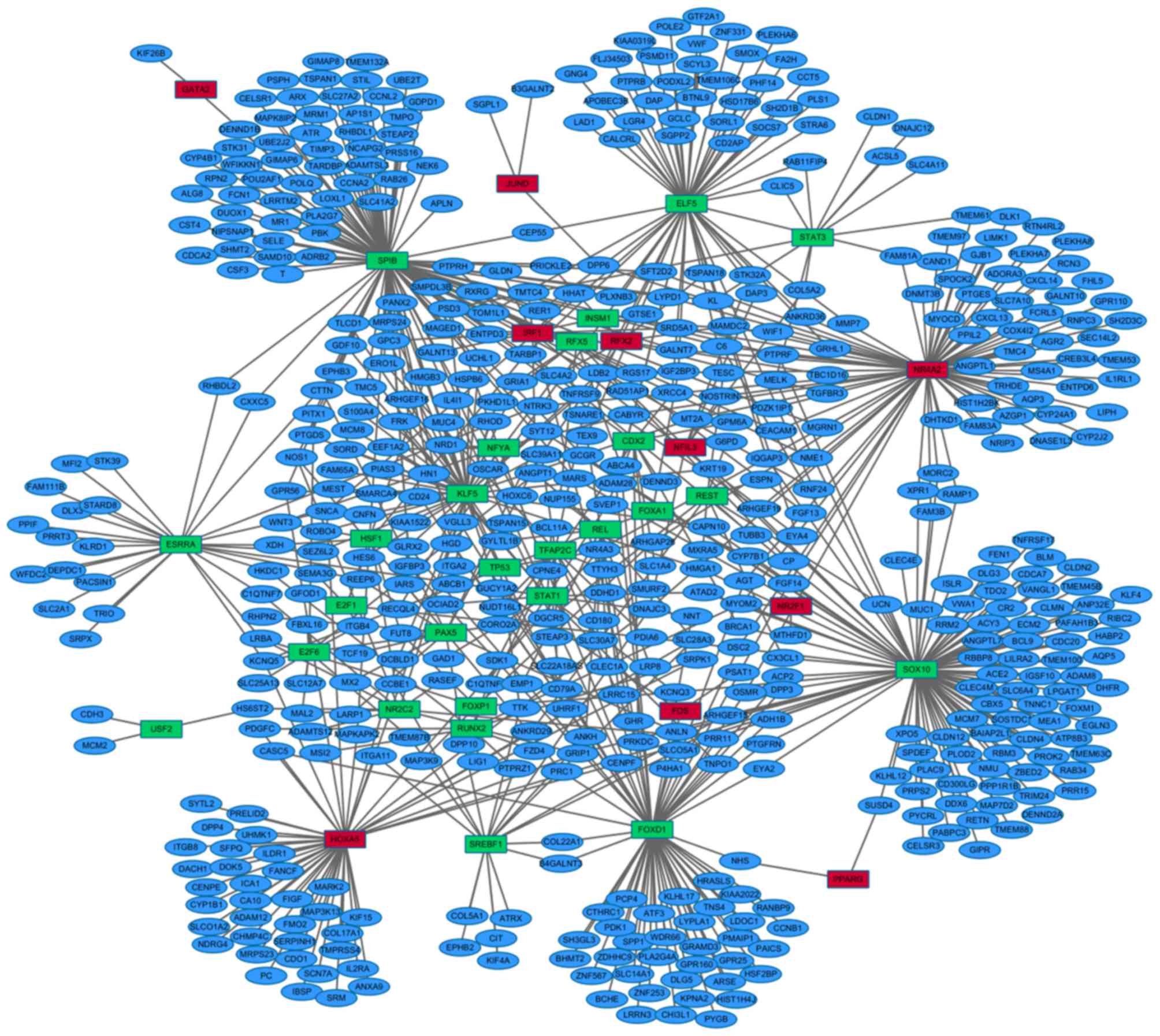
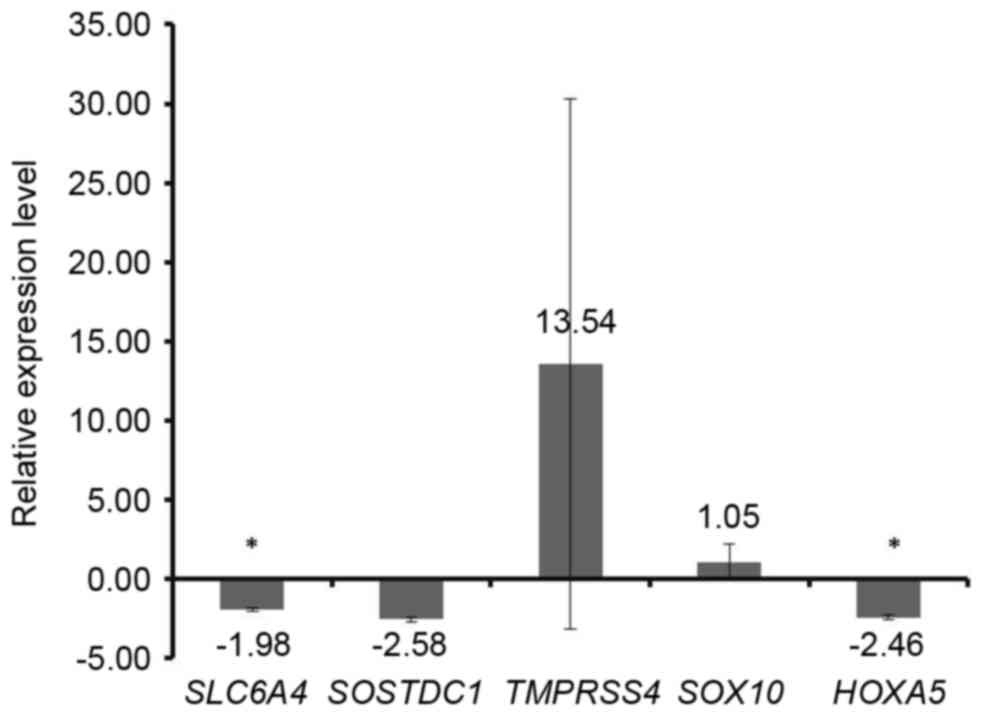
| SOX10 | 9.43E-02 | Up | 109 | ANP32E, RIBC2,
GRIP1, ADH1B, RETN, FEN1, HOXA5, PAFAH1B3, MELK, DDX6, BAIAP2L1,
SYT12, TNNC1, DHFR, CLMN, PLAC9, FOXM1, CDC20, FAM3B, SLC6A4,
CELSR3, KLHL12, ANGPTL7, TMEM100, TDO2, EYA2, IGSF10, SOSTDC1,
ANLN, EGLN3, CLEC4E, RAMP1, ADAM8, CAPN10, KRT19, RBM3, C6, PABPC3,
ARHGEF15, MEA1, KLF4, DENND2A, SPDEF, AQP5, TRIM24, RAD51AP1, GCGR,
CBX5, VANGL1, SLCO5A1, IQGAP3, MARS, PTPRF, LILRA2, MYOM2, CD300LG,
ECM2, CLDN12, KCNQ3, LPGAT1, CLEC4M, CX3CL1, CR2, HABP2, ATP8B3,
MCM7, ATAD2, PTGFRN, CYP7B1, CDCA7, MAP7D2, DLG3, MUC1, RAB34,
RRM2, CLDN4, GIPR, DPP3, ISLR, CENPF, ACP2, MORC2, PDIA6, UCN,
XPO5, SUSD4, ACY3, PRPS2, BLM, TMEM45B, RBBP8, TNFRSF17, TMEM63C,
VWA1, XPR1, PPP1R1B, PROK2, BRCA1, TMEM88, HMGA1, ACE2, PLOD2,
TNFRSF9, PRR15, NMU, BCL9, PYCRL, ZBED2, CLDN2 |
| SPIB | 6.34E-02 | Up | 108 | PANX2, PTPRF,
CEP55, TLCD1, PBK, NIPSNAP1, ADAMTSL3, ARHGEF19, TSPAN15, ENTPD3,
ATR, CSF3, GALNT7, HGD, UBE2T, NOS1, MR1, SLC27A2, CDCA2, T,
GIMAP8, MAGED1, TOM1L1, PTPRH, PLXNB3, LYPD1, POLQ, RGS17, DPP6,
HMGB3, SHMT2, CST4, CYP4B1, CTTN, FRK, ALG8, KL, PITX1, TARDBP,
ERO1L, NEK6, CCNL2, WFIKKN1, RXRG, PSD3, RPN2, RAB26, RER1, PLA2G7,
C6, GDF10, SMPDL3B, MRPS24, CELSR1, RHBDL2, DUOX1, PDZK1IP1, TIMP3,
MRM1, ADRB2, TSPAN1, LOXL1, APLN, IGFBP3, PRSS16, GLRX2, NME1,
SYT12, GALNT13, TMC5, RAD51AP1, POU2AF1, SAMD10, SFT2D2, UBE2J2,
RHBDL1, SELE, MAPK8IP2, TMEM132A, GATA2, HN1, STEAP2, MAPKAPK2,
FCN1, PSPH, MGRN1, TMPO, GPC3, GDPD1, GPR56, MARS, LRRTM2, GUCY1A2,
GIMAP6, SLC41A2, CEACAM1, STK31, TNFRSF9, CCNA2, NCAPG2, HOXC6,
AP1S1, DENND1B, GCGR, STIL, ARX, CD24, CXXC5 |
| NR4A2 | 1.45E-02 | Down | 81 | CABYR, AQP3,
RTN4RL2, MORC2, DPP6, SEC14L2, CAND1, MMP7, TRHDE, PLEKHA8,
ANGPTL1, DHTKD1, PLEKHA7, MAMDC2, NRIP3, FAM83A, ARHGAP26, LIPH,
TMC4, RGS17, ENTPD6, NME1, GALNT10, PTPRF, MYOM2, LYPD1, DNAJC3,
COX4I2, DNMT3B, GALNT7, AGR2, GJB1, TMEM97, MGRN1, AGT, SRD5A1,
LIMK1, TMEM61, CEACAM1, MT2A, CYP2J2, SFT2D2, GRHL1, DSC2, PTGES,
FHL5, CYP24A1, TMEM53, FAM81A, CREB3L4, OSMR, SLC7A10, CXCL13,
DNASE1L3, GPR110, PDZK1IP1, AZGP1, RAMP1, FAM3B, SPOCK2, RCN3,
MS4A1, NR4A3, C6, CXCL14, ADORA3, HIST1H2BK, MYOCD, ANKRD36, WIF1,
RNPC3, NOSTRIN, DLK1, FCRL5, IL1RL1, PPIL2, FGF14, XPR1, NTRK3,
SH2D3C, ARHGEF19 |
| FOXD1 | 8.42E-02 | Up | 68 | PAICS, TNPO1,
PRR11, HRASLS, ZNF253, DSC2, COL22A1, EYA2, PRKDC, ZDHHC9, DDHD1,
HSF2BP, DPP3, SPP1, ABCA4, BHMT2, ADH1B, AGT, PLA2G4A, DLG5,
RANBP9, UHRF1, CLEC1A, TNS4, ADAM28, LDOC1, ANLN, NNT, ZNF567,
CCNB1, GUCY1A2, NHS, ARSE, CTHRC1, CD24, PTGFRN, CHI3L1, CASC5,
PYGB, ATF3, PMAIP1, HOXC6, HIST1H4J, LYPLA1, GPR25, KIAA2022, PRC1,
HN1, GLRX2, IGFBP3, TSPAN15, PDK1, WDR66, ACP2, KPNA2, PCP4,
B4GALNT3, SLC14A1, GPR160, HGD, FGF14, P4HA1, SH3GL3, OSMR, GRAMD3,
LRRN3, KLHL17, BCHE |
| ELF5 | 5.48E-02 | Up | 59 | PTPRB, RNF24,
POLE2, PLXNB3, ANKRD36, TMEM106C, CLIC5, FA2H, APOBEC3B, SGPP2,
GNG4, ZNF331, TSPAN18, DAP3, SORL1, FAM81A, TOM1L1, STRA6, LDB2,
DAP, GTF2A1, VGLL3, LAD1, BTNL9, MMP7, PHF14, TESC, ABCA4, XRCC4,
TARBP1, SMOX, SOCS7, GRHL1, STK32A, PLS1, PODXL2, VWF, TEX9,
IGF2BP3, CD2AP, KIAA0319L, FLJ34503, PSMD11, CCT5, GCLC, CEP55,
RAB11FIP4, LGR4, SNCA, IQGAP3, HSD17B6, SCYL3, CALCRL, EYA4,
TSNARE1, PLEKHA6, RER1, SH2D1B, ADAM28 |
| HOXA5 | 4.13E-04 | Down | 50 | NR4A3, CENPF,
CENPE, SERPINH1, CYP1B1, IBSP, ICA1, IL2RA, ITGB8, SNCA, SFPQ,
ITGA11, CDO1, NDRG4, FANCF, SYTL2, VGLL3, PRELID2, MRPS23, FMO2,
DNAJC3, ILDR1, DACH1, SLC28A3, CHMP4C, ADAM12, DCBLD1, TMPRSS4,
ANKRD29, MAP3K13, KIF15, SRPK1, DOK5, COL17A1, MARK2, PC, SRM,
CASC5, DPP4, ANXA9, PDGFC, SCN7A, CA10, SLCO1A2, FIGF, KCNQ5,
UHMK1, GPR56, GRIP1, MAPKAPK2 |
| KLF5 | 9.34E-02 | Up | 45 | SORD, GPR56, PSD3,
HMGA1, SMARCA4, CNFN, TLCD1, PKHD1L1, GYLTL1B, SEZ6L2, TNFRSF9,
HSPB6, PTGDS, RECQL4, KRT19, MUC4, SLC4A2, ROBO4, ABCB1, DCBLD1,
EEF1A2, PANX2, UHRF1, LDB2, PIAS3, MT2A, CAPN10, FAM65A, EPHB3,
S100A4, SLC39A11, GLRX2, MEST, GDF10, UCHL1, GRIA1, HMGB3, HES6,
RHOD, ARHGEF16, IL4I1, NR2C2, MRPS24, MCM8, KIAA1522 |
| ESRRA | 7.63E-02 | Up | 27 | WFDC2, MFI2,
PACSIN1, C1QTNF7, XDH, KCNQ5, RHBDL2, EPHB3, DEPDC1, STK39, CXXC5,
SLC2A1, PRRT3, HKDC1, PPIF, STARD8, KLRD1, DLX3, SRPX, SNCA, TRIO,
NNT, SEZ6L2, FAM111B, LRBA, NTRK3, REEP6 |
| SREBF1 | 5.74E-02 | Up | 18 | COL22A1, SLC28A3,
DPP10, CIT, ATRX, EPHB2, CCBE1, TMEM87B, DNAJC3, ITGA11, KIF4A,
B4GALNT3, TTK, PTPRZ1, ARHGEF15, COL5A1, ANKH, SLCO5A1 |
| REL | 2.16E-02 | Up | 14 | CLEC1A, NUP155,
LRP8, ANGPT1, ANKH, GHR, KCNQ5, EMP1, FOXA1, TEX9, SLC28A3,
SLC30A7, GYLTL1B, LRRC15 |