Introduction
The senescence of vascular smooth muscle cells
(VSMCs) serves a causal role in the pathogenesis and development of
vascular aging, that is closely associated with cardiovascular
diseases (1). Senescent cells
exhibit increased oxidative stress, telomere dysfunction and stable
growth arrest (2) as well as
increased expression of senescence-associated β-galactosidase
(SA-β-gal), p53, p21 and p16 (3,4).
High intracellular levels of reactive oxygen species (ROS) are
known to induce premature senescence (5). Recent studies suggested that
ROS-induced senescence involves reinforcement of positive feedback
signaling, further amplifying senescent signals, and ultimately the
senescent phenotype (5,6).
Ang II serves an important role in the pathogenesis
of various human diseases including atherosclerosis, and inhibition
of Ang II activity has been demonstrated to reduce the morbidity
and mortality due to cardiovascular diseases (7). Ang II increases ROS production and
reduces cellular antioxidant capacity, leading to vascular
senescence through a p53/p21-dependent mechanism in VSMCs (4). A growing body of evidence has
demonstrated that treatment with biological substances and
pharmaceuticals can reduce premature cellular senescence, providing
a potential treatment for aging-associated diseases (8,9).
Celastrol is a quinone methide triterpenoid isolated
from the traditional Chinese medicinal plant Tripterygium
wilfordii Hook f. Celastrol has attracted increasing attention
due to its potential therapeutic effects on inflammation and
metabolic disorders (10,11). There is accumulating evidence that
celastrol can also favorably affect cardiovascular function by
preventing circulatory failure (12), atherosclerosis (13) or myocardial ischemic injury
(14). Our previous study
demonstrated that celastrol exerted inhibitory effects on platelet
activation which could contribute to both thrombotic and
inflammatory events (15). A
recent study reported that a short-term treatment using tripterine
(celastrol) reduced ox-low-density lipoprotein (LDL)-induced ROS
generation in endothelial progenitor cells (16). It remains unknown whether celastrol
counteracts cellular senescence by reducing ROS.
Among the mechanisms regulating ROS, autophagy
serves an important role (17).
Autophagy is a type of evolutionarily conserved mechanism that is
linked to several cellular signaling pathways and can impact VSMC
survival and function. Upregulation of autophagy with stimuli,
including intracellular or extracellular factors, can protect VSMCs
against cell death (18,19). A recent study reported that
defective autophagy in VSMCs accelerates senescence and promotes
ligation-induced neointima formation and diet-induced
atherogenesis, implying that stimulating autophagy could be a novel
strategy in the treatment of arterial diseases (20). In the present study, it was
hypothesized that celastrol serves an anti-senescence role by
inducing autophagy to reduce ROS production, using VSMCs.
Materials and methods
Chemicals, reagents and
antibodies
Celastrol, N-acetyl-L-cysteine (NAC),
3-methyladenine (3Ma), bafilomycin A1 (Baf A1), rapamycin (Rapa)
and dimethyl sulfoxide (DMSO) were provided by Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). Angiotensin II (Ang II) was obtained
from EMD Millipore (Billerica, MA, USA). A Senescence
β-galactosidase staining kit (cat. no. 9860), polyclonal rabbit
antibodies against LC3A/B (cat. no. 4108), phosphorylated
(p)-protein kinase B (Akt; cat. no. 9271), Akt (cat. no. 9272),
p-mechanistic target of rapamycin (mTOR; cat. no. 2971), mTOR (cat.
no. 2972), p-ribosomal protein S6 kinase β1 (p70S6K; cat. no.
9205), p70S6K (cat. no. 9202) and β-Actin (cat. no. 4967), and the
monoclonal rabbit antibody against GAPDH (cat. no. 2118) were all
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Monoclonal mouse antibodies against p21 (cat. no. sc-6246) and p53
(cat. no. sc-99) were from Santa Cruz Biotechnology, Inc. (Dallas,
TX, USA). Polyclonal rabbit antibodies against p-phosphoinositide 3
kinase (PI3K; cat. no. ab182651) and PI3K (cat. no. ab40755) were
obtained from Abcam (Shanghai, China). A polyclonal goat
CY5-conjugated secondary antibody (cat. no. A10523) was from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). An ROS assay kit and a
Cell Counting kit-8 (CCK-8) were from the Beyotime Institute of
Biotechnology (Haimen, China). Fetal bovine serum (FBS) and
Dulbecco's modified Eagle's medium (DMEM) were provided by Gibco;
Thermo Fisher Scientific, Inc. (Waltham, MA, USA).
Ethics and animal protocols
A total of 20 Sprague-Dawley rats (150–180 g, 6
weeks of age) were obtained from the Laboratory Animal Center of
the Third Military Medical University (Chongqing, China). The
animals had free access to food and water and were housed at a
temperature of 25°C with 45–70% humidity, under a 12-h light/dark
cycle in the specific pathogen free facility at Laboratory Animal
Center of Third Military Medical University (Chongqing, China). All
procedures were conducted in accordance with the animal care
guidelines approved by the Animal Ethics Committee of the Third
Military Medical University. The animal care and procedures were in
accordance with the guidelines of the National Institutes of Health
(Bethesda, MD, USA). Thoracic aortas were isolated from euthanized
rats, and all efforts were made to minimize any pain to the
animals.
Isolation and culture of VSMCs
Primary rat VSMCs were isolated from the thoracic
aorta of Sprague-Dawley rats by an explant method as previously
described (21). Briefly, male
Sprague-Dawley rats were anaesthetized with 5% chloral hydrate (1
ml/100 g body weight) and sacrificed to obtain VSMCs from the
thoracic aorta. VSMCs were cultured in DMEM supplemented with 10%
FBS at 37°C in a humidified atmosphere containing 5%
CO2. The medium was changed every 2–3 days, and VSMCs at
passages 3–8 were used for all experiments.
Analysis of cellular senescence
Senescent cells were identified using a Senescence
β-Galactosidase Staining kit. Briefly, VSMCs were seeded into
6-well plates (NEST, Wuxi, China) at 2×105/well. At 80%
confluence, VSMCs were pretreated with celastrol at 10, 50 or 100
nM for 12 h before the addition of 100 nM Ang II for 2 days.
Subsequently, cells were fixed with the fixative solution, rinsed
twice with PBS and incubated with the β-galactosidase staining
solution at 37°C overnight in a dry incubator (without
CO2). For 3Ma or Baf A1 stimulation, cells were
pretreated with 3 mM 3Ma or 50 nM Baf A1 for 2 h.
Cell cycle analysis
The cell cycle analysis was performed according to
the manufacturer's protocol. Briefly, after serum starvation for 12
h, VSMCs were treated with 50 nM celastrol for 12 h before
incubation with Ang II for 2 days at 37°C. Subsequently, cells were
harvested and fixed in 70% ethanol, stained for DNA content with
propidium iodide for 30 min at room temperature and detected using
a flow cytometer (BD FACScan, BD Biosciences, Franklin Lakes, NJ,
USA). Fluorescence dot plots were analyzed and reconstructed with
FlowJo version 7.2.5 software (FlowJo LLC, Ashland, OR, USA).
Western blot analysis
Western blot assays were performed as previously
described (22). Briefly, VSMCs
were harvested and lysed in RIPA buffer (Beyotime Institute of
Biotechnology, Haimen, China), which contained a cocktail of both
protease and phosphatase inhibitors (Roche Diagnostics, Basel,
Switzerland). Following extraction from VSMCs, the protein
concentration was determined by the BCA protein assay (Beyotime
Institute of Biotechnology). Samples containing 30 µg of protein
were subjected to 10% SDS-PAGE and separated. Following protein
transfer to nitrocellulose membranes (EMD Millipore), the membranes
were blocked and incubated with primary antibodies overnight (12 h)
at 4°C, then washed three times with TBST (Tris-buffered saline; 10
mM Tris-HCl pH 7.5, 150 mM NaCl and 0.1% Tween-20) for 30 min and
incubated with secondary antibodies for 1 h at room temperature.
Membranes were washed four times with TBST for 40 min. The protein
signals were detected using the SuperSignal West Pico
Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific,
Inc.). The relative protein levels were analyzed by Image J
software version 1.6.0_24 (National Institutes of Health, Bethesda,
MD, USA).
Determination of intracellular
ROS
A ROS assay kit was used to determine the
intracellular ROS generation. In brief, quiescent cells were
incubated with 50 nM celastrol for 12 h before stimulation with 100
nM Ang II for 12 h, and then incubated with 2, 7-DCFH-DA in DMEM
without FBS at 37°C for 30 min and rinsed three times with PBS. ROS
levels were measured by using a flow cytometer.
Immunofluorescent staining of LC3
Cells were fixed with 4% paraformaldehyde for 20
min, rinsed three times with PBS, blocked with 1% bovine serum
albumin (Beyotime Institute of Biotechnology) for 1 h, and
incubated with a rabbit anti-LC3 A/B antibody for 1 h at room
temperature. Subsequently, the cells were washed and incubated with
a CY5-conjugated secondary antibody for 1 h at room temperature.
Immunofluorescence was detected by using a confocal microscope
(TCS-SP5, Leica Microsystems GmbH, Wetzlar, Germany).
Statistical analysis
Data analysis was performed using GraphPad Prism 5.0
software package (GraphPad Software, Inc., La Jolla, CA, USA). All
experimental data are expressed as the mean ± standard error with
each experiment repeated at least three times. The statistical
analysis was performed with SPSS software version 20.0 (IBM Corp.,
Armonk, NY, USA). The data comparisons were analyzed using one-way
analysis of variance followed by Bonferroni's multiple comparisons
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Celastrol treatment counteracts Ang
II-induced cellular senescence in VSMCs
Senescent cells are characterized by stable growth
arrest (2) and increased
expression of SA-β-gal, p53, p21 and p16 (3,4).
Treatment with celastrol alone had little effect on VSMC senescence
compared with treatment with the vehicle control (DMSO). A
significant increase in VSMC senescence upon stimulation with Ang
II was evidenced by the increase in the number of SA-β-gal-positive
cells. In this setting, celastrol demonstrated a dose-dependent
inhibition of VSMC senescence with its maximal effect achieved at
50 nM (Fig. 1A). Protein
expression levels of p53 and p21 were significantly increased in
VSMCs after a 2-day period of stimulation with Ang II. Pretreatment
with celastrol significantly counteracted this effect of Ang II on
(Fig. 1B). Flow cytometry was also
used to investigate the effect of celastrol on the cell cycle of
VSMCs. The senescent cells were defined as arrested primarily in
the G0/G1 phase but occasionally at the S or G2 phase. After
stimulation with 100 nM Ang II for 2 days, the percentage of G0/G1
phase cells was significantly increased compared with that of the
DMSO control group. In contrast, in VSMCs treated with Ang II, the
number of G0/G1-arrested cells was decreased while the number of
cells in S/G2 phase was increased by celastrol (50 nM)-pretreatment
(Fig. 1C), indicating that the
proliferative capacity of senescent VSMCs induced by Ang II was
restored by celastrol.
 | Figure 1.Celastrol treatment reduces Ang
II-induced senescence by reducing ROS activity in VSMCs. (A)
Senescence-associated β-galactosidase staining of VSMCs. The blue
puncta indicate the senescent cells. Scale bar=250 µm. (B) Western
blot measuring the effects of celastrol treatment on the protein
expression of p53 and p21 induced by Ang II in VSMCs. (C) Cell
cycle analysis using flow cytometry. The bars show the percentage
of cells in the different phases (G1/G0, S, and G2/M phase)
compared with control. (D) Flow cytometric analysis of ROS in
celastrol-treated cells. The representative flow cytometry results
and statistical analysis are presented. Data are presented as the
mean ± standard error. *P<0.05 (n=3). CeT, celastrol. Ang II,
angiotensin II; NAC, N-acetyl-L-cysteine; ROS, reactive
oxygen species; FITC, fluorescein isothiocyanate. VSMCs, vascular
smooth muscle cells. SAβ-gal, senescence-associated
β-galactosidase; SAβ-gal, senescence-associated β-galactosidase;
VSMCs, vascular smooth muscle cells; DMSO, dimethyl sulfoxide. |
Celastrol alleviates senescence
through reducing ROS activity in VSMCs
Intracellular ROS serves a major role in the
development of Ang II-induced cellular senescence (23), which can be alleviated by the
specific ROS scavenger NAC in VSMCs (8). A ROS assay kit was used to
investigate whether celastrol attenuated the Ang II-induced ROS
activity in VSMCs. Treatment with celastrol alone had little effect
on basal ROS generation, similar to that of the DMSO control
(Fig. 1D). ROS production was
increased in the Ang II-treated group and this was significantly
decreased in the celastrol-pretreated group, although such
inhibitory efficacy was less potent compared with cells treated
with NAC (Fig. 1D). These results
suggested that the effects of celastrol on cellular senescence in
VSMCs are potentially mediated through the inhibition of ROS
production.
Celastrol-induced autophagy reduces
Ang II-mediated senescence in VSMCs
Immunofluorescence assay demonstrated that the level
of total LC3 was significantly increased in the
celastrol-pretreated group compared with the control group
(Fig. 2A). Western blot assay
further demonstrated that compared with the DMSO control, the
protein expression level of LC3 II was increased in the
celastrol-treated group, which could be further increased by
bafilomycin A1, or decreased by 3Ma (Fig. 2B). Furthermore, the p62 protein
level was decreased in the celastrol-treated group, and treatment
with the two autophagy inhibitors reduced these effects (Fig. 2B). Ang II stimulation significantly
increased cellular ROS levels in VSMCs, and celastrol reduced the
Ang II-stimulated ROS levels, similar to the effects of the
positive control rapamycin; however, pretreating with the autophagy
inhibitor 3Ma reversed the effect of celastrol (Fig. 2C), suggesting that celastrol at
least partially inhibits intracellular ROS by activating autophagy
in VSMCs.
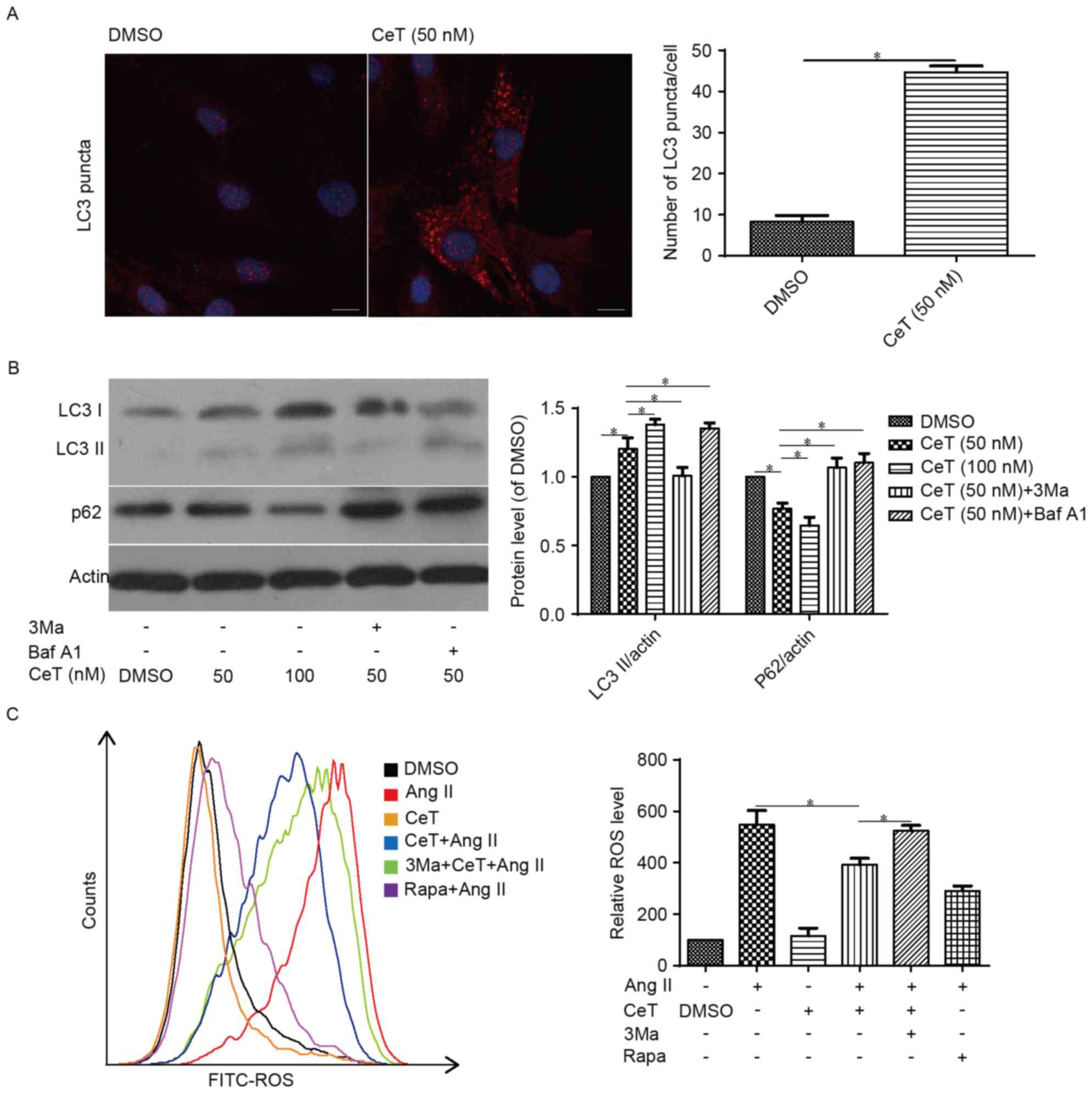 | Figure 2.Celastrol induces autophagy in VSMCs.
Serum-starved VSMCs were pre-incubated with varying concentrations
of either celastrol or vehicle (0.1% DMSO) for 12 h and then (A)
examined using immunofluorescence assay to identify LC3 puncta or
(B) subjected to western blotting to determine the protein levels
of LC3 and p62. Scale bar=10 µm. (C) Flow cytometric analysis of
ROS in celastrol-treated cells. Data are presented as the mean ±
standard error. *P<0.05 (n=3). CeT, celastrol; Ang II,
angiotensin II. LC3, microtubule-associated protein light chain 3;
3Ma, 3-methyladenine. Baf A1, bafilomycin A1; Rapa, rapamycin;
FITC, fluorescein isothiocyanate; ROS, reactive oxygen species;
VSMCs, vascular smooth muscle cells; DMSO, dimethyl sulfoxide. |
To further confirm whether celastrol-induced
autophagy can reduce the Ang II-induced senescence of VSMCs,
senescence β-galactosidase staining and western blot analysis were
performed. The results demonstrated that, just like in the
autophagy inducer rapamycin-pretreated group, the SA-β-gal positive
cell counts were significantly decreased in the
celastrol-pretreated group, which was reversed by the autophagy
inhibitors 3Ma and Baf A1 (Fig.
3A). Western blot assay further demonstrated that the autophagy
inhibitor 3Ma largely abolished the effects of celastrol on p53 and
p21 expression (Fig. 3B).
Celastrol inhibits the PI3K/Akt/mTOR
signaling pathway
The present study then assessed tested whether
celastrol could suppress the PI3K/Akt/mTOR signaling pathway. The
results demonstrated that, compared with the control group, the
phosphorylation levels of PI3K, Akt, mTOR and p70S6K in the
celastrol group were downregulated (Fig. 4), indicating that celastrol
treatment suppressed the PI3K/Akt/mTOR signaling pathway.
 | Figure 4.Celastrol suppresses the
PI3K/Akt/mTOR signaling pathway. Representative western blot images
and quantification of protein expression levels of p-PI3K, p-Akt,
p-mTOR and p-p70S6K in VSMCs. GAPDH served as an internal control.
Data are presented as the mean ± standard error. *P<0.05 (n=3).
CeT, celastrol; Ang II, angiotensin II; VSMCs, vascular smooth
muscle cells; p, phosphorylated; Akt, protein kinase B; mTOR,
mechanistic target of rapamycin; PI3K, phosphoinositide 3 kinase;
DMSO, dimethyl sulfoxide; p70s6k, ribosomal protein S6 kinase
β1. |
Discussion
It has been reported that treatment with tripterine
(celastrol) decreases ox-LDL-induced oxidative stress, apoptosis
and senescence of endothelial progenitor cells (16). However, whether celastrol exerts
any effects on VSMC senescence remains unknown. The present study
provided, to the best of our knowledge, the first evidence that
celastrol upregulates autophagy which reduces ROS levels, and
subsequently counteracts Ang II-induced cellular senescence.
Meanwhile, celastrol inhibited the PI3K/Akt/mTOR signaling pathway
in VSMCs. Senescence contributes to the development of
aging-associated pathologies such as cardiovascular disorders,
diabetes and neurodegenerative diseases (20,24).
ROS is an important senescence-inducing stressor for
vascular cells and can induce certain DNA lesions and drive
senescence, either dependent or independent of telomere shortening
(25). ROS has been implicated in
Ang II-induced cellular senescence (7) and interventions to decrease ROS
production by both scavenging free radicals and enhancing
antioxidant defense have been widely proposed as an anti-aging
strategy (24). To the best of our
knowledge, the present study revealed for the first time that
pretreatment with celastrol reduces the level of Ang II-induced
ROS.
Autophagy has attracted growing interest because it
affects almost every organ system and modulates an expanding list
of disease processes (18).
Autophagy contributes to general homeostasis through the turnover
of long-lived proteins and organelles via lysosomal degradation.
Recent studies have suggested that autophagy is relevant to human
diseases (20,26). Researchers have investigated the
association between autophagy and aging over the years. The
induction of autophagy by caloric restriction, spermidine,
resveratrol or rapamycin can extend the life span of different
organisms (27). The autophagy
inducers rapamycin and resveratrol suppress cellular senescence by
partially preventing the loss of proliferative potential (28,29),
whereas defective autophagy in VSMCs accelerates senescence and
promotes ligation-induced neointima formation and diet-induced
atherogenesis (20). It has been
demonstrated that autophagy reduces ROS (17). By upregulation of autophagy,
celastrol protects the SH-SY5Y human neuroblastoma cell line from
rotenone-induced injury (30) and
ameliorates experimental colitis in interleukin-10-deficient mice
(31). However, whether celastrol
can induce autophagy and subsequently exert a protective effect on
VSMCs is still unknown. In the present study, novel evidence is
presented for a prominent role of celastrol-activated autophagy in
regulating ROS to reduce VSMC senescence.
In addition, celastrol has been reported to affect a
diverse range of cellular functions that involves various signaling
pathways. For example, celastrol protects against obesity and
metabolic dysfunction through activation of a HSF1-PGC1a
transcriptional axis (11).
Celastrol attenuates hypertension-induced inflammation and
oxidative stress in VSMCs via induction of heme oxygenase-1
(32). However, there have been no
reports about the direct target or binding receptor of celastrol in
a cell yet, which makes it difficult to explain the mechanisms for
what the reported studies observed. It is well known that the
PI3K/Akt/mTOR pathway mediates an inhibitory signal on autophagy
(33). Several studies have
implicated that celastrol induces autophagy via the suppression of
the PI3K/Akt/mTOR pathway in the proximal colon of mice and in
tumor cells (31,34). The present study demonstrated that
celastrol significantly suppressed the PI3K/Akt/mTOR signaling
pathway and upregulated autophagy; celastrol reduces ROS by
inducing autophagy in VSMCs by using an autophagy inhibitor. Thus,
though the direct target or receptor of celastrol in VSMCs remains
unknown, the present study revealed the close association between
celastrol and the PI3K/Akt/mTOR-autophagy-ROS axis, i.e. that
celastrol possibly promotes autophagy via the PI3K/Akt/mTOR pathway
in VSMCs. However, it cannot be concluded that the effect of
autophagy upregulation by celastrol is precisely PI3K/Akt/mTOR
dependent, because specific inhibitors or specific small
interfering RNAs of PI3K/Akt/mTOR were not used in this study.
Therefore, there may be other possible mechanisms through which
celastrol may function, as opposed to a direct effect on
PI3K/Akt/mTOR. Nevertheless, this issue could be addressed in the
future by investigating the effects of celastrol on autophagy with
or without administration of the specific inhibitors or siRNAs of
PI3K/Akt/mTOR axis in the same cell model.
As one of the biomarkers of autophagy used in these
experiments, LC3 protein is involved in phagophore formation, which
is the primary autophagy-related protein (Atg)8 homolog examined in
mammalian cells (35), and p62 is
constantly degraded via autophagy through its LIR domain that binds
to LC3 on the membranes of autophagosomes (36). The concomitant increase in LC3-II
and degradation of p62 typically reflect the potent induction of
autophagy (35). The present study
observed increased LC3-II and decreased p62 levels in VSMCs by
celastrol treatment, which may be due to the inhibition of
PI3K/Akt/mTOR signaling pathway by celastrol, as mTOR is a classic
negative regulator of autophagy (33). In addition, though there are
several autophagy markers such as LC3, Atg5, Atg7, Atg9,
Atg12-Atg5, ATG14, ATG16L1 and p62 (35), only LC3 and p62 were selected as
representative markers, as investigated previously (37,38)
to assess the induction of autophagy by celastrol, thus; whether
other autophagy markers are also affected by celastrol has to be
further verified in future work.
The present study demonstrated that celastrol
upregulated autophagy, which reduced ROS and subsequently
counteracted the cellular senescence of VSMCs as a consequence of
Ang II stimulation. However, it has also been revealed that
celastrol suppresses the viability of cancer cells through AMPK
activation, which is also caused by ROS generation (39). In this context, celastrol was
demonstrated to induce prolonged and excessive autophagy in tumor
cells, leading to cell death (31). In these studies, celastrol was used
at a much higher dose compared with the dose (50 nM) tested in the
present study, which is consistent with some reports, in which
either a moderate dose or short duration treatment of celastrol
induced an anti-inflammatory or anti-autoimmune effect (10,40).
To avoid inducing intracellular ROS and excessive autophagy, which
can lead to apoptosis and cell death, the present study conducted a
cytotoxic experiment to optimize the celastrol dose to a moderate
level. The results demonstrated that celastrol did not influence
the viability of VSMCs at a dose up to 50 nM (data not shown). In
general, these results demonstrated that treatment with a moderate
dose of celastrol (50 nM) on VSMCs in vitro can
significantly exert a positive effect on VSMCs.
In conclusion, the present study demonstrated that
celastrol upregulates autophagy, which confers resistance to Ang
II-induced superoxide generation and the resultant senescence.
Notably, the effects of celastrol on stimulating autophagy may be
associated with its inhibition of mTOR activity (Fig. 5). As VSMC senescence is present in
atherosclerotic plaques and contributes to the pathogenesis of
atherosclerosis, celastrol may hold promise for the prevention and
treatment of cardiovascular diseases.
Acknowledgements
The present study was supported by the grants from
the National Natural Science Foundation of China (grant nos.
81270362 and 81470561) and the State Project for Essential Drug
Research and Development (grant no. 2013ZX09103003-001). The
manuscript language has been corrected by American Journal Experts,
(certificate verification key: 9290-E20B-063D-B6AD-8954).
Glossary
Abbreviations
Abbreviations:
|
CeT
|
celastrol
|
|
SAβ-gal
|
senescence-associated
β-galactosidase
|
|
NAC
|
N-acetyl-L-cysteine
|
|
3Ma
|
3-methyladenine
|
|
Baf A1
|
bafilomycin A1
|
|
LC3
|
microtubule-associated protein light
chain 3
|
References
|
1
|
Wang JC and Bennett M: Aging and
atherosclerosis: Mechanisms, functional consequences, and potential
therapeutics for cellular senescence. Circ Res. 111:245–259. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Childs BG, Durik M, Baker DJ and van
Deursen JM: Cellular senescence in aging and age-related disease:
From mechanisms to therapy. Nat Med. 21:1424–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O,
et al: A biomarker that identifies senescent human cells in culture
and in aging skin in vivo. Proc Natl Acad Sci USA. 92:9363–9367.
1995; View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kunieda T, Minamino T, Nishi J, Tateno K,
Oyama T, Katsuno T, Miyauchi H, Orimo M, Okada S, Takamura M, et
al: Angiotensin II induces premature senescence of vascular smooth
muscle cells and accelerates the development of atherosclerosis via
a p21-dependent pathway. Circulation. 114:953–960. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wiley CD, Velarde MC, Lecot P, Liu S,
Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A,
et al: Mitochondrial dysfunction induces senescence with a distinct
secretory phenotype. Cell Metab. 23:303–314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Correia-Melo C, Hewitt G and Passos JF:
Telomeres, oxidative stress and inflammatory factors: Partners in
cellular senescence? Longev Healthspan. 3:12014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herbert KE, Mistry Y, Hastings R, Poolman
T, Niklason L and Williams B: Angiotensin II-mediated oxidative DNA
damage accelerates cellular senescence in cultured human vascular
smooth muscle cells via telomere-dependent and independent
pathways. Circ Res. 102:201–208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao L, Li AQ, Zhou TF, Zhang MQ and Qin
XM: Exendin-4 alleviates angiotensin II-induced senescence in
vascular smooth muscle cells by inhibiting Rac1 activation via a
cAMP/PKA-dependent pathway. Am J Physiol Cell Physiol.
307:C1130–C1141. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee KY, Kim JR and Choi HC:
Genistein-induced LKB1-AMPK activation inhibits senescence of VSMC
through autophagy induction. Vascul Pharmacol. 81:75–82. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bian M, Du X, Cui J, Wang P, Wang W, Zhu
W, Zhang T and Chen Y: Celastrol protects mouse retinas from bright
light-induced degeneration through inhibition of oxidative stress
and inflammation. J Neuroinflammation. 13:502016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma X, Xu L, Alberobello AT, Gavrilova O,
Bagattin A, Skarulis M, Liu J, Finkel T and Mueller E: Celastrol
protects against obesity and metabolic dysfunction through
activation of a HSF1-PGC1α transcriptional axis. Cell Metab.
22:695–708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang YL, Lam KK, Cheng PY and Lee YM:
Celastrol prevents circulatory failure via induction of heme
oxygenase-1 and heat shock protein 70 in endotoxemic rats. J
Ethnopharmacol. 162:168–175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu L, Bai W, Li S, Zhang Y, Han Y, Gu Y,
Meng G, Xie L, Wang J, Xiao Y, et al: Celastrol prevents
atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS
One. 8:e654772013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Der Sarkissian S, Cailhier JF, Borie M,
Stevens LM, Gaboury L, Mansour S, Hamet P and Noiseux N: Celastrol
protects ischaemic myocardium through a heat shock response with
up-regulation of haeme oxygenase-1. Br J Pharmacol. 171:5265–5279.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hu H, Straub A, Tian Z, Bassler N, Cheng J
and Peter K: Celastrol, a triterpene extracted from Tripterygium
wilfordii Hook F, inhibits platelet activation. J Cardiovasc
Pharmacol. 54:240–245. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu C, Zhang X, Zhang D, Pei E, Xu J, Tang
T, Ye M, Uzan G, Zhi K, Li M and Zuo K: Short time tripterine
treatment enhances endothelial progenitor cell function via heat
shock protein 32. J Cell Physiol. 230:1139–1147. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and Autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tai S, Hu XQ, Peng DQ, Zhou SH and Zheng
XL: The roles of autophagy in vascular smooth muscle cells. Int J
Cardiol. 211:1–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nussenzweig SC, Verma S and Finkel T: The
role of autophagy in vascular biology. Circ Res. 116:480–488. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Grootaert MO, da Costa Martins PA, Bitsch
N, Pintelon I, De Meyer GR, Martinet W and Schrijvers DM: Defective
autophagy in vascular smooth muscle cells accelerates senescence
and promotes neointima formation and atherogenesis. Autophagy.
11:2014–2032. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Feng S, Hu Y, Peng S, Han S, Tao H, Zhang
Q, Xu X, Zhang J and Hu H: Nanoparticles responsive to the
inflammatory microenvironment for targeted treatment of arterial
restenosis. Biomaterials. 105:167–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen Q, Bei JJ, Liu C, Feng SB, Zhao WB,
Zhou Z, Yu ZP, Du XJ and Hu HY: HMGB1 induces secretion of matrix
vesicles by macrophages to enhance ectopic mineralization. PLoS
One. 11:e01566862016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsai IC, Pan ZC, Cheng HP, Liu CH, Lin BT
and Jiang MJ: Reactive oxygen species derived from NADPH oxidase 1
and mitochondria mediate angiotensin II-induced smooth muscle cell
senescence. J Mol Cell Cardiol. 98:18–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davalli P, Mitic T, Caporali A, Lauriola A
and D'Arca D: ROS, cell senescence, and novel molecular mechanisms
in aging and age-related diseases. Oxid Med Cell Longev.
2016:35651272016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin H and Pickering JG: Cellular
senescence and vascular disease: Novel routes to better
understanding and therapy. Can J Cardiol. 32:612–623. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhong Z, Sanchez-Lopez E and Karin M:
Autophagy, inflammation, and immunity: A troika governing cancer
and its treatment. Cell. 166:288–298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rubinsztein DC, Marino G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shan H, Guo D, Li X, Zhao X, Li W and Bai
X: From autophagy to senescence and apoptosis in Angiotensin
II-treated vascular endothelial cells. APMIS. 122:985–992. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mei Y, Thompson MD, Cohen RA and Tong X:
Autophagy and oxidative stress in cardiovascular diseases. Biochim
Biophys Acta. 1852:243–251. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deng YN, Shi J, Liu J and Qu QM: Celastrol
protects human neuroblastoma SH-SY5Y cells from rotenone-induced
injury through induction of autophagy. Neurochem Int. 63:1–9. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhao J, Sun Y, Shi P, Dong JN, Zuo LG,
Wang HG, Gong JF, Li Y, Gu LL, Li N, et al: Celastrol ameliorates
experimental colitis in IL-10 deficient mice via the up-regulation
of autophagy. Int Immunopharmacol. 26:221–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yu X, Tao W, Jiang F, Li C, Lin J and Liu
C: Celastrol attenuates hypertension-induced inflammation and
oxidative stress in vascular smooth muscle cells via induction of
heme oxygenase-1. Am J Hypertens. 23:895–903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shinojima N, Yokoyama T, Kondo Y and Kondo
S: Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in
curcumin-induced autophagy. Autophagy. 3:635–637. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH
and Chun KH: Celastrol inhibits gastric cancer growth by induction
of apoptosis and autophagy. BMB Rep. 47:697–702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Arozena A Acevedo, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moscat J and Diaz-Meco MT: p62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rodriguez-Arribas M, Yakhine-Diop SM,
González-Polo RA, Niso-Santano M and Fuentes JM: Turnover of
lipidated LC3 and autophagic cargoes in mammalian cells. Methods
Enzymol. 587:55–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang S, Wang C, Yan F, Wang T, He Y, Li H,
Xia Z and Zhang Z: N-acetylcysteine attenuates diabetic myocardial
ischemia reperfusion injury through inhibiting excessive autophagy.
Mediators Inflamm. 2017:92572912017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kim JH, Lee JO, Lee SK, Kim N, You GY,
Moon JW, Sha J, Kim SJ, Park SH and Kim HS: Celastrol suppresses
breast cancer MCF-7 cell viability via the AMP-activated protein
kinase (AMPK)-induced p53-polo like kinase 2 (PLK-2) pathway. Cell
Signal. 25:805–813. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yu Y, Koehn CD, Yue Y, Li S, Thiele GM,
Hearth-Holmes MP, Mikuls TR, O'Dell JR, Klassen LW, Zhang Z and Su
K: Celastrol inhibits inflammatory stimuli-induced neutrophil
extracellular trap formation. Curr Mol Med. 15:401–410. 2015.
View Article : Google Scholar : PubMed/NCBI
|
















