Introduction
Naturally occurring dietary compounds derived from
medicinal plants serve important roles against inflammation and
cancer (1–3). Coffee, the most commonly consumed
beverage worldwide, is a rich source of dietary phenolic
phytochemicals (4).
Epidemiological studies have indicated that the dietary intake of
coffee lowers the risk of developing cardiovascular disorders,
diabetes, Parkinson's disease and cancer of the colon, liver,
breast, prostate and endometrium (5–7).
Chlorogenic acid (CA) is one of the most abundant
phenolic phytochemicals purified from various plants, fruits and
beverages, such as coffee. Previous in vitro and in
vivo studies have reported that CA demonstrates significant
anti-inflammatory, antioxidative and anticarcinogenic effects
(8–10). This suggests that the use of CA may
be a useful strategy for the treatment and/or control of
inflammation and cancer. However, the underlying molecular
mechanisms and targets responsible for the health benefits of CA
remain unknown.
Although the pathogenesis of inflammatory bowel
disease (IBD) has not yet been elucidated, the currently accepted
pathogenic scenario suggests that IBD may occur as a result of
dysregulated innate and adaptive immune responses against the
commensal gut flora, thus generating an excessive production of
pro-inflammatory mediators (11).
As part of the dysregulated immune response, macrophages serve a
major role in IBD (12–14).
Lipopolysaccharide (LPS), a component of
Gram-negative bacterial cell walls, is involved in the barrier
function of the intestinal epithelium and activates the immune
system, leading to the production of pro-inflammatory mediators,
including nitric oxide (NO), enzymes including
interleukin-1-receptor-associated kinases or
interleukin-receptor-associated kinases (IRAKs), inflammatory
cytokines, chemokines and adhesion molecules (15,16).
The production of pro-inflammatory mediators induced by LPS has
been implicated in the activation of a number of signaling pathways
involving the phosphorylation of Janus kinase/signal transducer and
activator of transcription (JAK/STAT), mitogen-activated protein
kinases (MAPKs) and nuclear factor-κB (NF-κB). Among LPS-induced
signaling pathways, the JAK/STAT pathway is crucial for the
expression of pro-inflammatory mediators, including inducible
nitric oxide synthase (iNOS), NO and interleukin (IL)-1β (17–19).
Thus, activation of JAK/STAT serves a critical role in
inflammation-associated injuries, such as IBD, and has been
proposed to be a major causative event and therapeutic target for
IBD (20,21). In addition, the use of a JAK/STAT
inhibitor is being investigated as a target immunomodulator for IBD
treatment (22,23).
The aim of the present study was to evaluate the
effects of CA on LPS-induced inflammation and its associated
intracellular signaling pathway in macrophages.
Materials and methods
Cell culture
RAW264.7 murine macrophage-like cells were obtained
from the American Type Culture Collection (Manassas, VA, USA; ref.
no. CRL-1589). The cells were cultured in Dulbecco's modified
Eagle's medium (Hyclone; GE Healthcare Life Sciences, Logan, UT,
USA) supplemented with 10% fetal bovine serum (Hyclone; GE
Healthcare Life Sciences) and 1% (v/v) penicillin/streptomycin
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used
at 37°C in a humidified atmosphere of 5% CO2. CA and LPS
from Escherichia coli (Serotype 0111:B4) were purchased from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). STAT3 inhibitor
(STAT3i) S3I-201 was obtained from Sigma-Aldrich; Merck KGaA and
dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA).
Cells were pretreated with 2 mM CA or 50 µM STAT3i prior to
stimulation with 100 ng/ml LPS for 24 h.
Cell viability assay
RAW264.7 cells were plated on a 96-well plate at a
density of 1×104 cells/well and were incubated for 24 h
at 37°C prior to treatment with CA. Cells were then treated with
various concentrations of CA (0, 0.05, 0.1, 0.2, 0.5, 1, 2 mM) or
LPS (50, 100 and 200 ng/ml) for 24 h. Following treatment, cell
viability was determined using an EZ-Cytox (WST-1 tetrazolium salt)
Colorimetric Cell Viability assay kit (Daeil Lab Service Co., Ltd,
Seoul, Republic of Korea) according to the manufacturer's
protocols. Following incubation of cells with the WST-1 reagent
(100 µl/well) diluted 1:20 for 90 min at 37°C, the absorbance at
450 nm was measured using a microplate reader (Infinite M200; Tecan
Trading AG., Männedorf, Switzerland). Each experiment was performed
in triplicate wells and repeated at least three times.
Semi-quantitative reverse
transcription-polymerase chain reaction (Semi-qRT-PCR)
RAW264.7 cells were plated on a 12-well plate at a
density of 2×105 cells/well, and then treated with 2 mM
CA or 100 ng/ml LPS for 0 to 4 or 16 h. Total RNA was isolated from
cells by using the TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocols.
The quantity and purity of total RNA was determined using a
NanoDrop spectrophotometer (NanoDrop Technologies, LLC; Thermo
Fisher Scientific, Inc., Wilmington, DE, USA). Total RNA (1 µg) was
reverse transcribed to cDNA using the Moloney murine leukemia virus
reverse transcriptase (Promega Corporation, Madison, WI, USA)
according to manufacturer's protocols. Semi-qRT-PCR was
performed to measure the mRNA level of target genes and reference
gene (GAPDH) using gene-specific primers (Table I) and the GoTaq DNA Polymerase
(Promega Corporation) according to manufacturer's protocols. The
Semi-qRT-PCR was performed by initial incubation at 94°C for
5 min followed by 30 cycles of 94°C for 20 sec, 58°C for 30 sec and
72°C for 30 sec, with a final extension step of 72°C for 5 min. The
PCR products were separated by electrophoresis on a 1% agarose gel
containing ethidium bromide. The signals were quantified by
densitometric analysis using Multi-Gauge software version 3.0
(Fujifilm Holdings Corporation). Experiments were performed in
triplicate.
 | Table I.Primers employed for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primers employed for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene | Sequence
(5′-3′) |
|---|
| IL-6 |
|
|
Forward |
GGATACCACCCACAACAGACC |
|
Reverse |
GGTCCTTAGCCACTCCTTCTG |
| TNF-α |
|
|
Forward |
GCACAGAAAGCATGATCCGCG |
|
Reverse |
GACAGAAGAGCGTGGTGGCCC |
| MIP-2 |
|
|
Forward |
GACTTCAAGAACATCCAGAGCT |
|
Reverse |
GTTAGCCTTGCCTTTGTTCAG |
| IL-1β |
|
|
Forward |
GGAGAACCAAGCAACGACAAA |
|
Reverse |
TGGGGAACTCTGCAGACTCAAAC |
| iNOS |
|
|
Forward |
CTGCAGCACTTGGATCAGGA |
|
Reverse |
GAGTAGCCTGTGTGCACCTG |
| COX-2 |
|
|
Forward |
CCTTCTCCAACCTCTCCTACTA |
|
Reverse |
GATACACCTCTCCACCAATG |
| GAPDH |
|
|
Forward |
ACCACAGTCCATGCCATCAC |
|
Reverse |
TCCACCACCCTGTTGCTGTA |
Western blot analysis
RAW264.7 cells plated on a 6-well plate at a density
of 3×105 cells/well and then treated with 2 mM CA or 100
ng/ml LPS. Cells were lysed in radioimmunoprecipitation extraction
solution containing 1X Halt™ Phosphatase inhibitor and
1X Halt™ Protease inhibitor cocktail (Thermo Fisher
Scientific, Inc.) for 15 min in an ice bath. The protein
concentration of lysates from treated cells was measured using a
bicinchoninic protein assay kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. Equal quantities of
protein (20 µg) were separated using SDS-PAGE (10–12%) and
transferred to a polyvinylidene difluoride membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Antibodies against the
following proteins were employed: iNOS (cat. no. 2977),
cyclooxygenase-2 (COX-2; cat. no. 4842), STAT3 (cat. no. 9139),
phosphorylated (p)-STAT3 (cat. no. 8336), JAK2 (cat. no. 3230),
p-JAK2 (cat. no. 4406), extracellular signal-regulated kinase1/2
(ERK1/2; cat. no. 4695), p-ERK1/2 (cat. no. 4370), p38 (cat. no.
9212), p-p38 (cat. no. 4511), c-Jun NH2-terminal kinase (JNK; cat.
no. 9252), p-JNK (cat. no. 4668), nuclear factor of κ light
polypeptide gene enhancer in B-cells inhibitor, α (IκBα; cat. no.
9242), p-IκBα (cat. no. 9246), p65 (cat. no. 4764), p-p65 (cat. no.
3033) all from Cell Signaling Technology, Inc. (Danvers, MA, USA),
and lamin A (cat. no. sc-20680) and GAPDH (cat. no. sc-25778) from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). The primary
antibodies were diluted 1:1,000 and incubated with membranes for 15
h at 4°C. Following a final rinsing with Tris-buffered saline-Tween
(TBST) each membrane was incubated with 1:1,000 diluted secondary
horseradish peroxidase (HRP)-linked anti-rabbit (cat. no. 7074) or
anti-mouse (cat. no. 7076) immunoglobulin (Ig)G from Cell Signaling
Technology, Inc. for 1 h at room temperature. Protein bands were
analyzed using an enhanced chemiluminescence detection system
(Amersham; GE Healthcare Life Sciences) and the ImageQuant LAS-4000
luminescent image analyzer (Fujifilm Holdings Corporation, Tokyo,
Japan) Immunoblots were quantified using Multi-Gauge software
version 3.0 (Fujifilm Holdings Corporation). Experiments were
performed in triplicate.
Determination of IL-6, tumor necrosis
factor-α (TNF-α), macrophage inflammatory protein-2 (MIP-2) and
IL-1β levels
RAW264.7 cells were plated on a 12-well plate at a
density of 2×105 cells/well and pretreated with CA (1–2
mM) for 2 h followed by stimulation with 100 ng/ml LPS for 24 h.
The cell culture medium was then collected and centrifuged at
15,000 × g at 4°C for 7 min. The levels of IL-6, TNF-α, MIP-2 and
IL-1β in the culture medium were determined using Quantikine ELISA
mouse MIP-2 (cat. no. MM200) and mouse IL-1b kits (cat. no. MLB00C)
from R&D Systems Inc. (Minneapolis, MN, USA), and the OptEIA™
kits for mouse interleukin-6 (IL-6; cat. no. 555240) and mouse TNF
(cat. no. 558534) from BD Biosciences (San Jose, CA, USA) according
to the manufacturer's protocols. The quantity of IL-6, TNF-α, MIP-2
and IL-1β was determined based on the optical density values (read
at 450 nm) obtained using a Bio-Rad Model 550 Microplate Reader
(Bio-Rad Laboratories, Inc.) and a standard curve. Experiments were
performed in triplicate.
NO analysis
RAW264.7 cells were plated on a 12-well plate at a
density of 2×105 cells/well and pretreated with varying
concentrations of CA (0 to 2 mM) for 2 h followed by stimulation
with 100 ng/ml LPS for 24 h. NO synthesis was determined by
analyzing the level of nitrite in cell culture supernatants using
Griess Reagent (1% sulfanilamide, 0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride in 5% phosphoric
acid solution) following incubation at room temperature for 30 min.
The absorbance was measured at 540 nm and the nitrite concentration
was determined using sodium nitrite as a standard. Three replicates
were performed for each of the different treatments.
Preparation of nuclear extracts
RAW264.7 cells were plated on a 60 mm dish at a
density of 2×106 cells and pretreated with CA (2 mM) for
2 h followed by stimulation with 100 ng/ml LPS for 24 h. The cells
were washed with phosphate-buffered saline and lysis the pellet
cells by centrifugation at 250 × g for 5 min at 4°C. Nuclear
protein extracts were prepared using the ProteoJET™
Cytoplasmic and Nuclear Protein Extraction kit (Fermentas; Thermo
Fisher Scientific, Inc., Pittsburgh, PA, USA) according to the
manufacturer's protocols.
Statistical analysis
The results are presented as the mean ± standard
deviation. Differences between groups were determined by Student's
t-test. The statistical analysis was performed with the SPSS
version 15.0 (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
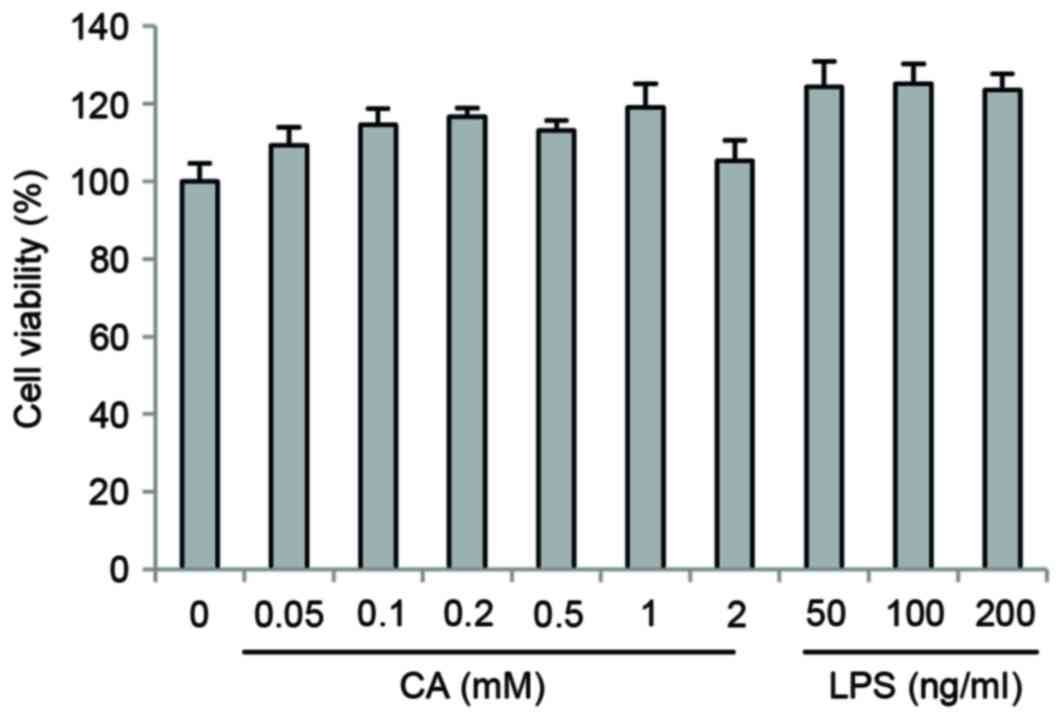
Impact of CA on the viability of
RAW264.7 cells
RAW264.7 cells were treated with CA and LPS at a
range of different concentrations (CA, 0 to 2 mM; LPS, 0 to 200
ng/ml) for 24 h. As shown in Fig.
1, CA and LPS treatment did not exhibit cytotoxic effects on
RAW264.7 cells.
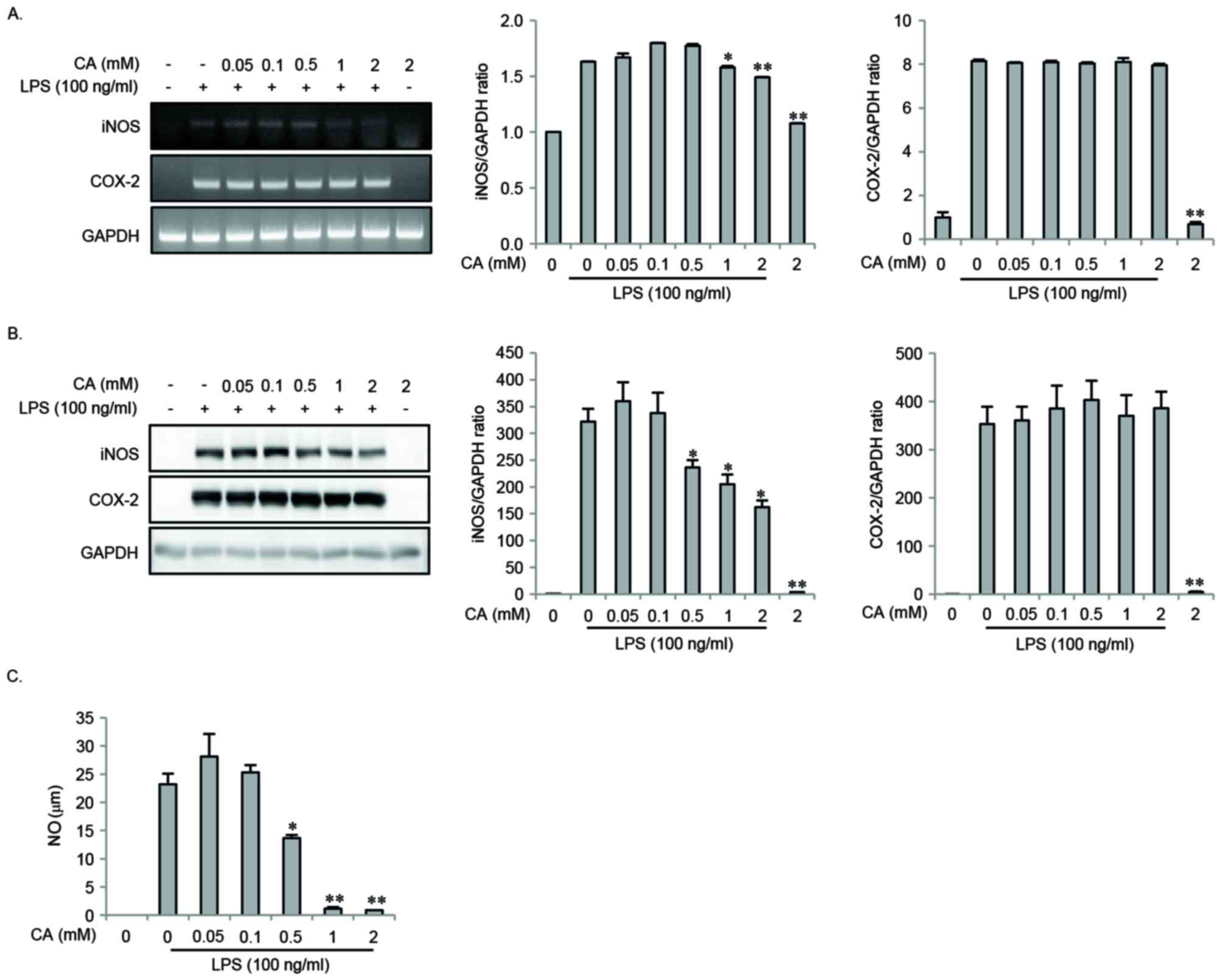
Effect of CA on LPS-induced expression
of iNOS, NO and COX-2 in RAW264.7 cells
The present study investigated the effects of CA on
the expression of iNOS and COX-2 in response to LPS challenge of
RAW264.7 cells. The cells were pretreated with varying
concentrations of CA for 2 h, and then stimulated with LPS for 16
or 24 h. LPS-induced mRNA and protein expression of iNOS and COX-2
was measured by Semi-qRT-PCR and western blotting. LPS-induced
expression of iNOS mRNA and protein was significantly inhibited in
a dose-dependent manner by CA pretreatment, whereas COX-2
expression was not (Fig. 2A and
B). In addition, LPS-induced NO production was inhibited in a
dose-dependent manner by CA pretreatment (Fig. 2C).
Impact of CA on LPS-induced
pro-inflammatory mediator expression
The effect of CA on the production of
pro-inflammatory mediators was investigated. Cells were pretreated
with CA for 2 h, and were then stimulated with LPS for 0, 1, 4 or
24 h. CA pretreatment significantly inhibited the LPS-induced
expression of IL-6, TNF-α, MIP-2 and IL-1β at the mRNA and protein
levels (Fig. 3A and B).
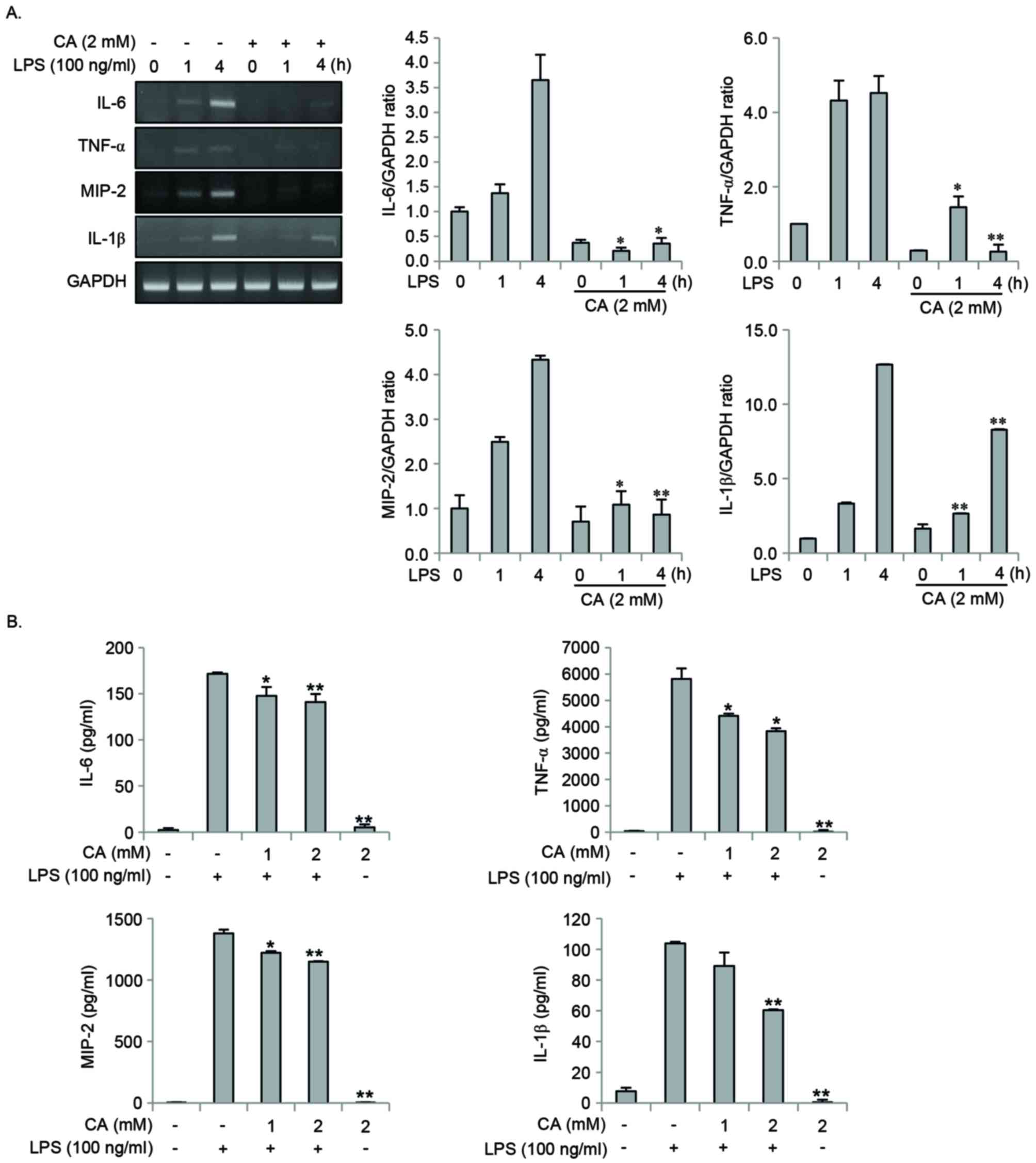 | Figure 3.CA inhibits LPS-induced expression of
pro-inflammatory mediators in RAW264.7 cells. (A) RAW264.7 cells
were pretreated with 2 mM CA for 2 h, followed by stimulation with
100 ng/ml LPS for 0 to 4 h. Total RNA was isolated using the TRIzol
procedure and the expression of IL-6, TNF-α, MIP-2 and IL-1β was
detected by reverse transcription-quantitative polymerase chain
reaction. Gene expression levels were normalized to those of GAPDH.
(B) RAW264.7 cells were pretreated with 0, 1 or 2 mM CA for 2 h
prior to stimulation with 100 ng/ml LPS. Following 24 h, the levels
of IL-6, TNF-α, MIP-2 and IL-1β in the culture medium were measured
using an ELISA kit. *P<0.05 and **P<0.01 vs. LPS stimulation
alone. CA, chlorogenic acid; LPS, lipopolysaccharide; IL,
interleukin; TNF-α, tumor necrosis factor-α; MIP-2, macrophage
inflammatory protein-2. |
Effect of CA on LPS-induced JAK2 and
STAT3 activation
The expression of pro-inflammatory mediators is
regulated by the activation of the MAPKs, NF-κB, JAK2 and STAT3
signaling pathways (17).
Therefore, the present study determined the effect of CA on the
LPS-induced MAPKs, NF-κB, JAK2 and STAT3 by western blotting. Cells
were pretreated with specific concentrations of CA for 2 h, prior
to stimulation with LPS for various lengths of time. CA
pretreatment resulted in a decrease in the level of p-JAK2 and
p-STAT3 (Fig 4A and B), whereas
the levels of ERK1/2, JNK, p38, IκBα and p65 remained unchanged
(Fig. 4C).
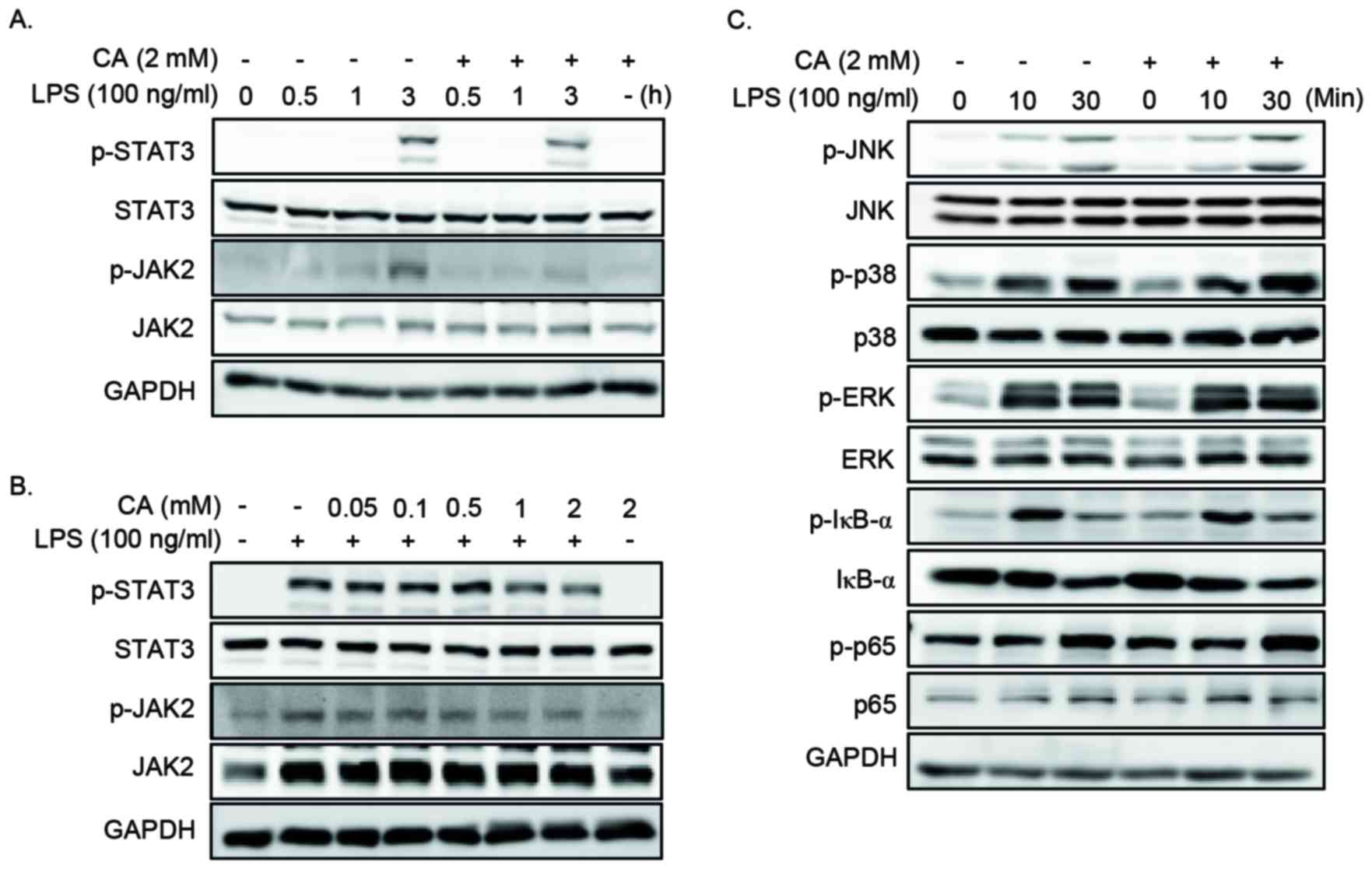 | Figure 4.CA inhibits the LPS-induced activation
of JAK2 and STAT3 in RAW264.7 cells. (A) Cells were pretreated with
2 mM CA for 2 h and then stimulated with 100 ng/ml LPS for 0, 0.5,
1 or 3 h. Cell lysates were prepared and subjected to western
blotting analysis. CA pretreatment was associated with decreased
levels of p-JAK2 and p-STAT3 at 3 h following LPS stimulation. (B)
RAW264.7 cells were pretreated with 0 to 2 mM CA for 2 h prior to
stimulation with 100 ng/ml LPS for 3 h. Cell lysates were prepared
and subjected to western blotting analysis. CA pretreatment with 1
or 2 mM was associated with a decrease in the level of p-JAK2 and
p-STAT3. (C) Cells were pretreated with 0 or 2 mM CA for 2 h prior
to stimulation with 100 ng/ml LPS for 0, 10 or 30 min. Cell lysates
were prepared and subjected to western blotting analysis. The
levels of p-ERK1/2, p-JNK, p-p38, p-IκBα and p-p65 remained
unchanged with CA pretreatment. CA, chlorogenic acid; LPS,
lipopolysaccharide; p-STAT3, phosphorylated signal transducer and
activator of transcription 3; p-JAK2, phosphorylated Janus kinase
2; p-JNK, phosphorylated c-Jun NH2-terminal kinase; p-ERK,
phosphorylated extracellular signal-regulated kinase; p-IkBa,
phosphorylated nuclear factor of k light polypeptide gene enhancer
in B-cells inhibitor, α. |
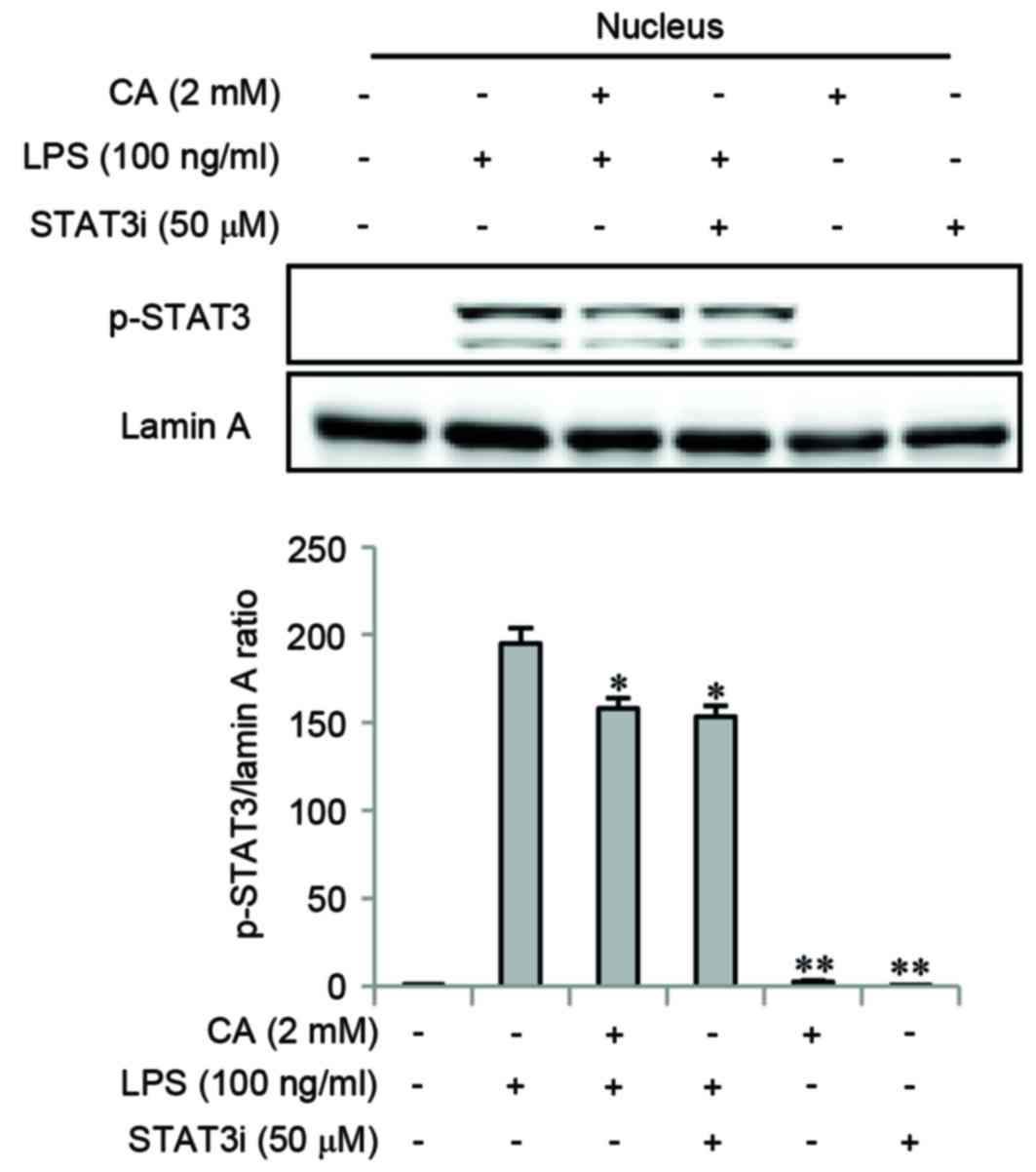
Impact of CA on nuclear translocation
of phosphorylated STAT3
A STAT3i was used to determine whether the STAT3
signaling pathway may be involved in the anti-inflammatory effects
of CA. Cells were pretreated with CA or STAT3i for 2 h, and then
stimulated with LPS for 3 h. The level of p-STAT3 was determined by
western blotting. CA and STAT3i pretreatments significantly
inhibited LPS-induced nuclear translocation of p-STAT3 (Fig. 5).
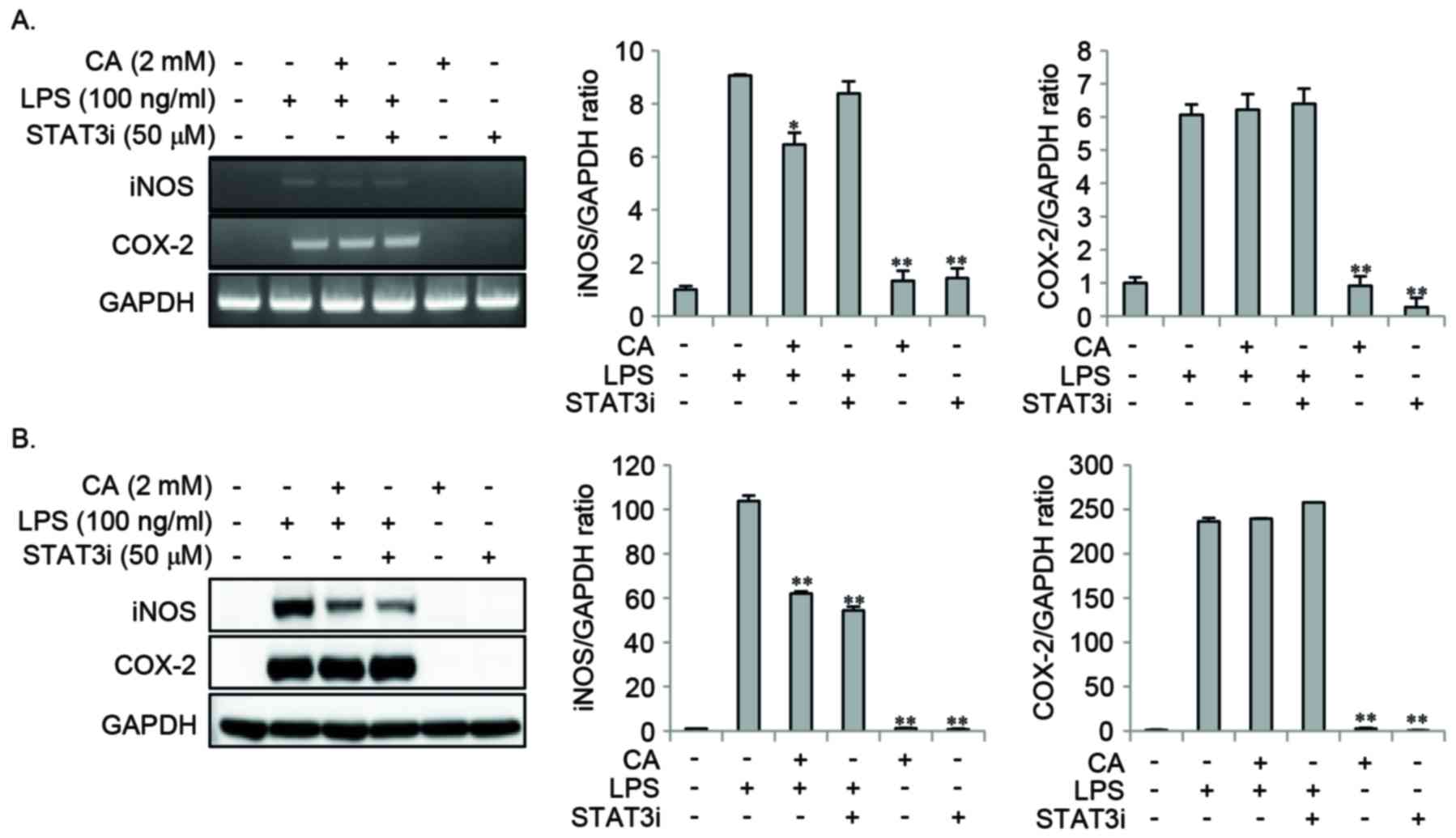
Effect of STAT3i on LPS-induced NO and
the expression of pro-inflammatory mediators
In order to examine the effect of STAT3 signaling on
LPS-induced NO and pro-inflammatory mediator expression, cells were
pretreated with CA or STAT3i for 2 h, and then stimulated with LPS
for 16 or 24 h. STAT3i pretreatment inhibited the LPS-induced
expression of iNOS protein, whereas the LPS-induced mRNA and
protein expression of COX-2 remained unchanged (Fig. 6). Pretreatment with CA
significantly reduced the LPS-induced expression of iNOS mRNA and
protein, whereas no effect on COX-2 mRNA and protein was observed
(Fig. 6). In addition, STAT3i
pretreatment significantly inhibited the LPS-induced expression of
NO and IL-1β when compared to LPS treatment alone (Fig. 7A and B); however, the expression
levels of IL-6, TNF-α, and MIP-2 remained unaffected (Fig. 7C-E). Pretreatment with CA
significantly inhibited LPS-induced expression of NO and all of the
pro-inflammatory mediators investigated (Fig. 7). These results suggest that the
inhibitory effect of CA on LPS-induced NO and IL-1β expression may
be mediated by inhibiting the STAT3 signaling pathway.
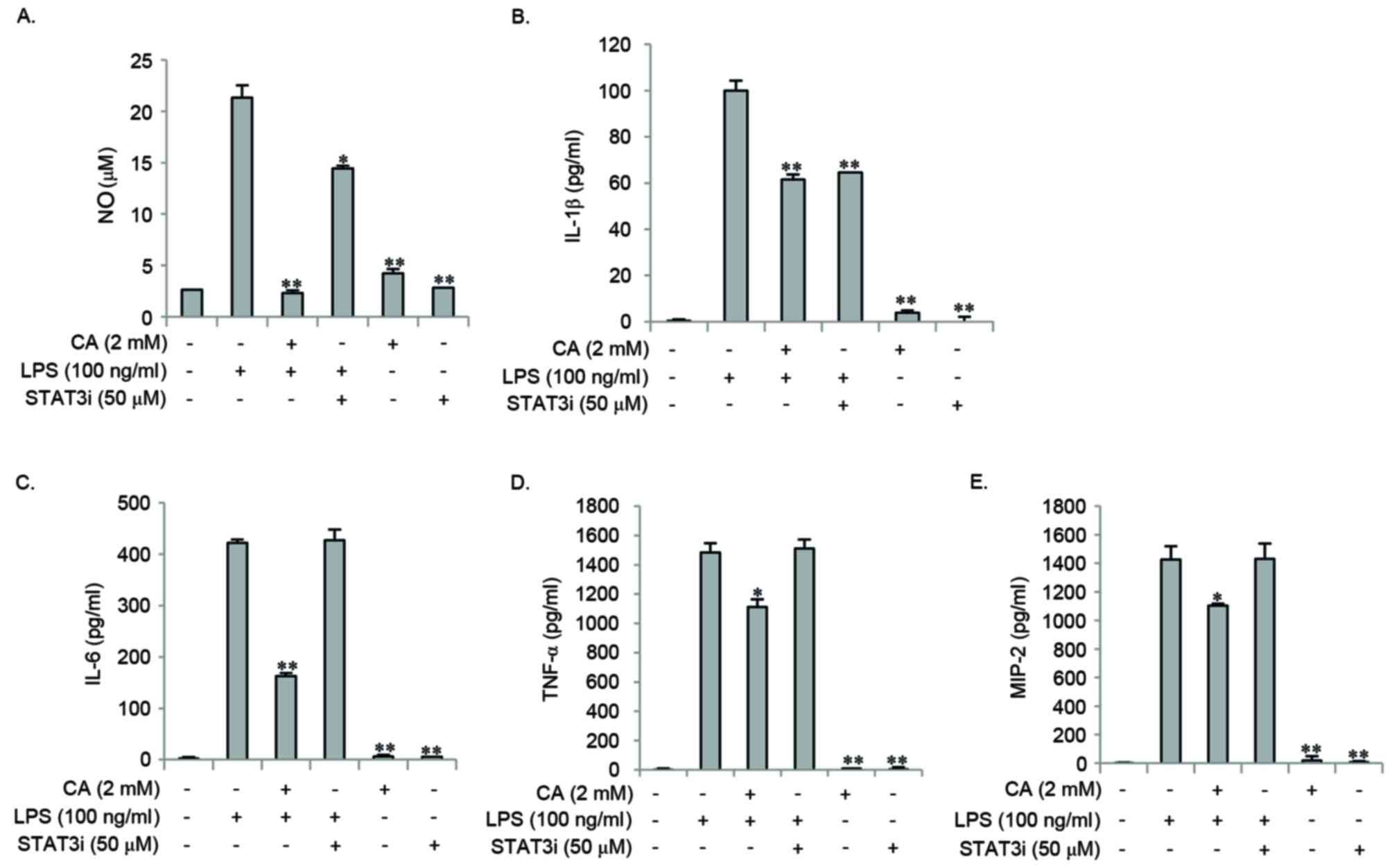 | Figure 7.STAT3i inhibited LPS-induced
expression of NO and IL-1β, and not the expression of IL-6, TNF-α
and MIP-2 in RAW264.7 cells. RAW264.7 cells were pretreated with 2
mM CA or STAT3i for 2 h, and then stimulated with 100 ng/ml LPS for
24 h. The levels of NO, IL-6, TNF-α, MIP-2 and IL-1β were measured
in the culture medium using Griess reagents or ELISA kits.
Pretreatment with STAT3i and CA inhibited LPS-induced (A) NO and
(B) IL-1β expression. Unlike pretreatment of cells with CA, STAT3i
did not inhibit the LPS-induced expression of (C) IL-6, (D) TNF-α
and (E) MIP-2. Data are expressed as the mean ± standard deviation
of three independent experiments. *P<0.05 and **P<0.01 vs.
LPS stimulation alone. STAT3i, STAT3 inhibitor; LPS,
lipopolysaccharide; NO, nitric oxide; IL-6, interleukin-6; TNF-α,
tumor necrosis factor-α; MIP-2, macrophage inflammatory protein-2;
CA, chlorogenic acid. |
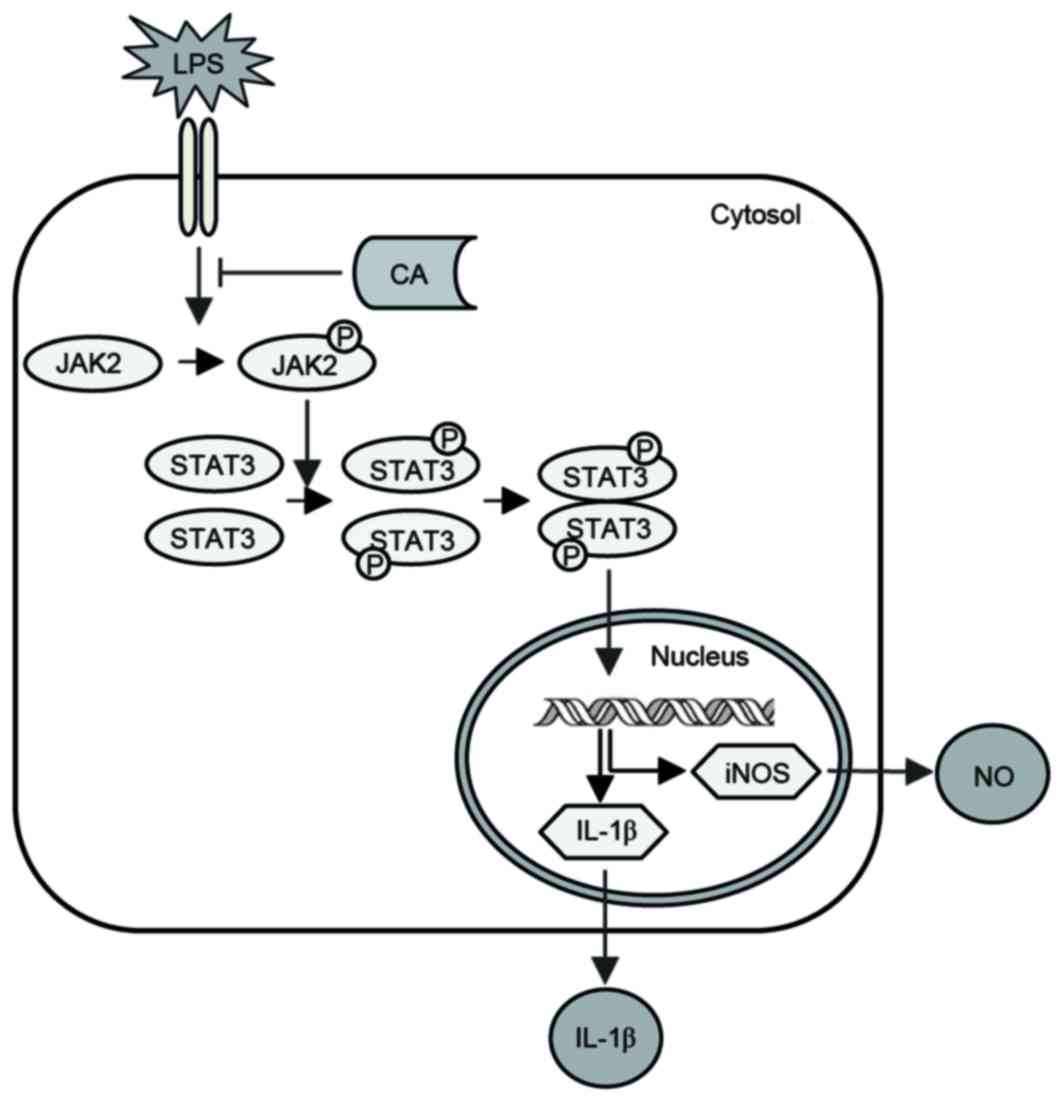
Potential mechanisms underlying the
anti-inflammatory actions of CA
CA inhibited LPS-induced JAK2 and STAT3
phosphorylation, the nuclear translocation of p-STAT3 and the
expression of NO and IL-1β in RAW264.7 cells (Fig. 8). These results indicate that CA
may suppress LPS-induced NO and IL-1β expression by inhibiting
JAK2/STAT3 activation in RAW264.7 cells.
Discussion
CA is one of the most abundant polyphenol compounds
present in the human diet and is a major constituent of coffee.
Previous studies have demonstrated that CA possesses a number of
biological properties including anti-inflammatory, antioxidant and
anticarcinogenic activities (8).
These beneficial effects of CA may be due to its ability to
scavenge free radicals, such as reactive nitrogen and oxygen
species, and downregulate pro-inflammatory mediators (8–10).
NO is an inorganic free radical, synthesized by NOS.
Two major isoforms of NOS have been identified in the intestine,
including the constitutive (endothelial NOS) and inducible forms
(iNOS). Endothelial NOS is constitutively expressed and is
responsible for the maintenance of housekeeping and physiological
functions. By contrast, iNOS is overexpressed under pathological
conditions, such as inflammation, in a number of cells, which leads
to the production of high levels of NO. The production of NO by
iNOS and reactive nitrogen species disrupts intestinal barrier
function, leading to the increased uptake of luminal antigens, as
well as the activation of a dysregulated immune response (24–26).
Two different isoforms of COX catalyze the synthesis of
prostaglandins from arachidonic acid. COX-1 is known as the
housekeeping enzyme, while COX-2 is induced in response to a
variety of pro-inflammatory mediators, growth factors and hormones
(24–27).
The present study first investigated the effect of
CA on the expression of reactive nitrogen species and COX-2 in
LPS-stimulated macrophages. The results demonstrated that CA
inhibited LPS-induced iNOS expression and NO production in
macrophages, whereas COX-2 expression remained unchanged.
A number of studies have revealed that the excessive
production of pro-inflammatory cytokines and chemokines, including
IL-1β, IL-6, TNF-α, monocyte chemotactic protein (MCP) and MIP,
leads to uncontrolled inflammation and tissue injury (12–16).
In the present study, CA inhibited the LPS-induced expression of
IL-6, TNF-α, MIP-2 and IL-1β in macrophages. Similarly, a previous
study demonstrated that CA inhibited the staphylococcal
exotoxin-induced production of IL-1β, TNF-α, IL-6, interferon-γ,
MCP-1, MIP-1α and MIP-1β in human peripheral blood mononuclear
cells (28). These results
indicate that CA may exert anti-inflammatory activities via the
suppression of reactive nitrogen species and the downregulation of
pro-inflammatory cytokines and chemokines, irrespective of cell
type.
The production of various pro-inflammatory mediators
is controlled by the activity of transcription factors and protein
kinases, including NF-κB, MAPKs and JAK/STAT. Consequently, the
activation of these transcription factors and protein kinases is
important in inflammation-associated diseases, such as IBD, and
have been proposed as potential therapeutic targets for IBD
(20,21,29–31).
A previous study revealed that CA exhibits anti-inflammatory
activities by modulating important metabolic pathways (8). It has been previously demonstrated
that CA inhibits LPS-induced inflammation in macrophages by
suppressing NF-κB and MAPK signaling pathways (32,33).
In addition, CA exerted beneficial effects in LPS-, dextran sulfate
sodium- and trinitrobenzene sulfonic acid-induced colitis in animal
models (34–36). Therefore, the results from in
vitro and in vivo studies indicate that part of the
anti-inflammatory effects of CA may be attributed to the inhibition
of NF-κB and MAPK activities. However, the effect of CA on JAK/STAT
signaling remains to be elucidated.
The present study then investigated the impact of CA
on LPS-induced NF-κB, MAPKs and JAK/STAT signaling pathways in
macrophages. CA reduced the level of p-JAK2 and p-STAT3, whereas
the phosphorylation levels of ERK1/2, JNK, p38, IκBα and p65
remained unchanged. The JAK/STAT signaling pathway is an essential
inflammatory pathway that mediates immune responses. A previous
study demonstrated that JAK/STAT are involved in inflammatory
signaling pathways in response to various external stimuli,
including LPS, hormones, growth factors and cytokines (19). The binding of ligands to its
associated receptors induces JAK phosphorylation and activation.
Activated JAK then phosphorylates STAT, which subsequently forms a
homo- or heterodimer. These dimers translocate to the nucleus and
bind to specific sequences in the promoter regions of target genes
encoding pro-inflammatory mediators, including cytokines,
chemokines and inducible enzymes, such as iNOS and COX-2 (16–20).
In the present study, STAT3i was used to determine
whether the STAT3 signaling pathway may be involved in the
anti-inflammatory effects of CA. The results demonstrated that CA
and STAT3i inhibited the LPS-induced nuclear translocation of
p-STAT3. In addition, LPS-induced expression of iNOS, NO and IL-1β
protein was inhibited by STAT3i and CA pretreatments. However,
LPS-induced expression of IL-6, TNF-α and MIP-2 was not inhibited
by STAT3i. These results indicate that induction of IL-6, TNF-α and
MIP-2 by LPS may not be directly affected by the STAT3
transcription factor in the nucleus. Alternatively, the binding of
IL-6 to its receptor may induce phosphorylation of JAK/STAT and its
subsequent translocation to the nucleus (37,38).
In conclusion, the results of the present study
indicate that CA may suppress LPS-induced NO and IL-1β expression
by inhibiting JAK2/STAT3 activation in RAW264.7 cells. Therefore,
modulation of this cell signaling pathway by CA may be beneficial
in inflammation-associated diseases, such as IBD.
References
|
1
|
Trendowski M: Recent advances in the
development of antineoplastic agents derived from natural products.
Drugs. 75:1993–2016. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Joo YE: Natural product-derived drugs for
the treatment of inflammatory bowel diseases. Intest Res.
12:103–109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SB, Park SJ, Chung SH, Hahn KY, Moon
DC, Hong SP, Cheon JH, Kim T and Kim WH: Vaccination and
complementary and alternative medicine in patients with
inflammatory bowel disease. Intest Res. 12:124–130. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang NJ, Lee KW, Kim BH, Bode AM, Lee HJ,
Heo YS, Boardman L, Limburg P, Lee HJ and Dong Z: Coffee phenolic
phytochemicals suppress colon cancer metastasis by targeting MEK
and TOPK. Carcinogenesis. 32:921–928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Svilaas A, Sakhi AK, Andersen LF, Svilaas
T, Ström EC, Jacobs DR Jr, Ose L and Blomhoff R: Intakes of
antioxidants in coffee, wine, and vegetables are correlated with
plasma carotenoids in humans. J Nutr. 134:562–567. 2004.PubMed/NCBI
|
|
6
|
Ludwig IA, Clifford MN, Lean ME, Ashihara
H and Crozier A: Coffee: Biochemistry and potential impact on
health. Food Funct. 5:1695–1717. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bøhn SK, Blomhoff R and Paur I: Coffee and
cancer risk, epidemiological evidence, and molecular mechanisms.
Mol Nutr Food Res. 58:915–930. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liang N and Kitts DD: Role of chlorogenic
acids in controlling oxidative and inflammatory stress conditions.
Nutrients. 8:pii: E162015. View Article : Google Scholar
|
|
9
|
Upadhyay R and Rao LJ Mohan: An outlook on
chlorogenic acids-occurrence, chemistry, technology, and biological
activities. Crit Rev Food Sci Nutr. 53:968–984. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weng CJ and Yen GC: Chemopreventive
effects of dietary phytochemicals against cancer invasion and
metastasis: Phenolic acids, monophenol, polyphenol, and their
derivatives. Cancer Treat Rev. 38:76–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim YJ, Chang SY and Ko HJ:
Myeloid-derived suppressor cells in inflammatory bowel disease.
Intest Res. 13:105–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strober W, Fuss I and Mannon P: The
fundamental basis of inflammatory bowel disease. J Clin Invest.
117:514–521. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Podolsky DK and Xavier RJ: Unravelling the
pathogenesis of inflammatory bowel disease. Nature. 448:427–434.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SH: Intestinal permeability regulation
by tight junction: Implication on inflammatory bowel diseases.
Intest Res. 13:11–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Morris MC, Gilliam EA and Li L: Innate
immune programing by endotoxin and its pathological consequences.
Front Immunol. 5:6802015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Han DS: Current status and prospects of
intestinal microbiome studies. Intest Res. 12:178–183. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu Z, Zhang W and Kone BC: Signal
transducers and activators of transcription 3 (STAT3) inhibits
transcription of the inducible nitric oxide synthase gene by
interacting with nuclear factor kappaB. Biochem J. 367:97–105.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okugawa S, Ota Y, Kitazawa T, Nakayama K,
Yanagimoto S, Tsukada K, Kawada M and Kimura S: Janus kinase 2 is
involved in lipopolysaccharide-induced activation of macrophages.
Am J Physiol Cell Physiol. 285:C399–C408. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Villarino AV, Kanno Y, Ferdinand JR and
O'Shea JJ: Mechanisms of Jak/STAT signaling in immunity and
disease. J Immunol. 194:21–27. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zundler S and Neurath MF: Integrating
immunologic signaling networks: The JAK/STAT pathway in colitis and
colitis-associated cancer. Vaccines (Basel). 4:pii: E52016.
View Article : Google Scholar
|
|
21
|
Coskun M, Salem M, Pedersen J and Nielsen
OH: Involvement of JAK/STAT signaling in the pathogenesis of
inflammatory bowel disease. Pharmacol Res. 76:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Danese S, Grisham M, Hodge J and Telliez
JB: JAK inhibition using tofacitinib for inflammatory bowel disease
treatment: A hub for multiple inflammatory cytokines. Am J Physiol
Gastrointest Liver Physiol. 310:G155–G162. 2016.PubMed/NCBI
|
|
23
|
Vuitton L, Koch S and Peyrin-Biroulet L:
Janus kinase inhibition with tofacitinib: Changing the face of
inflammatory bowel disease treatment. Curr Drug Targets.
14:1385–1391. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim SF: The nitric oxide-mediated
regulation of prostaglandin signaling in medicine. Vitam Horm.
96:211–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Salvemini D, Kim SF and Mollace V:
Reciprocal regulation of the nitric oxide and cyclooxygenase
pathway in pathophysiology: Relevance and clinical implications. Am
J Physiol Regul Integr Comp Physiol. 304:R473–R487. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim SF: The role of nitric oxide in
prostaglandin biology; update. Nitric Oxide. 25:255–264. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gądek-Michalska A, Tadeusz J, Rachwalska P
and Bugajski J: Cytokines, prostaglandins and nitric oxide in the
regulation of stress-response systems. Pharmacol Rep. 65:1655–1662.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Krakauer T: The polyphenol chlorogenic
acid inhibits staphylococcal exotoxin-induced inflammatory
cytokines and chemokines. Immunopharmacol Immunotoxicol.
24:113–119. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei J and Feng J: Signaling pathways
associated with inflammatory bowel disease. Recent Pat Inflamm
Allergy Drug Discov. 4:105–117. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karrasch T and Jobin C: NF-kappaB and the
intestine: Friend or foe? Inflamm Bowel Dis. 14:114–124. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Broom OJ, Widjaya B, Troelsen J, Olsen J
and Nielsen OH: Mitogen activated protein kinases: A role in
inflammatory bowel disease? Clin Exp Immunol. 158:272–280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hwang SJ, Kim YW, Park Y, Lee HJ and Kim
KW: Anti-inflammatory effects of chlorogenic acid in
lipopolysaccharide-stimulated RAW 264.7 cells. Inflamm Res.
63:81–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shan J, Fu J, Zhao Z, Kong X, Huang H, Luo
L and Yin Z: Chlorogenic acid inhibits lipopolysaccharide-induced
cyclooxygenase-2 expression in RAW264.7 cells through suppressing
NF-kappaB and JNK/AP-1 activation. Int Immunopharmacol.
9:1042–1048. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ruan Z, Liu S, Zhou Y, Mi S, Liu G, Wu X,
Yao K, Assaad H, Deng Z, Hou Y, et al: Chlorogenic acid decreases
intestinal permeability and increases expression of intestinal
tight junction proteins in weaned rats challenged with LPS. PLoS
One. 9:e978152014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Shin HS, Satsu H, Bae MJ, Zhao Z, Ogiwara
H, Totsuka M and Shimizu M: Anti-inflammatory effect of chlorogenic
acid on the IL-8 production in Caco-2 cells and the dextran
sulphate sodium-induced colitis symptoms in C57BL/6 mice. Food
Chem. 168:167–175. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zatorski H, Sałaga M, Zielińska M,
Piechota-Polańczyk A, Owczarek K, Kordek R, Lewandowska U, Chen C
and Fichna J: Experimental colitis in mice is attenuated by topical
administration of chlorogenic acid. Naunyn Schmiedebergs Arch
Pharmacol. 388:643–651. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shen X, Tian Z, Holtzman MJ and Gao B:
Cross-talk between interleukin 1beta (IL-1beta) and IL-6 signalling
pathways: IL-1beta selectively inhibits IL-6-activated signal
transducer and activator of transcription factor 1 (STAT1) by a
proteasome-dependent mechanism. Biochem J. 352:913–919. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi D, Wang Q, Zheng H, Li D, Shen Y, Fu
H, Li T, Mei H, Lu G, Qiu Y, et al: Paeoniflorin suppresses
IL-6/Stat3 pathway via upregulation of Socs3 in dendritic cells in
response to 1-chloro-2,4-dinitrobenze. Int Immunopharmacol.
38:45–53. 2016. View Article : Google Scholar : PubMed/NCBI
|