJ, Yoon KC and Park SH: The ER
stress-mediated decrease in DDAH1 expression is involved in
formaldehyde-induced apoptosis in lung epithelial cells. Food Chem
Toxicol. 62:763–769. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lundman P, Eriksson MJ, Stühlinger M,
Cooke JP, Hamsten A and Tornvall P: Mild-to-moderate
hypertriglyceridemia in young men is associated with endothelial
dysfunction and increased plasma concentrations of asymmetric
dimethylarginine. J Am Coll Cardiol. 38:111–116. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Surdacki A, Nowicki M, Sandmann J, Tsikas
D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F,
Dubiel JS and Froelich JC: Reduced urinary excretion of nitric
oxide metabolites and increased plasma levels of asymmetric
dimethylarginine in men with essential hypertension. J Cardiovasc
Pharmacol. 33:652–658. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Alpoim PN, Sousa LP, Mota AP, Rios DR and
Dusse LM: Asymmetric dimethylarginine (ADMA) in cardiovascular and
renal disease. Clin Chim Acta. 440:1–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tousoulis D, Georgakis MK, Oikonomou E,
Papageorgiou N, Zaromitidou M, Latsios G, Papaioannou S and Siasos
G: Asymmetric dimethylarginine: Clinical significance and novel
therapeutic approaches. Curr Med Chem. 22:2871–2901. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santilli F, Liani R, Di Fulvio P, Formoso
G, Simeone P, Tripaldi R, Ueland T, Aukrust P and Davì G: Increased
circulating resistin is associated with insulin resistance,
oxidative stress and platelet activation in type 2 diabetes
mellitus. Thromb Haemost. 116:1089–1099. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kielstein JT, Böger RH, Bode-Böger SM,
Schäffer J, Barbey M, Koch KM and Frölich JC: Asymmetric
dimethylarginine plasma concentrations differ in patients with
end-stage renal disease: Relationship to treatment method and
atherosclerotic disease. J Am Soc Nephrol. 10:594–600.
1999.PubMed/NCBI
|
|
34
|
Wang LY, Zhang DL, Zheng JF, Zhang Y,
Zhang QD and Liu WH: Apelin-13 passes through the ADMA-damaged
endothelial barrier and acts on vascular smooth muscle cells.
Peptides. 32:2436–2443. 2011. View Article : Google Scholar : PubMed/NCBI
|