Introduction
Macrophages serve different roles in homeostasis,
innate immunity against microbes and tissue repair (1–3).
Macrophages are differentiated from monocytes, and maybe polarized
into the inflammatory M1 (classically activated) lineage or the
immunomodulatory M2 (alternatively activated) lineage when they
leave the blood vessels, depending on differential tissue
microenvironment signaling, specific pathogens or cytokine
stimulation (2,4). M1 polarization is induced by
lipopolysaccharide (LPS) and/or interferon-γ (IFN-γ) during acute
infections to release pro-inflammatory cytokines and chemokines
that mount an immune response against various intracellular
pathogens, whereas M2 polarization is primarilyactivated by
interleukin-4 (IL-4) to produce anti-inflammatory cytokines (e.g.,
IL-10), provide tissue remodeling and enhance adaptive T helper 2
cell immunity (4,5).
Previous studies intomacrophage function suggested
that the activation of serine/threonine-protein kinase mTOR complex
1 (mTORC1) and mTORC2 may selectively control differential
macrophage polarization (6–8).
Macrophages with constitutive mTORC1 activation were resistant to
M2 polarization, although they displayed elevated M1 inflammation
via mTORC1-mediated downregulation of RAC-α
serine/threonine-protein kinase (Akt) signaling under IL-4
stimulation (6). Conversely, the
mTORC2-interferon regulatory factor (IRF)4 pathway was important
for IL-4 induced alternative M2 activation (9). Additionally, mTORC2-Akt suppressed
LPS-Toll like receptor 4 (TLR4) induced inflammation in innate
immunity (10). Paradoxically,
IL-4 may cooperate with the Akt-mTORC1 pathway to regulate M2
activation via increased M2 gene-specific histone acetylation
(11). Whether mTORC1 and mTORC2
are integrally linked during the polarization of macrophages
remains unknown. In addition, macrophage polarization may be
controlled by other regulatory factors.
Tumor necrosis factor α-induced protein 8-like
protein 2 (TIPE2) is a member of the tumor necrosis
factor-α-induced protein 8 family which performs diverse functions,
including the negative regulation of innate and adaptive immunity,
transcription factor AP-1 and nuclear factor (NF)-κB activation and
tumor suppression (12,13). Consequently, TIPE2-deficient mice
have been demonstrated to exhibit increased M1 inflammation and
resistance to M2 polarization, and the deficiency may retard
systemic lupus erythematosus autoimmunity via macrophage
polarization (14,15). Additionally, a cell-permeable TIPE2
protein exerted inhibitory effects on mitogen-activated protein
kinase kinase kinase 7 (TAK1) phosphorylation (16). The present study assessed the role
of TIPE2 in determining commitment to the process of macrophage
activation and polarization. M2 polarization was hallmarked by
increased TIPE2 expression during M2-like differentiation.
Exogenous overexpression of TIPE2 in M2 macrophages drove global
expression of M2-specific cytokines via inhibition of mTORC1.
Conversely, the induction of an M1-specific phenotype was impaired
in M1 macrophages with ectopic TIPE2 expression by disturbing
TAK1-inhibitor of NF-κB (IKK) and TLR4-mTORC1 activation. The
results of the present study provided a basis for understanding how
TIPE2 may control macrophage activation and polarization.
Materials and methods
Bone marrow-derived macrophages
(BMDMs)
All animal procedures were pre-approved by the
Institutional Animal Care and Use Committee of Zhejiang University
(Hangzhou, China). The present study used 6-week-old male C57BL/6J
mice (supplied by Beijing Vital River Laboratory, Beijing, China;
n=12; 18–25 g). Mice were housed at room temperature with ad
libitum access to food and water, relative humidity 50–60% and 12-h
day/night cycles. The femurs were removed from the mice following
euthanasia with sodium pentobarbital (250 mg/kg; Sigma-Aldrich,
Merck KGaA, Darmstadt, Germany), and BM cells were released by
gently crushing the bones with a sterile mortar and pestle. For
macrophage differentiation, BM-derived cells were plated in petri
dishes with RPMI 1640 medium (10% fetal calf serum;
penicillin/streptomycin; 2 mM L-glutamine; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with macrophage
colony-stimulating factor (M-CSF; 20 ng/ml; Sigma-Aldrich; Merck
KGaA) containing L929 cell supernatant for 7 days (Fig. 1A). M-CSF differentiated macrophages
were harvested and plated in tissue culture dishes for subsequent
experiments. For M1-like activation, 0.5–0.7×106 BMDMs
were plated in 12-well plates and treated with 10 ng/ml LPS
(Sigma-Aldrich; Merck KGaA) and 100 U/ml IFN-γ (Sigma-Aldrich;
Merck KGaA) and incubated overnight. For M2 polarization, the cells
were treated with 10 ng/ml IL-4 (Sigma-Aldrich; Merck KGaA) and
incubated overnight.
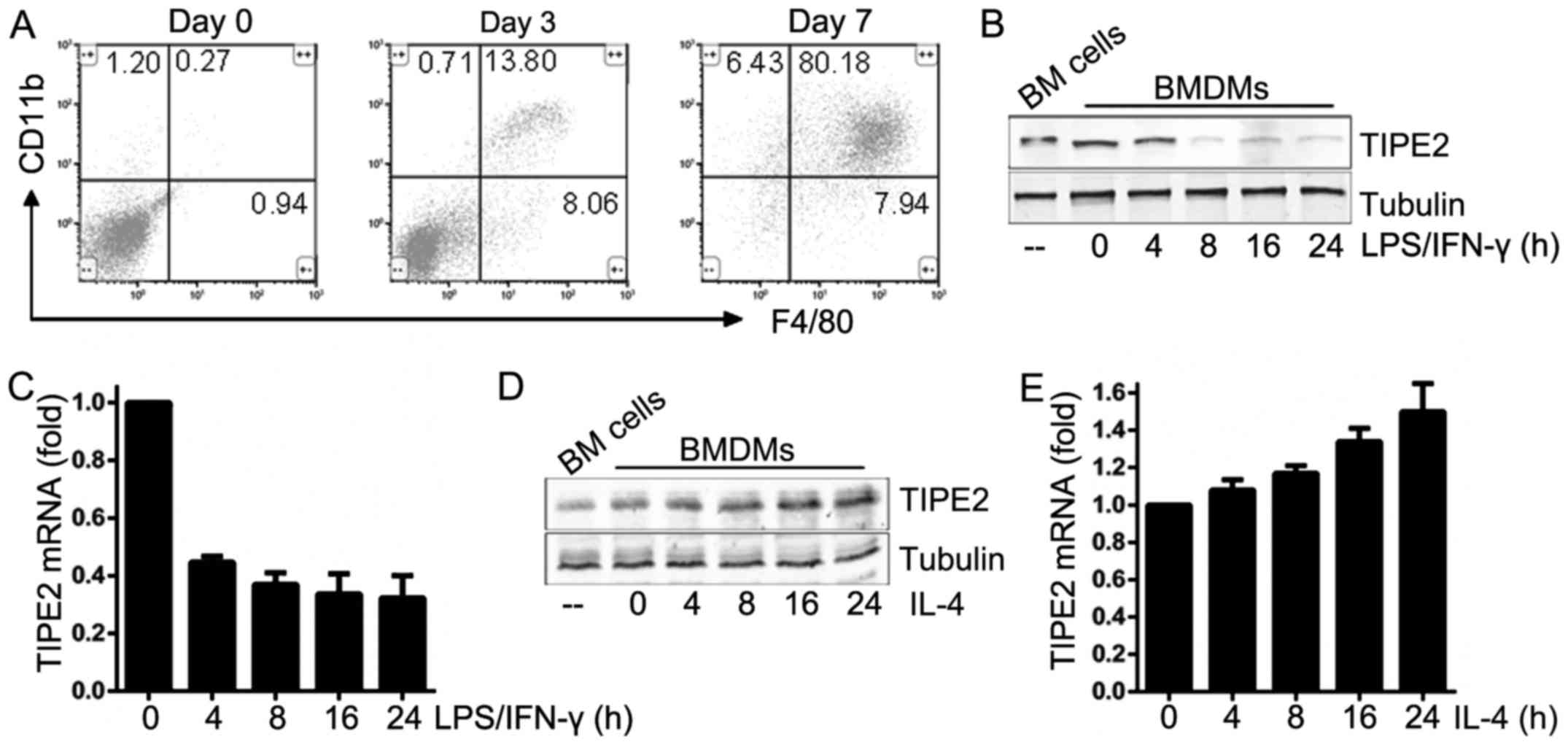 | Figure 1.Expression pattern of TIPE2 in murine
M1 and M2 macrophages. (A) Flow cytometric analysis of the
CD11b+, F4/80+ macrophage population in BM
cells at days 0, 3 and 7 in the presence of macrophage colony
stimulating factor (20 ng/ml). WTBMDMs were stimulated with LPS (10
ng/ml) and IFN-γ (100 U/ml) for 24 h. TIPE2 expression levels were
determined by (B) immunoblotting and (C) RT-qPCR analysis. WTBMDMs
were stimulated with IL-4 (20 ng/ml) for 24 h. TIPE2 expression
levels were determined by (D) immunoblotting and (E) RT-qPCR.
Statistics were performed on pooled data from three independent
experiments. Error bars represent the standard deviations of the
means. TIPE2, tumor necrosis factor α-induced protein 8-like
protein 2; CD, cluster of differentiation; BMDMs, bone
marrow-derived macrophages; LPS, lipopolysaccharide; IFN-γ,
interferon-γ; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; WT, wild-type; IL-4, interleukin-4. |
Western blotting
Cells were washed twice with cold PBS and lysed in
1% nonidet P-40 buffer with EDTA-free protease inhibitor tablets
(Roche Diagnostics, Indianapolis, IN, USA) and aphosphatase
inhibitor cocktail (Sigma-Aldrich; Merck KGaA). The protein
concentration in the cell lysates was determined using a Bradford
protein concentration assay kit (Beyotime Institute of
Biotechnology, Haimen, China). Equal amounts of protein (10 µg)
were loaded onto 10% SDS-PAGE gels (Sigma-Aldrich; Merck KGaA) and
subsequently transferred to polyvinylidene fluoride membranes
(Thermo Fisher Scientific, Inc.) for immunoblotting with the
indicated primary antibodies following 1 h of blocking with 5%
bovine serum albumin (cat. no. KGY00810; Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China) at room temperature, the proteins
transferred onto polyvinylidene fluoride membrane were incubated
overnight with primary antibody solution against the target protein
at 4°C and then in the IRDye®680RD or IRDye®
800CW conjugated secondary antibody solution for 1 h in a dark at
room temperature. Immunoprecipitation was performed by using
Pierce™ Co-Immunoprecipitation kit (cat. no. 26149;
Thermo Fisher Scientific, Inc.). Images of protein bands were
visualized using Odyssey® Fc imaging system (LI-COR
Biosciences, Lincoln, NE, USA) and analyzed using Odyssey software
(version 1.2; LI-COR Biosciences).
Antibodies and reagents
Primary antibodies were purchased from Abcam
(Cambridge, UK), including: TIPE2 (cat. no. ab110389), IKKβ (cat.
no. ab124957), phospho-Y188-IKKβ (cat. no. ab194519), IRF5 (cat.
no. ab181553), p65 (cat. no. ab16502), phosphor (p)-S536-p65 (cat.
no. ab86299), S6K1 (cat. no. ab32529), p-T389-S6K1 (cat. no.
ab2571), TSC1 (cat. no. ab217328), TLR4 (cat. no. ab30667), BCAP
(cat. no. ab155077), STAT6 (cat. no. ab32520), p-Y641-STAT6 (cat.
no. ab28829), AKT (cat. no. ab8805), p-T308-AKT (cat. no. ab38449),
p-S473-AKT (cat. no. ab81283), 4E-BP1 (cat. no. ab2606),
phospho-T70-4E-BP1 (cat. no. ab75831), IRS1 (cat. no. ab52167),
IRS2 (cat. no. ab134101); all 1:1,000 dilution in 5% bovine serum
albumin (cat. no. KGY00810; Nanjing KeyGen Biotech Co., Ltd.),
except for α-tubulin (Sigma-Aldrich; Merck KGaA; 1:5,000).
Immunoglobulin G-conjugated secondary antibodies were purchased
from LI-COR Biosciences (cat. nos. 925-32210 and 925-68070; all
1:10,000 dilution in 5% bovine serum albumin. For flow cytometry,
CD11b-phycoerythrin (BD Biosciences, Franklin Lakes, NJ, USA) and
F4/80-fluorescein isothiocyanate (BioLegend, Inc., San Diego, CA,
USA) antibodies were used. Rapamycin used at a dose of 100 nM for
an overnight incubation was purchased from Sigma-Aldrich (Merck
KGaA). Sodium nitroprusside (cat. no. S4059, 0.5 mM, overnight), as
the chemically distinct nitric oxide (NO) donor, was purchased from
Selleck Chemicals (Houston, TX, USA). The target sequences for the
TIPE2 gene short hairpin (sh)RNA (5′-CCGTGTAAACCACTCAACTCATCTT-3′)
and hamartin (TSC1) gene shRNA (5′-TACGTCCAAGGTCGGGCAGGAAGA-3′)
were designed based on the sequence of mouse TIPE2 cDNA (accession
no. NM_027206) and the sequence of mouse TSC1 cDNA (accession no.
NM_001289575). The negative control shRNA (20 µM), TIPE2 (20 µM)
and TSC1 shRNAs (20 µM) were sequence-confirmed and cloned into the
pLL3.7-green fluorescent protein lentiviral vector (Clontech,
Mountain View, CA USA). Rescue experiments were performed using the
pLV-yellow fluorescent protein lentiviral vector (Addgene, Inc.,
Cambridge, MA USA) expressing the wild-type TIPE2 gene and the
K171T-IKK subunit β (IKKβ) gene mutation. Lentivirus-based TIPE2
shRNA expression plasmid, wild-type TIPE2 expression plasmid and
IKKβ mutant expression plasmid construction, lentivirus harvesting,
and lentivirus titer testing were made purchased from IBSBIO
Company (Shanghai, China). The negative control lentiviral
particles were also provided by IBSBIO Company.
ViraDuctin™ Lentivirus Transduction kit (LTV-201; Cell
Biolabs, San Diego, CA, USA) was used to enhance the transduction
of BMDMs with lentiviral particles.
Arginase assay
BMDMs were lysed in 0.1% Triton X-100 lysis buffer
(Sigma-Aldrich; Merck KGaA) with EDTA-free protease inhibitor
tablets (Roche Diagnostics). Lysates containing equal amounts of
protein (3 µg) were incubated with 500 mM L-arginine for 45 min at
37°C, followed by acid stop solution. The degradation of L-arginine
to urea was measured by adding 9% isonitrosopropiophenone in 100%
ethanol. Absorbance was read at 540 nm usinga microplate reader
(BioTek Instruments, Inc., Winooski, VT, USA).
ELISA analysis
Cytokine concentration was determined for
IL-6 (cat. no. BMS603-2), IL-10 (cat. no. BMS614-2TWO), IL-12b
(cat. no. EMIL12B) and tumor necrosis factor-α (TNFα, cat. no.
BMS607-3) using ELISA kits purchased from Thermo Fisher Scientific,
Inc. Experimental supernatants were collected and centrifuged at
4°C at 3,000 × g for 5 min. Supernatants were analyzed according to
the manufacturer's protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from BMDMs using an RNeasy
kit (Qiagen, Inc., Valencia, CA, USA). For RT-qPCR analysis, total
RNA (2 µg) was converted to cDNA using the SuperScript First-Strand
Synthesis system (Takara Bio, Inc., Otsu, Japan). The following
thermocycling conditions were used for qPCR: Initial denaturation
at 95°C for 5 min; followed by 22 cycles of, 95°C for 20 sec, 55°C
for 20 sec and 72°C for 20 sec; and final extension for 1 min. The
reactions were performed in triplicate with appropriate primers
using SYBR-Green (Roche Diagnostics) and an ABI Prism 7900 Sequence
Detector (Applied Biosystems; Thermo Fisher Scientific, Inc.).
β-actin was used as the loading control and data were analyzed by
the 2−ΔΔCq method (17). Sequences for all RT-qPCR primers
are presented in Table I.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene name | Forward primer
sequence (5′→3′) | Reverse primer
sequence (5′→3′) |
|---|
| β-actin |
TGCGTGACATCAAAGAGAAG |
TCCATACCCAAGAAGGAAGG |
| ARG1 |
AAGAAAAGGCCGATTCACCT |
CACCTCCTCTGCTGTCTTCC |
| ARG2 |
GGATCCAGAAGGTGATGGAA |
AGAGCTGACAGCAACCCTGT |
| CD38 |
ACTGGAGAGCCTACCACGAA |
AGTGGGGCGTAGTCTTCTCT |
| EGR2 |
GATCACAGGCAGGAGAGACTG |
GCGAAGCTACTCGGATACGG |
| FIZZ1 |
GGAACTTCTTGCCAATCCAGC |
CAGTGGTCCAGTCAACGAGT |
| FPR2 |
TTCATGGGCCAGGACTTTCG |
CACAGACTTCATGGGGCCTT |
| GPR18 |
CTTCGTGCCCTTCCACATCT |
TGCGAACACTGCGAAGGTAA |
| MGL1 |
AGCCTGGTAAAGCTCCTTCC |
GGGATCCAATCACGGAGACG |
| MGL2 |
GGAGTCTCCAAAGTTTGCTCT |
GGTGCCTAGGTCCCTCCTTA |
| MRC1 |
GTGGAGTGATGGAACCCCAG |
CTGTCCGCCCAGTATCCATC |
| PGC1β |
CTTCCGTTGGCCCAGATAC |
CTGCTGGGCCTCTTTCAGTA |
| YM1 |
AGAAGGGAGTTTCAAACCT |
GTCTTGCTCATGTGTGTAAGTGA |
| TIPE2 |
CGGTTGCCTCTTCCAGTGA |
GCTGCTGGTCTCGTCGATAA |
Statistical analysis
Statistical analysis was performed using Prism 6
(GraphPad Software, Inc., La Jolla, CA, USA) software. The
Student's t-test and repeated measures analysis of variance
followed by Tukey's post hoc test for multiple comparisons were
used to determine statistical significance. Data are presented as
the mean ± standard deviation, n=3. P<0.05 was considered to
indicate a statistically significant difference.
Results
Opposite expression patterns of TIPE2
in murine M1 and M2 macrophages
To assess the expression pattern of TIPE2 in the
differential polarization of macrophages, BM cells were isolated
from C57BL/6J mice. Murine BM cells were exposed to M-CSF under
appropriate culture conditions to induce differentiation into
mature BMDMs. These murine BMDMs were used as the models of
undifferentiated (M0) macrophages. RT-qPCR analysis and
immunoblotting demonstrated that TIPE2 mRNA and protein expression
was impaired 24 h following LPS/IFNγ stimulation of M0 macrophages
to induce polarization into M1 macrophages (Fig. 1B and C), whereas IL-4-challenged M0
macrophages polarized into M2 macrophages exhibited elevated TIPE2
expression levels (Fig. 1D and E).
Therefore, it was hypothesized that the expression of TIPE2 was
associated with M1 and M2 polarization of macrophages.
TIPE2 regulates the differential
polarization status of BMDMs
As presented in Fig.
1, TIPE2 expression was selectively inhibited during the M1
polarization of macrophages by LPS/IFNγ, although it was elevated
in M2 macrophages. Subsequently, the role of TIPE2 in the process
of differential polarization of BMDMs was investigated, and it was
observed that BMDMs with TIPE2 shRNA exhibited decreased expression
levels of TIPE2 (Fig. 2A). TIPE2
deletion negatively regulated the expression levels of IL-10 and M2
genes in BMDMs stimulated by IL-4 (Fig. 2B and C). M1 cytokines (TNFα, IL-6
and IL-12b) and M1 genes were significantly elevated in BMDMs
treated with TIPE2 shRNA and LPS/IFNγ (Fig. 2D and E). Exogenous TIPE2
overexpression in BMDMs impeded LPS/IFNγ-induced M1 inflammation
(Fig. 3A-C), whereas these cells
exhibited enhanced responses to IL-4 stimulation (Fig. 3D and E).
 | Figure 2.BMDMs with TIPE2 deletion have
defective M2 polarization and enhanced responses to LPS/ IFN-γ
challenge. (A) BMDMs were transduced with TIPE2 shRNA. TIPE2
expression levels were determined by immuno blotting and reverse
transcription-quantitative polymerase chain reaction analysis. (B)
Measurement of IL-10 secretion in BMDMs by ELISA analysis following
treatment with IL-4 (20 ng/ml) for 3 and 6 h. (C) Expression of M2
genes in BMDMs following treatment with IL-4 (20 ng/ml) for 24 h.
(D) Measurement of TNFα, IL-6 and IL-12b secretion in BMDMs by
ELISA analysis following treatment with LPS (10 ng/ml) and IFN-γ
(100 U/ml) for 3 and 6 h. (E) Expression of M1 genes in BMDMs
following treatment with LPS (10 ng/ml) and IFN-γ (100 U/ml) for 24
h. Statistics were performed on pooled data from three independent
experiments. Error bars represent the standard deviations of the
means. *P<0.05, **P<0.01. shRNA, short hairpin RNA; CTRL,
control; TIPE2, tumor necrosis factor α-induced protein 8-like
protein 2; BMDMs, bone marrow-derived macrophages; LPS,
lipopolysaccharide; IFN-γ, interferon-γ; EGR2, E3 SUMO protein
ligase EGR2; MGL1, C-type lectin domain family 10 member A; MGL2,
monoglyceride lipase; FIZZ1, resistin-like α; YM1, chitinase-like
protein 3; MRC1, macrophage mannose receptor 1; PGC1β, peroxisome
proliferator-activated receptor gamma coactivator 1-β; CD38,
cluster of differentiation 38; FPR2, formyl peptide receptor 2;
GPR18, N-arachidonyl glycine receptor; TNFα, tumor necrosis
factor-α; IL, interleukin. |
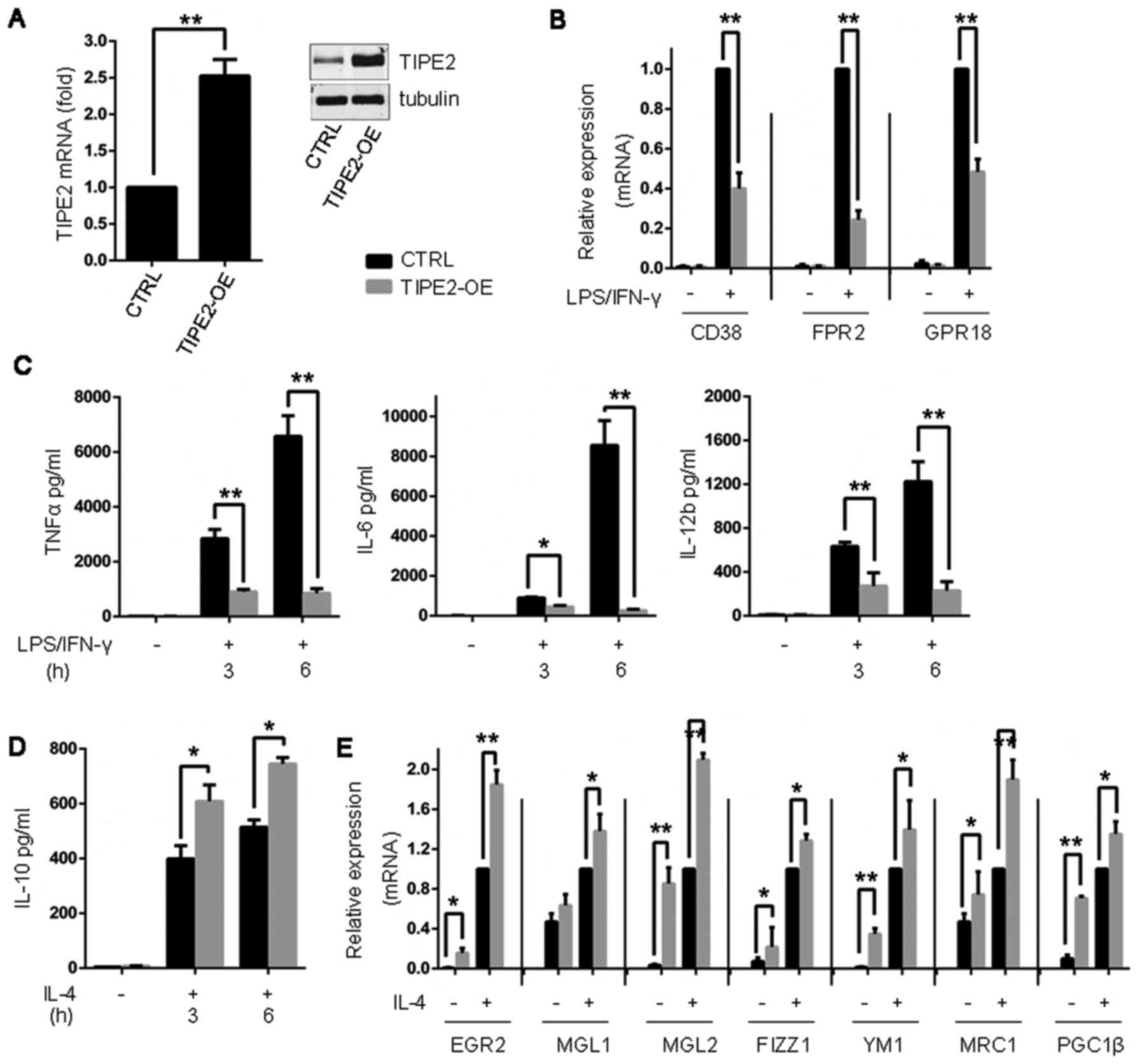 | Figure 3.TIPE2 overexpression promotes M2
polarization and disturbs M1 inflammation. (A) BMDMs were
transduced with exogenous TIPE2. TIPE2 expression levels were
determined by immunoblotting and reverse transcription-quantitative
polymerase chain reaction analysis. (B) Expression of M1 genes in
BMDMs following treatment with LPS (10 ng/ml) and IFN-γ (100 U/ml)
for 24 h. (C) Measurement of TNFα, IL-6 and IL-12b secretion in
BMDMs by ELISA analysis following treatment with LPS (10 ng/ml) and
IFN-γ (100 U/ml) for 3 and 6 h. (D) Measurement of IL-10 secretion
in BMDMs by ELISA following treatment with IL-4 (20 ng/ml) for 3 h
and 6 h. (E) Expression of M2 genes in BMDMs following treatment
with IL-4 for 24 h. Statistics were performed on pooled data from
three independent experiments. Error bars represent the standard
deviations of the means. *P<0.05, **P<0.01. BMDMs, bone
marrow-derived macrophages; TIPE2, tumor necrosis factor α-induced
protein 8-like protein 2; CTRL, control vector; TIPE2-OE, TIPE2
overexpression; LPS, lipopolysaccharide; CD38, cluster of
differentiation 38; FPR2, formyl peptide receptor 2; GPR18,
N-arachidonyl glycine receptor; IFNγ, interferon-γ; TNFα, tumor
necrosis factor-α; IL, interleukin; EGR2, E3 SUMO protein ligase
EGR2; MGL1, C-type lectin domain family 10 member A; MGL2,
monoglyceride lipase; FIZZ1, resistin-like α; YM1, chitinase-like
protein 3; MRC1, macrophage mannose receptor 1; PGC1β, peroxisome
proliferator-activated receptor gamma coactivator 1-β. |
TIPE2 impedes M1 polarization by
interfering with TAK1-IKKβ and BCAP-mTORC1 activation
The TAK1-IKKβ pathway promotes M1 macrophage
polarization via IRF5 and NF-κB activation (18–20).
It was observed that exogenous TIPE2 was able toinhibit IKKβ, and
downstream IRF5 and NF-κB activation (Fig. 4A), which was responsible for the
negative regulation of TIPE2 in M1 polarization. M0 macrophages
with mutant (mt)-IKKβ (K171T) were additionally constructed
(Fig. 4B), and these cells
exhibited M1-like functional activities either prior to or during
LPS/IFNγ stimulation (Fig. 4C and
D). Ectopic TIPE2 expression partially antagonized the LPS/IFNγ
induced expression of M1 cell markers and cytokines during
exogenous co-expression of mt-IKKβ and TIPE2 in macrophages
(Fig. 4C and D), suggesting the
presence of alternative pathways for M1 polarization interrupted by
TIPE2.
 | Figure 4.Ectopic TIPE2 attenuates M1
polarization via disturbance of the IKKβ and mTORC1 pathways. (A)
The expression levels of IKKβ, phospho-Y188-IKKβ, IRF5, p65,
phospho-S536-p65, S6K1, phospho-T389-S6K1 and tubulin were
determined by immunoblotting. (B) The expression levels of IKKβ,
IRF5, p65, phospho-S536-p65, TSC1, S6K1, phospho-T389-S6K1 and
tubulin were determined by immunoblotting. (C) Measurement of TNFα
and IL-6 secretion by ELISA following treatment with LPS (10 ng/ml)
and IFN-γ (100 U/ml) for 3 and 6 h. (D) Expression of M1 genes
following treatment with LPS (10 ng/ml) and IFN-γ (100 U/ml) for 24
h. (E) BCAP formed a complex with TLR4 and TLR4 formed a complex
with BCAP in BMDMs, prior to and following LPS (10 ng/ml) and IFN-γ
(100 U/ml) stimulation. Statistics were performed on pooled data
from three independent experiments. Error bars represent the
standard deviations of the means. *P<0.05, **P<0.01. phospho,
phosphorylated; CTRL, control vector; TIPE2, tumor necrosis factor
α-induced protein 8-like protein 2; OE, overexpression; sh, short
hairpin RNA; IKKβ, inhibitor of nuclear factor-κB subunit β;
mt-IKKβ, IKKβ with K171T mutation; IRF5, interferon regulatory
factor 5; p65, transcription factor p65; mTORC,
serine/threonine-protein kinase mTOR complex 1; S6K1, ribosomal
protein s6 kinase β-1; TSC1, hamartin; IL-6, interleukin-6; TNFα,
tumor necrosis factor-α; IFNγ, interferon-γ; LPS,
lipopolysaccharide; BCAP, B-cell receptor-associated protein; TLR4,
Toll-like receptor 4; BMDMs, bone marrow-derived macrophages; CD38,
cluster of differentiation 38; FPR2, formyl peptide receptor 2;
GPR18, N-arachidonyl glycine receptor. |
In addition to the canonical IRF5 and NF-κB
pathways, TLR4-BCAP-mTORC1 signaling is involved in the M1
polarization of macrophages with LPS/IFNγ stimulation (7). It was observed that exogenous TIPE2
disturbed the formation of the TLR4-BCAP complex (Fig. 4E) and subsequently inhibited mTORC1
activation, signifiedby diminished S6K phosphorylation (Fig. 4A), in macrophages following
LPS/IFNγ stimulation. Following TSC1 deletion in BMDMs (Fig. 4B) with mt-IKKβ, the cells displayed
hyperactivated mTORC1, IRF5 and NF-κB (Fig. 4B), and were resistant to the
effects of disturbance of ectopic TIPE2 on M1 polarization
following LPS/IFNγ stimulation (Fig.
4C and D). These data suggested that mTORC1 and IKKβ activation
coordinated M1 polarization.
TIPE2 enhances M2 polarization
The above data indicated that TIPE2 attenuated M1
polarization stimulated by LPS/IFNγ via the inhibition of IKKβ and
mTORC1 activation. IL-4 induced M2 polarization was subsequently
examined, which relied on tyrosine-protein kinase JAK-signal
transducer and activator of transcription (STAT)6, and insulin
receptor substrate (IRS)1/2-mTORC2-Akt signaling. Constitutive
mTORC1 activation impairs M2 polarization by IL-4 (6). It was observed that the activation of
STAT6 was unaltered in wild-type (WT) and TIPE2-overexpressing
BMDMs (Fig. 5A). However, mTORC2
induced AKT S473 phosphorylation. AKT T308 phosphorylation in BMDMs
with TIPE2 overexpression was more marked compared with WT BMDMs
(Fig. 5A). IL-4 induced BMDMs with
TIPE2 overexpression additionally exhibited weaker phosphorylation
of the mTORC1 downstream molecules ribosomal protein s6 kinase β-1
and eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP1) compared with WT BMDMs (Fig.
5B). TIPE2 overexpression promoted M2 polarization and
expression of M2 genes (Fig. 3D and
E). Arginase I and arginase II expression was significantly
increased in BMDMs with ectopic TIPE2 expression following IL-4
challenge (Fig. 5C), which could
be responsible for the arginine-urea cycle dependence on mTORC1
inhibition (Fig. 5D) (21). Exogenous nitric oxide (NO) partly
decreased IL-10 production under treatment with IL-4, whereas NO
additionally promoted M1 inflammation induced by LPS and IFNγ.
TIPE2 overexpression was able to reverse these phenotypes induced
by exogenous NO (Fig. 5E).
Conversely, in BMDMs with TSC1 deletion, constitutive mTORC1
activity impaired IL-4-induced M2 polarization (Fig. 5C and E-G), and obstructed IRS-1/2
degradation, and AktT308 and S473 activation (Fig. 5H). BMDMs with TSC1 deletion through
treatment with rapamycin and IL-4, or BMDMs with TSC1 deletion and
TIPE2 overexpression, were able torescue M2 responses (Fig. 5C-E). The results of the present
study demonstrated that TIPE2 overexpression weakened the
activation of mTORC1 pathway, which accelerated IL-4-induced M2
polarization.
 | Figure 5.TIPE2 overexpression promotes M2
polarization induced by IL-4. (A) The expression levels of AKT,
phospho-T308-AKT, phospho-S473-AKT, STAT6 and phospho-Y641-STAT6
were determined by immunoblotting. (B) The expression levels of
4E-BP1, phospho-T70-4E-BP1, S6K1 and phospho-T389-S6K1 were
determined by immunoblotting. (C) The expression levels of the ARG
I and II genes following treatment with IL-4 (20 ng/ml) for 24 h
were determined by RT-qPCR analysis. (D) Measurement of urea
production normalized to the total protein in BMDMs, Following
treatment with IL-4 (20 ng/ml) for 6 h. (E) Measurement of IL-10,
IL-6 and TNFα secretion by ELISA following treatment with IL-4 (20
ng/ml), NO (100 µM) or LPS (10 ng/ml) and IFN-γ (100 U/ml) for 6 h.
(F) The expression levels of M2 genes following treatment with IL-4
(20 ng/ml) for 24 h were determined by RT-qPCR. (G) Measurement of
IL-10 secretion by ELISA following treatment with IL-4 (20 ng/ml)
for 6 h. (H) The expression levels of IRS1, IRS2, AKT,
phospho-T308-AKT, phospho-S473-AKT and tubulin were determined by
immunoblotting. Statistics were performed on pooled data from three
independent experiments. Error bars represent the standard
deviations of the means. *P<0.05, **P<0.01. phospho,
phosphorylated; CTRL, control vector; OE, overexpression; sh, short
hairpin RNA; RAPA, rapamycin; STAT6, signal transducer and
activator of transcription 6; AKT, RAC-α serine/threonine-protein
kinase; TIPE2, tumor necrosis factor α-induced protein 8-like
protein 2; IL, interleukin; 4E-BP1, eukaryotic translation
initiation factor 4E-binding protein 1; S6K1, ribosomal protein S6
kinase β-1; ARG, arginase; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; LPS,
lipopolysaccharide; IFNγ, interferon-γ; NO, nitric oxide; TNFα,
tumor necrosis factor-α; EGR2, E3 SUMO protein ligase EGR2; MGL1,
C-type lectin domain family 10 member A; MGL2, monoglyceride
lipase; FIZZ1, resistin-like α; YM1, chitinase-like protein 3;
MRC1, macrophage mannose receptor 1; PGC1β, peroxisome
proliferator-activated receptor gamma coactivator 1-β; IRS, insulin
receptor substrate; TSC1, hamartin. |
Discussion
In response to numerous microenvironmental signals,
including different intracellular pathogens or direct interactions
between inflammatory cells, macrophages primarily undergo M1
(classical) or M2 (alternative) activation, and exhibit different
functional states of polarization (4). TIPE2 acts as a negative regulator of
innate immunity (14,15,22,23),
althoughits role in macrophage polarization remains largely
unknown. In the present study, an inverse expression pattern of
TIPE2 in murine M1 and M2 macrophages was observed. BMDMs following
LPS and IFNγ stimulation exhibited markedly decreased TIPE2
expression, which was consistent with a previous report (23). However, IL-4-induced M0 macrophages
polarized into M2 macrophages in line with elevated TIPE2
expression levels. The present study additionally identified the
regulatory role of TIPE2 as an activator of M2 marker expression,
and demonstrated that alterations in TIPE2 expression led to an
interchange between macrophage subtypes.
TIPE2 deficiency facilitates M1 inflammation
(14), whereas exogenous TIPE2
overexpression enhances M2 polarization. The TLR-TAK1-IKKβ pathway
promotes the transcription of M1-polarizing genes via IRF5 and
NF-κB activation (18–20). TIPE2 disrupted TAK1-TGF-β-activated
kinase 1 and MAP3K7-binding protein (TAB)1-TAB2 formation, and
subsequently inhibited the activation of TAK1 and its downstream
molecules (16). In the present
study, BMDMs with mt-IKKβ (K171T), which represent constitutive
activation of IKKβ, exhibited increased M1-like functional
activation, even prior to LPS/IFNγ challenge. Exogenous TIPE2 and
mt-IKKβ expression in M0 macrophages illustrated that ectopic TIPE2
partially reduced glycolytic flux and reversed the course of M1
polarization stimulated by LPS/IFNγ.
Except for the TAK1-IKKβ pathway, TIPE2 disturbed
the activation of TLR4-BCAP-mTORC1 signaling and inhibited the
transcription of M1 genes. Therefore, mTORC1 and IKKβ may be able
to coordinate M1 polarization following stimulation with
LPS/IFNγ.
The role of TIPE2 in inhibiting the activation of M1
inflammation identified in the present study is important, due
toits well-documented suppression of TAK1-IKKβ (16) and TLR4 activation (22). In the present study, the expression
of TIPE2 was elevated in IL-4-induced M2 polarization. Exogenous
TIPE2 expression accelerated the process of M2 polarization by
IL-4. Overexpression of TIPE2 elevated the arginase I and arginase
II levels, which may accelerate the conversion of L-arginine into
urea (24,25). Exogenous NO partially inhibited the
production of IL-10 under treatment with IL-4, and was able to
promote M1 inflammation induced by LPS and IFNγ, which may be
caused by the activation of mTORC1 (21). TIPE2 was able to reverse these
processes induced by NO. These observations were consistent with
the hypothesis that arginases and NO synthases may compete for
L-arginine, the common substrate of urea, or NO synthesis, which
determines the fate of macrophage polarization (24,26–28).
In addition, arginine metabolism was demonstrated to servean
important role in manipulating the distinct M1/M2 phenotypes
(26,27,29).
In conclusion, TIPE2 serves an important role in
orchestrating the differential polarization status of macrophages,
via negative regulation of mTORC1 activation and accelerated
conversion of arginine to urea. The influence of TIPE2 on the
elevation of arginase expression and the effects of NO or urea on
M1/M2 polarization remain unclear. Further investigation of how
signal transduction downstream of TIPE2 affects arginine metabolism
may reveal the mechanism through which TIPE2 enhances IL-4 induced
M2 polarization.
Acknowledgements
The present study was supported by grants from the
Projects of Medical and Health Technology Development Program in
Zhejiang Province, China (grant nos. 2016KYB135 and
2016KYB171).
References
|
1
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ginhoux F and Jung S: Monocytes and
macrophages: Developmental pathways and tissue homeostasis. Nat Rev
Immunol. 14:392–404. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Varol C, Mildner A and Jung S:
Macrophages: Development and tissue specialization. Annu Rev
Immunol. 33:643–675. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Italiani P and Boraschi D: From Monocytes
to M1/M2 Macrophages: Phenotypical vs. functional differentiation.
Front Immunol. 5:5142014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saeed S, Quintin J, Kerstens HH, Rao NA,
Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van
der Ent MA, et al: Epigenetic programming of monocyte-to-macrophage
differentiation and trained innate immunity. Science.
345:12510862014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Byles V, Covarrubias AJ, Ben-Sahra I,
Lamming DW, Sabatini DM, Manning BD and Horng T: The TSC-mTOR
pathway regulates macrophage polarization. Nat Commun. 4:28342013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Covarrubias AJ, Aksoylar HI and Horng T:
Control of macrophage metabolism and activation by mTOR and Akt
signaling. Semin Immunol. 27:286–296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weichhart T, Hengstschläger M and Linke M:
Regulation of innate immune cell function by mTOR. Nat Rev Immunol.
15:599–614. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang SC, Smith AM, Everts B, Colonna M,
Pearce EL, Schilling JD and Pearce EJ: Metabolic reprogramming
mediated by the mTORC2-IRF4 signaling axis is essential for
macrophage alternative activation. Immunity. 45:817–830. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brown J, Wang H, Suttles J, Graves DT and
Martin M: Mammalian target of rapamycin complex 2 (mTORC2)
negatively regulates Toll-like receptor 4-mediated inflammatory
response via FoxO1. J Biol Chem. 286:44295–44305. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Covarrubias AJ, Aksoylar HI, Yu J, Snyder
NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V,
Polynne-Stapornkul T, et al: Akt-mTORC1 signaling regulates Acly to
integrate metabolic input to control of macrophage activation.
Elife. 5:pii: e11612. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun H, Gong S, Carmody RJ, Hilliard A, Li
L, Sun J, Kong L, Xu L, Hilliard B, Hu S, et al: TIPE2, a negative
regulator of innate and adaptive immunity that maintains immune
homeostasis. Cell. 133:415–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu Y, Tao M, Wu J, Meng Y, Xu C, Tian Y,
Zhou X, Xiang J, Zhang H and Xie Y: Adenovirus-directed expression
of TIPE2 suppresses gastric cancer growth via induction of
apoptosis and inhibition of AKT and ERK1/2 signaling. Cancer Gene
Ther. 23:98–106. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lou Y, Zhang G, Geng M, Zhang W, Cui J and
Liu S: TIPE2 negatively regulates inflammation by switching
arginine metabolism from nitric oxide synthase to arginase. PLoS
One. 9:e965082014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li F, Zhu X, Yang Y, Huang L and Xu J:
TIPE2 alleviates systemic lupus erythematosus through regulating
macrophage polarization. Cell Physiol Biochem. 38:330–339. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oho M, Nakano R, Nakayama R, Sakurai W,
Miyamoto A, Masuhiro Y and Hanazawa S: TIPE2 (tumor necrosis factor
alpha-induced protein 8-like 2) is a novel negative regulator of
TAK1 signal. J Biol Chem. 291:22650–22660. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren J, Chen X and Chen ZJ: IKKβ is an IRF5
kinase that instigates inflammation. Proc Natl Acad Sci USA.
111:17438–17443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lopez-Pelaez M, Lamont DJ, Peggie M,
Shpiro N, Gray NS and Cohen P: Protein kinase IKKβ-catalyzed
phosphorylation of IRF5 at Ser462 induces its dimerization and
nuclear translocation in myeloid cells. Proc Natl Acad Sci USA.
111:17432–17437. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mihaly SR, Morioka S, Ninomiya-Tsuji J and
Takaesu G: Activated macrophage survival is coordinated by TAK1
binding proteins. PLoS One. 9:e949822014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chantranupong L, Scaria SM, Saxton RA,
Gygi MP, Shen K, Wyant GA, Wang T, Harper JW, Gygi SP and Sabatini
DM: The CASTOR proteins are arginine sensors for the mTORC1
pathway. Cell. 165:153–164. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun H, Zhuang G, Chai L, Wang Z, Johnson
D, Ma Y and Chen YH: TIPE2 controls innate immunity to RNA by
targeting the phosphatidylinositol 3-kinase-Rac pathway. J Immunol.
189:2768–2773. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang Z, Fayngerts S, Wang P, Sun H,
Johnson DS, Ruan Q, Guo W and Chen YH: TIPE2 protein serves as a
negative regulator of phagocytosis and oxidative burst during
infection. Proc Natl Acad Sci USA. 109:15413–15418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Munder M: Arginase: An emerging key player
in the mammalian immune system. Br J Pharmacol. 158:638–651. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nissim I, Luhovyy B, Horyn O, Daikhin Y,
Nissim I and Yudkoff M: The role of mitochondrially bound arginase
in the regulation of urea synthesis: Studies with [U-15N4]arginine,
isolated mitochondria, and perfused rat liver. J Biol Chem.
280:17715–17724. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Comalada M, Yeramian A, Modolell M,
Lloberas J and Celada A: Arginine and macrophage activation.
Methods Mol Biol. 844:223–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rath M, Müller I, Kropf P, Closs EI and
Munder M: Metabolism via arginase or nitric oxide synthase: Two
competing arginine pathways in macrophages. Front Immunol.
5:5322014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McNeill E, Crabtree MJ, Sahgal N, Patel J,
Chuaiphichai S, Iqbal AJ, Hale AB, Greaves DR and Channon KM:
Regulation of iNOS function and cellular redox state by macrophage
Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2
activation. Free Radic Biol Med. 79:206–216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thomas AC and Mattila JT: ‘Of mice and
men’: Arginine metabolism in macrophages. Front Immunol. 5:4792014.
View Article : Google Scholar : PubMed/NCBI
|














