Introduction
Obesity, as defined by an excess of adipose tissue,
has increased globally in most of the westernized world over recent
decades. This condition is associated with several chronic
complications, such as hypertension, coronary heart disease,
hyperglycemia, hypertriglyceridemia, dyslipidemia and insulin
resistance (1). Although over 30
gene loci combinations have been identified as being associated
with the development of obesity and metabolic disease (2), these loci are responsible for only
2–3% of the incidence of these conditions. Other causes of obesity
are known to include energy-dense diets high in saturated fat and
sugar, and the sedentary lifestyles common in the modern world
(3–5). In recent years, animal models have
been developed to study such things as diet-induced obesity to
study the underlying mechanisms of obesity and related
diseases.
It is further generally accepted that obesity and
metabolic disorders are associated with chronic, low-grade systemic
inflammation (6). This kind of
inflammation originates predominantly in adipose tissue, which
leads to greater numbers of immune cells, resulting in the
activation of other cells, such as adipocytes, which cause adipose
tissue remodeling and further incitement of the inflammatory
process (7). Interestingly,
malnutrition, including excessive energy intake, increase fat
accumulation, and lipotoxicity, can activate the expression of
pro-inflammatory effector molecules in metabolic tissues and cells
involved in innate immunity (8,9).
Although many recent studies have focused on the role of adipose
tissue macrophages (ATMs), which are known to be the main factor
initiating systemic inflammatory promotion in obesity, the recently
identified innate lymphoid cells (ILCs) have also been reported to
play a role in the inflammatory process in obese adipose tissue
(10).
In this article, we review the current understanding
of the complex interplay between the types of ILCs that have been
found to link inflammation to obesity. We also discuss the cellular
and molecular basis of obesity-induced inflammation and the
functional role of each type of ILC. Finally, we open a new
discussion by noting that even if adipose tissue is accepted as a
major cause of systemic inflammation, it is still unclear whether
any metabolic-related organs can influence obesity-related
pathologies.
Interplay between diets, inflammation and
gut microbiota in obesity
In the last decade, various mechanisms linking
immunity, metabolic abnormalities, and intestinal microbiota have
been proposed (11). Although the
cause of metabolic inflammation in obesity has not been fully
clarified, some studies have shown that diet-induced dysbiosis may
be the origin of this inflammation (9,12),
and this scenario is related to increased intestinal permeability
caused by changes of normal flora and their metabolites (13,14).
The studies found that diet can be associated with structural
variations in gut microbiota, especially the ‘Western’ diet, which
has various effects on host gastrointestinal tract (GI) metabolism,
microbiota and immune homeostasis (15,16).
Changes in microbiota can affect gut metabolic
activity in various way, such as increasing the production of
short-chain fatty acids (SCFAs), which leads to alterations of
intestinal homeostasis. Decreased bacterial diversity and
alteration of representative bacterial genes and metabolic pathways
can be found in obese individuals (17). In particular, high-fat diets or
diets low in fiber have been associated with a higher abundance of
Firmicutes (18), while
studies comparing obese individuals and their lean twins have also
shown a higher predominance of Firmicutes and lower
abundance of Bacteroidetes (17,19)
in the obese subjects. It must be noted, however, that other
studies have not found similar differences (20,21).
Further investigations have been shown that the
complex interplay between diet and the intestinal microbiota in the
context of obesity can lead to the release of gut-derived
inflammatory factors into the circulation, resulting in the
development of obesity (22).
Lipopolysaccharide (LPS), a potent inflammatory mediator of
Gram-negative bacteria, has been recently shown to trigger
inflammation in obese and metabolic syndrome individuals by
signaling through the CD14/TLR4 pathway (23). Such LPS-induced systemic
inflammation may result from intestinal permeability mediated by a
high-fat diet since increases in the translocation of intestinal
Gram-negative bacteria (which produce LPS) to the mesenteric lymph
nodes (mLNs) and mesenteric fat can be found in high-fat diet-fed
mice (24). One recent study found
that antibiotic treatment or CD14 suppression appeared to reduce
inflammatory cytokine expression and improve weight gain in
high-fat diet mice, indicating a role of the microbiota in the
inflammatory process (25).
Therefore, it is possible that intestinal inflammation may lead to
GI permeability, resulting in an increase in circulating LPS and
bacterial DNA, which promote systemic inflammation and insulin
resistance in both mice and humans (26).
This metabolic inflammation, which does not
necessarily involve pathogens, is associated with inflammatory
adipose tissue and higher immune cell accumulation in fatty tissue
regions (Fig. 1) (6). ATMs appear to play a major function
in the regulation of obesity-induced inflammation, and different
types of macrophage can cause the different effects in adipose
tissue. Currently, macrophages are divided into 2 groups, the M1
and M2 types. M1 macrophages characterized by the expression of
F4/80+ CD11b+ CD11c+
iNOS+ (inducible nitric oxide synthase) and the
production of pro-inflammatory cytokines [IL-1β, IL-6, IL-12, tumor
necrosis factor (TNF)-α, MCP-1] is considered to be involved in
adipose tissue inflammation, while the M2 type macrophages, which
express F4/80+ CD11c− CD301+
Arg1+ CD206+ and produce anti-inflammatory
cytokines [IL-1 receptor antagonist, IL-4, IL-10, transforming
growth factor (TGF)-β1], have been known to suppress inflammation
in adipose tissue (27,28). Other studies have also suggested
that high levels of inflammatory cytokines in adipose tissue during
obesity are consistent with increasing macrophage numbers, which
can be described as a metabolic activation instead of the classical
activation related to infections (29,30).
In addition to macrophages, lymphocytes are strongly associated
with inflammatory processes in obesity. Although there are several
types of lymphocytes that are related to obesity and metabolic
syndrome, pro-inflammatory Th1, Th17 and CD8+ T cells
predominate over anti-inflammatory regulatory T (Treg) cells and
Th2 cells, which are found in higher proportions in lean adipose
tissue (7,31). One study found that mice fed a
high-fat diet displayed more Th1 polarization and IFN-γ production,
which occurred several months after macrophage accumulation and
insulin resistance (32), while
the number of Treg cells was decreased in the adipose tissue of
obese mice; insulin sensitivity was also improved when these cells
were increased (Fig. 1) (31).
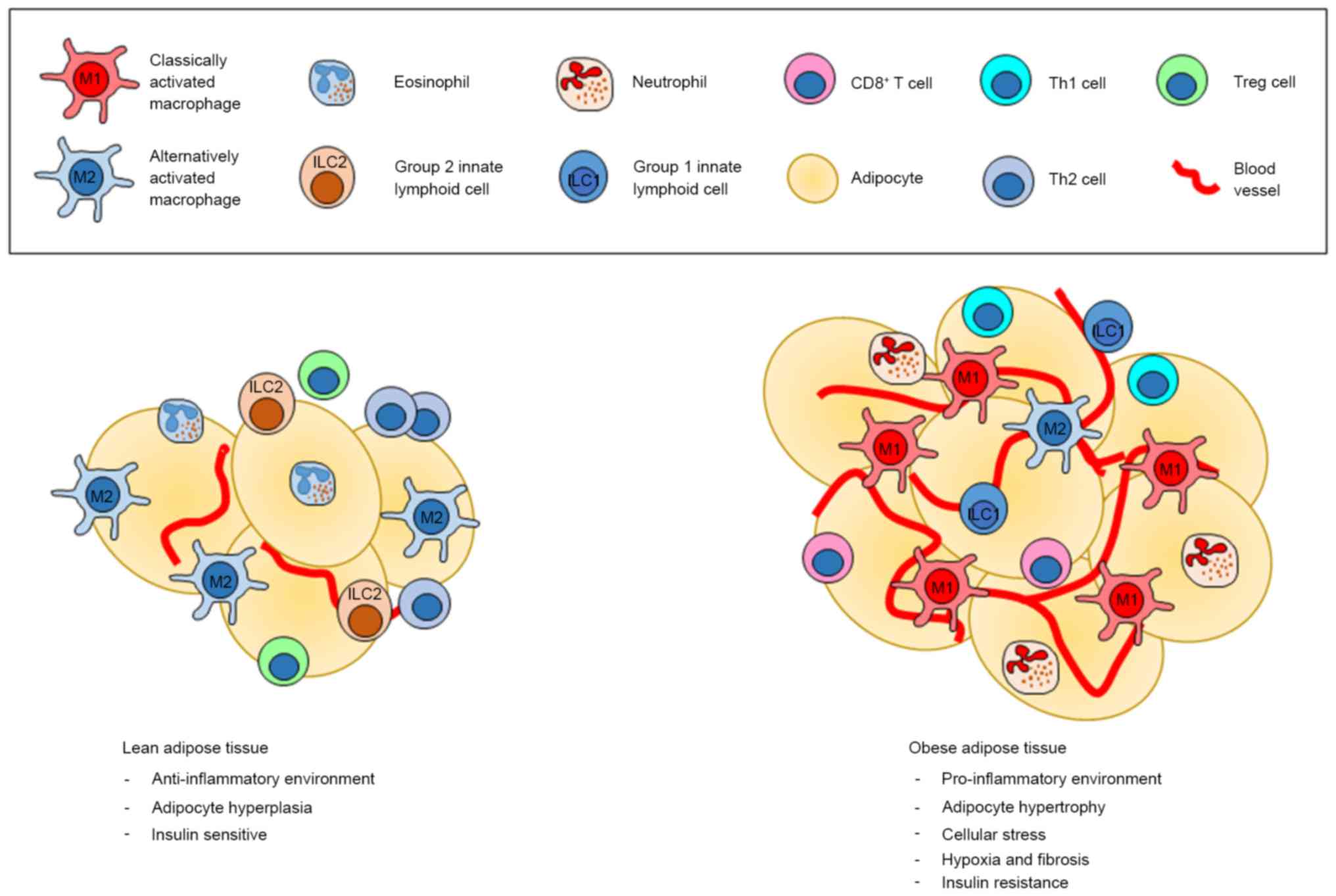 | Figure 1.The different environment between
lean and obese adipose tissues. The most common immune cells found
in lean adipose tissue are M2 macrophages, which create the
anti-inflammatory environment in cooperation with ILC2s,
eosinophils, Treg and Th2 cells. On the other hand, obese adipose
tissue is dominated by M1 macrophages, neutrophils, ILC1, Th1 as
well as CD8+ T cells, which all promote an inflammatory
condition, which in turn support insulin resistance. Treg,
regulatory T cells; ILC, innate lymphoid cell. |
Innate lymphoid cells (ILCs)
ILCs are a recently discover type of innate immune
cells, which have been identified as an important player in
lymphoid organogenesis, tissue defense, epithelial tissue
homeostasis and the amplification of immune responses (33,34).
Although it has been agreed that ILCs participate in the defense
against pathogens (35), some
studies have suggested they could also be involved in some systemic
conditions, such as chronic inflammation and autoimmune disorders
(36,37). ILCs have been defined as
lymphoid-derived, immune lineage negative (Lin−), Th
cytokine expressing cells (33).
Currently, three distinct groups of ILCs which exhibit a functional
diversity mirroring three types of effector CD4+ T-cells
have been identified: Group 1 ILCs, equivalent to Th1 T cells,
group 2 ILCs to Th2 cells, and group 3 ILCs to Th17 and Th22 cells
(Fig. 2) (35).
Group 1 ILCs
Group 1 ILCs are characterized based on their
ability to produce IFN-γ, and have been sub-grouped into two main
types, conventional NK (cNK) cells and ILC1 cells (38). cNK cells have cytotoxic ability and
can be found in numerous organs as they recirculate between the
blood and tissues while ILC1s have only limited cytotoxicity, but
have the ability to produce several types of inflammatory cytokines
mirroring Th1 cells (39,40). Both types of group 1 ILCs can
produce TNF and IFN-γ as a uniquely inflammatory profile when these
cells are stimulated with IL-12, IL-15, or IL-18, and rely on T-bet
transcription factor (T-bet) as a key transcription factor
(41,42). Generally, mice resting mature cNK
cells have been identified as Lin− CD3ε+
NK1.1+ NKp46+ CD49b(DX5)+
Eomes+ whereas Lin− CD3ε−
CD56+ NKp46+ NKp44− has been used
in humans (Fig. 2) (43). Two main groups of cNK cells have
been identified, including the majority of blood cNK cells, which
are CD56low and highly cytotoxic, and
CD56high low cytotoxicity cNK cells, which produce a
high number of inflammatory cytokines (44).
Unconventional NK cells or, also known as ILC1s, are
tissue-resident NK-like cells that are not derived from NK cell
precursors and are normally found in non-lymphoid organs (45). A defining distinction between NK
cells and ILC1s is the expression of the transcription factor,
Eomes (39). NK cells are
Eomes+ and require this transcription factor to develop
while ILC1s are Eomes− and do not need Eomes for their
development (39). Another
noticeable characteristic that differentiates ILC1s from NK cells
is their tissue resident markers, CD49a and CD69 (39). These tissue resident ILC1s have
been found in a variety of non-lymphoid tissues, including the
small intestine mucosa, liver, salivary glands, and female
reproductive tract (45).
Interestingly, recirculation of these tissue-resident ILC1s does
not seem to occur, and, in mice, the existence of these ILC1s is
likely maintained predominantly through local self-renewal instead
of replenishment from blood-derived ILC1s or their precursors
(46,47).
Emerging evidence in recent years has demonstrated
that ILC1s and NK cells in different organs show coincident but
different phenotypes and functions. Intestinal and hepatic ILC1s
share the CD49a+ CD49b− phenotype, produce
high IFN-γ levels and are Eomes− and less cytotoxic than
NK cells (48). The cytotoxicity
of hepatic resident ILC1s is regularly mediated by TRAIL rather
than perforin (48,49), which is found in NK cell-mediated
cytotoxicity, although mice and human NK cells can express TRAIL
(50). Moreover, various studies
have found that several types of group 1 ILCs that differ from cNK
cells are resident in the gastrointestinal mucosa, e.g.,
intraepithelial ILC1s (ieILC1s), which express surface markers
typical of intraepithelial T cells, such as CD49a, CD69, CD160, and
in humans, the integrin CD103 that makes them distinct from cNK
cells although ieILC1s also display canonical NK cell receptors and
need the transcription factors T-bet and Eomes in their
development, similar to cNK cells (42). Another type is lamina
propria-resident ILC1s (LP ILC1s), which express NKp46 and NK1.1,
which are found in both cNK cells and ieILC1s (45). However, mouse LP ILC1s express high
levels of the IL-7 receptor α chain (CD127) and are negative for
Eomes but positive for T-bet, which is not common in either cNK or
ieILC1 cells (Fig. 2) (45,48).
The major function of group 1 ILCs is the potent
expression of IFN-γ, which plays a crucial role in promoting
immunity against intracellular pathogens (51). cNK cells are known for their rapid
response after exposure to a variety of viral and bacterial
pathogens (52). It is now
suggested that ILC1s are the major source of IFN-γ and TNF in
orally Toxoplasma gondii-infected mice while cNK cells
contribute to a lesser extent (48). Another study found that T-bet
deficient mice were highly susceptible to T. gondii
infection, and adoptive transfer of ILC1s to Rag2−/−
Il2rg−/− mice boosted anti-bacterial immunity (48). Importantly, it seems likely that
some results from the previous studies regarded all NK-like
phenotypes with an ability to produce IFN-γ as cNK cells, which
make the un-recognition of ILC1s as a separate lineage (45). Studies that specifically
investigated the role of ILC1s, as compared to cNK cells, in host
defenses to pathogens are only now being conducted; using specific
markers for ILC1s, such as CD49a, CD127 and Eomes, may make it
possible to more accurately study the specific roles of ILC1s
compared to cNK cells (45).
Group 2 ILCs
ILC2s derive from common lymphoid progenitors like
most lymphoid cells but lack the common lineage marker expression
associated with T cells (CD3, CD4, and αβ/γδ TCR), B cells (CD19
and CD20), and other leukocytes including CD11c, CD14, CD16, CD56,
and FcεR1 (53). Moreover, they
are positive for CD90 (Thy1), CD25 (IL-2 receptor α), IL-25R
(IL-17RB), IL-33 receptor (IL-33R; ST2), and CD127 (IL-7 receptor
α) (Fig. 2) (54). Similar to Th2 cells, GATA3 acts as
a key transcription factor for development and function of ILC2s
(55), and transcription factors
Id2 and retinoic acid-related orphan receptor α (ROR)α have also be
recently include as important regulators in their development
(54,56,57).
ILC2s can respond to IL-25, IL-33 (57,58)
and thymic stromal lymphopoietin (TSLP) (59), and produce type 2 cytokines,
predominantly IL-5 and IL-13 (60). Several studies have shown that
ILC2s can produce high levels of IL-5 and IL-13, which contribute
to the immune response against helminth infection in the GI, lungs
and skin (61,62). ILC2s also produce IL-9, which one
study found supported the accumulation of mast cells and mucus
production (63). However, IL-9
expression by ILC2s was transient and dependent on IL-2 and intact
adaptive immunity, suggesting that IL-9 could amplify ILC2 function
(64). ILC2s can also activate
CD4+ T cells for the efficient induction of Th2 cell
development by presenting an antigen to non-activated
CD4+ T cells (65).
Another study also showed that OX40/OX40 L interactions and the
production of IL-4 by ILC2s contributed to CD4+ T-cell
responses in vitro supporting the role of
ILC2s-CD4+ T cell relation (66). Interestingly, lipid mediators, such
as CysLTs and PGD2, and the TNF-like ligand 1A (TL1A), which have
all been associated with Th2-driven diseases, have also been found
to be activators of ILC2s (67,68).
Furthermore, basophilic- and Th2-derived IL-4 also promote ILC2
activation (69). Cell-cell
interactions through molecular signaling of ICOS (binds ICOS-L) and
KLRG1 (binds cadherins) in ILC2s also promote ILC2 activation and
survival (70,71). A recent report also demonstrated
that ILC2 activation can be influenced by Treg cells, and activated
ILC2 also conversely promote Treg cell maintenance (72).
It has been proposed that ILC2-derived Th2 cytokines
contribute to several types of Th2-related diseases, such as
chronic rhinosinusitis, asthma, atopic dermatitis, and
gastrointestinal allergic disease, which have all been considered
as resident sites of ILC2s (Fig.
2) (64). Clinical experiments
with asthmatics have shown that IL-4, IL-5, and IL-13 inhibition
give beneficial effects to asthma patients, reflecting the
importance of Th2 cytokines, which are partly derived from ILC2s in
asthma (73). Notably, ILC2s seem
to play an important role in lung inflammation in responding to
protease-containing allergens. One study showed that papain, an
allergen used in the experiment, promoted allergic lung
inflammation even in RAG-deficient mice, suggesting a role of ILC2
in lung inflammation (74).
Moreover, airway hyper-reactivity mediated by ILC2s is found not
only in non-infectious inflammation but also after influenza viral
infections (75).
Another role of ILC2s in the metabolic pathway has
also been found, based on the knowledge that this kind of ILC can
regulate beige adipose tissue development and may promote the lean
phenotype (76). Moreover, ILC2s
can be detected in visceral adipose tissue (VAT) where they were
thought to be responsible for eosinophil accumulation (77). Another study found that
ILC2-depleted Rag1−/− mice became obese and
showed impaired glucose tolerance, but these problems improved when
ILC2 cells were transferred into these obese mice (78). Moreover, nutritional status also
likely influences ILC2 biology, as vitamins A and D are known to
skew the ILC3/ILC2 balance in the intestines (79).
Group 3 ILCs
There are currently two recognized sub-populations
of ILC3s, the CCR6+ lymphoid-tissue inducer (LTi) ILC3s
and the CCR6− ILC3s, of which the latter can be divided
into two groups based on the expression of NKp46 in mice (80) and NKp44 in humans (81) (Fig.
2). All groups of ILC3s need the nuclear hormone retinoic acid
receptor-related orphan receptor γt (RORγt) as a key regulator for
their development and function. These ILCs can be activated by
IL-23 or IL-1β stimulation to produce IL-17 or IL-22 mirroring Th17
and Th22 cells (82).
LTi cells appearing during embryonic development
were initially regarded as strictly required for prenatal lymph
node development and Peyer's patches (PPs), and also thought to be
important in the development of the adaptive immune system
(82,83). However, CCR6-expressing LTi-ILC3s
(CCR6+ ILC3s), which share several characteristics with
embryonic LTi cells, such as co-expression of CD4 and the
production of IL-17 (Fig. 2)
(84), can be found in mLNs and
the colon lamina propria (cLPL) of healthy adult mice (85).
CCR6− ILC3s, which produce only IL-22 and
account for a small proportion of ILC3s perinatally, increased
significantly within 4 weeks after birth (84,86),
and PLZF+ common helper-like ILC progenitor (CHILP)
cells seem likely to be a specific CCR6− ILC3
progenitor, which make CCR6− ILC3s differ from other
ILC3s (84). Moreover, some
studies have demonstrated that CCR6− ILC3s use distinct
transcriptional regulation pathways for their development (84,87),
and there are some differences within CCR6− ILC3s in
terms of their activation since CCR6−NKp46−
ILC3s do not require T-bet for cell differentiation and
maintenance, but CCR6−NKp46+ ILC3s do require
T-bet (48).
Although the proportion of ILC3s accounts for less
than 5% of lamina propria lymphocytes, it has been shown that this
cell type is one of the major sources of IL-22, which plays a
crucial role in mucosal immune defense. CCR6+ ILC3s
regulate immunity against infection and disease through the
secretion of IL-17 and IL-22, in addition to their key role in
organogenesis (82,88). For example, one study found that
intestinal tissue homeostasis could be supported by
CCR6+ ILC3-derived IL-22 in a graft-vs.-host disease
model (89). Another study showed
that this CCR6+ ILC3-derived IL-22 also promotes the
induction of intestinal fucosylation of the intestinal epithelium
with the cooperation of lymphotoxin, which is also derived from
CCR6+ ILC3s (90).
CCR6−NKp46+ ILC3s in the small intestine also
contribute to mucosal immunity against Citrobacter rodentium
(C. rodentium) through production of IL-22 in
Rag2−/− Il2rg−/− mice (80). In addition, ILC3s have been thought
to play a role in the intestinal defenses in Salmonella
typhimurium (91), Candida
albicans (92) and
Streptococcus pneumoniae (93) models. It has also been proposed
that IL-22-producing CCR6−NKp46+ ILC3s have
an impact on the resistance to bacterial invasion in the colitis
model (94,95), and modulate eosinophil infiltration
and lymphocyte invasion in allergic airway hyperreactivity (AHR) in
the lung (96). These all suggest
a role of ILC3s in homeostasis in multiple tissues following
inflammation or damage.
Relationship between ILCs and obesity
Although it is accepted that genetic and
environmental factors were originally thought to be the major
influences on the development of obesity, many researches have now
shown that immunological factors can also contribute to the
pathogenesis of obesity. Nowadays, several types of immune cells
have been recognized as critical regulators of metabolic
homeostasis (97,98), and this crosstalk likely involves
immune cells and low-grade inflammation, particularly in many
organs besides the adipose tissue, including the pancreas, liver
and intestines, which all showed as an emerging characteristics and
made a potentially regulatory force behind the development of
obesity (97,99).
ILCs are now recognized as a new regulator involved
in adipose tissue and in metabolic homeostasis (10,100). Most studies of ILCs and their
role in metabolism have focused on ILC2s, which have been reported
to play a role in maintaining metabolic homeostasis, since ILC2s
and eosinophils are predominantly resident in lean adipose tissue,
and are considered as ‘upstream’ regulators of M2 macrophages in
adipose tissue (77). Various
studies have shown that, with the cooperation of eosinophils and M2
macrophages, ILC2s regulate obesity, beige conversion of white
adipose tissue and beige fat biogenesis (Fig. 3) (76,77,101). The production of IL-5 and IL-13
from ILC2s in VAT has been shown to be essential for eosinophil and
M2 macrophage differentiation and activation, both of which act as
important regulators of obesity (77). In addition, ILC2 deficiency in
Rag1−/− mice resulted in significantly reduced
numbers of eosinophils and M2 macrophages, suggesting that ILC2s
alone can promote the development of eosinophils and M2 macrophages
in adipose tissue leading to obesity regulation (77,78).
IL-33 is another important inducer of ILC2s and can influence the
development of obesity. Recent studies have shown the effect of
ILC2s on the biogenesis of VAT under IL-33 stimulation, which might
regulate obesity (Fig. 3)
(76,102). In addition, the numbers of ILC2s
were decreased in obese murine epididyma, and this scenario is also
found in human abdominal subcutaneous white adipose tissue
(76). Notably, IL-33 knock-out
mice were shown to have weight gain and reduced frequency and
absolute numbers of ILC2s, even when the mice were fed a normal
diet (76). However, this
situation could be reversed by administration of IL-33, which led
to increased numbers of ILC2s, consequently promoting the recovery
of M2 macrophage numbers (76).
 | Figure 3.Influences of ILCs on obesity and
insulin resistance. In the healthy state, adipose tissue
inflammation is suppressed by IL-5 and IL-13, secreted by ILC2s
that contribute to the activation of M2 macrophages and
eosinophils. Moreover, the productions of IL-4 and IL-13 from ILC2s
also promote Th2 development. The inflammatory condition of adipose
tissue in obesity is associated with increased infiltration of M1
macrophages, neutrophils, Th1 and CD8+ T cells.
Recently, the role of ILC1 s in adipose tissue inflammation has
been identified, which are mediated by the secretion of IFN-γ that
promote M1 macrophage polarization and ILC2 suppression. ILC,
innate lymphoid cell; ILC1, group 1 ILC; ILC2, group 2 ILC; Treg,
regulatory T cells; TNF-α, tumor necrosis factor-α. |
Roles of cNK cells and ILC1s in obesity have also
been demonstrated. One study found that large quantities of IFN-γ,
which may trigger M1 macrophages in VAT, may be derived from cNK
cells in HFD-induced obesity (103). Moreover, systemic depletion of
NK1.1+ or NKp46+ cells decreased diet-induced
insulin resistance by restricting the polarization of M1
macrophages, but did not decrease obesity, suggesting that cNK
cells affect inflammation-related insulin resistance rather than
metabolism directly (103,104).
In addition, adoptive transfer of splenic NK cells into the VAT of
IFN-γ knock-out mice could restore insulin resistance following HFD
induction (103), implying that
this cell type may be a major regulator promoting insulin
resistance. Tissue resident group 1 ILCs have also been recently
reported to contribute to obesity-associated insulin resistance in
the absence of the influence of T and/or NKT cells (105). The same study also reported that
IL-12, which were undefined for upstream signals and cellular
sources, could activate adipose ILC1s, leading to the production of
IFN-γ, and the polarization of M1 macrophages in adipose tissue at
the early stage of HFD consumption (Fig. 3) (105). Mirroring the Th1-Th2 balance,
IFN-γ, which is partly derived from ILC1s, can counteract the IL-33
function and interfere with the activation of ILC2s in infected
tissues, as well as in healthy adipose tissues (72). Therefore, ILC1s may indirectly
affect ILC2-mediated regulation of obesity (Fig. 3).
Although there are currently no specific experiments
describing either the effects of ILC3s in adipose tissues or its
role or roles in HFD-induced obesity, it has been hypothesized that
IL-17 and/or IL-22 secreted from ILC3s might affect obesity or
metabolic homeostasis (10).
ILC3-derived IL-17 has been shown to be associated with
obesity-related diseases such as AHR (106). This same study also demonstrated
that AHR lesions were abundant with
CCR6+NKp46− ILC3s, which have the ability to
secrete excessive IL-17 (106).
Moreover, the same study found that Rag1−/−
IL-17A−/− mice did not develop AHR under HFD
conditions, but transferring CCR6+NKp46−
ILC3s into the Rag−/−IL-2Rγ−/−
mice resulted in AHR under HFD feeding, suggesting a role of IL-17
and ILC3s on AHR pathogenesis (106). However, it has also been reported
that IL-22, for which ILC3s were the major source, has been found
to alleviate metabolic abnormalities, including insulin resistance
and hyperglycemia via changes in liver metabolism (107). This same study showed that
IL-22R−/− mice were highly susceptible to
HFD-induced obesity and insulin resistance, but these events
improved when IL-22 was administrated to the obese mice. Another
study also demonstrated a protective role of IL-22 against
diabetes, partly through modulating oxidative stress and
inflammation pathways related to islet β cells, promoting the
secretion of insulin and fully restored glucose insulin sensitivity
in obese mice (108).
Determining whether intestinal ILCs
influence the development of obesity
As mentioned previously, energy balance and gut
homeostasis of the host may be influenced by diet and gut
microbiota composition, and, in metabolically abnormal conditions,
increased intestinal permeability and inflammation may play a role
in the adipose tissue inflammatory response (9,24).
The gut is resided by extensive immune system, and recent studies
have investigated changes in intestinal inflammatory and immune
cell populations with regard to their roles in obesity and insulin
resistance (109,110).
Early reports have shown increasing levels of gut
inflammatory cytokines, such as TNF-α, IL-1β and IL-12, which are
mostly produced by innate immune cells during HFD feeding, and
these increases are related to weight gain, adiposity, plasma
insulin and glucose levels (99,111). Interestingly, these expressions
were detected in bowel immune cells, especially in PPs and lymphoid
aggregates, in which ILCs are resident (112). However, this intestinal
inflammatory response was not found in HFD-fed germ-free mice,
suggesting the microbiota are a driving force behind intestinal
inflammation (111). Recent
evidence has also demonstrated high expression levels of several
pro-inflammatory cytokines, including IFN-γ and IL-1β, in the
duodenum of insulin-resistant obese patients (113), and IL-23, TNF-α, TGF-β, CCL5, and
IFN-γ in the combined lamina propria and epithelial fraction of
obese subjects (114).
Specifically, there are many studies which have investigated
changes of particular types of intestinal immune cells in obese
subjects. One study found that the intestinal frequency of γδT
cells was changed in mice with HFD feeding (109). This type of innate immune cell is
predominantly found in the intraepithelial lymphocyte fraction, and
the same study found that the numbers of IL-17-producing γδT cells
in the small intestine increased after 3 weeks of HFD consumption,
and after 12 weeks in both the colon and small bowel (109). Another study found that
intestinal eosinophils were also decreased in both number and
proportion after 1 week of HFD feeding (115). This reduction was correlated with
intestinal permeability of HFD mice predominantly in the ileum.
Another study also demonstrated that total macrophage density and
the numbers of mature DCs and NK cells were increased in obese
diabetic and obese non-diabetic humans (114).
HFD has been shown not only to be active in innate
immune cells, but it can also alter the proportions of adaptive
immune cells in the intestinal community. One study found that the
percentages of Treg cells, which play an important role in
inflammatory suppression, were decreased after 3 weeks of HFD
consumption, while the quantity of CD4+ and
CD8+ cells did not show any significant differences in
this period, but were higher after 12 weeks of HFD feeding
(109). Obese human subjects also
showed increased T-bet expression and CD8+ T cells and
reduced Foxp3 (Treg) cells in both the small bowel and colon
compared to lean subjects (109,114). Reduced numbers and frequency of
Th17 cells were also found in HFD-fed mice, apparently related to
alterations of the commensal bacteria populations, such as
segmented filamentous bacteria (SFB) and Porphyromonas
gingivalis (110). Another
study found that decreased numbers of Th17 and SFB were correlated
with an abnormal gut barrier and increased adiposity, linking the
role of microbiota and adaptive immunity to obesity (116).
Although the roles of ILCs on metabolic homeostasis
have been intensively studied, to date such studies have focused
only on adipose tissue ILCs, and these findings may not represent
the overall contributions of this cell type on obesity, since other
organs such as the intestine also displayed low-grade chronic
inflammatory changes in obese persons. There are few studies to
date which have examined the role of gut ILCs on intestinal
inflammation and homeostasis after HFD-feeding, and until now only
ILC3s, which are very abundant within the intestine, have been
reported to be associated with mucosal homeostasis during obesity
(107). This information is
consistent with the reduction of CCR6+NKp46+
ILC3s in the lamina propria of HFD-fed mice, and this seems to be
related to lower mucosal barrier integrity and increased serum LPS
(109). However, no current
reports have demonstrated a role for HFD in all types of intestinal
ILCs, which directly contact with diets and gut microbiota, and may
contribute to colon homeostasis in obesity.
Conclusion
Like macrophages, ILCs were first described as a
player in the innate immune system against several types of
pathogens, but new studies on these immune cells have proposed that
ILCs may also be a player in metabolic homeostasis. ILC2s appear to
be regulators of anti-inflammatory conditions in the lean state
while ILC1s seem to be involved in promoting the development of
obesity and other metabolic diseases by secreting inflammatory
factors in the obese state. Although these roles and the
characterizations of ILCs have been well documented in many
studies, most investigations to date have focused on the function
of this cell in adipose tissue, but since the primary functions of
this type of cell are carried out in barrier tissues such as the
liver, intestine and other abdominal viscera, which all affect the
progression of obesity and metabolic diseases, new research should
examine the influence of these cells in other organs on the
development of obesity and metabolic abnormalities.
References
|
1
|
Boulangé CL, Neves AL, Chilloux J,
Nicholson JK and Dumas ME: Impact of the gut microbiota on
inflammation, obesity and metabolic disease. Genome Med. 8:422016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lu Y and Loos RJ: Obesity genomics:
Assessing the transferability of susceptibility loci across diverse
populations. Genome Med. 5:552013. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Westerterp KR and Plasqui G: Physically
active lifestyle does not decrease the risk of fattening. PLoS One.
4:e47452009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
DiNicolantonio JJ, O'Keefe JH and Lucan
SC: Added fructose: A principal driver of type 2 diabetes mellitus
and its consequences. Mayo Clin Proc. 90:372–381. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DiNicolantonio JJ, Lucan SC and O'Keefe
JH: The evidence for saturated fat and for sugar related to
coronary heart disease. Prog Cardiovasc Dis. 58:464–472. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gregor MF and Hotamisligil GS:
Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sell H, Habich C and Eckel J: Adaptive
immunity in obesity and insulin resistance. Nat Rev Endocrinol.
8:709–716. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Purkayastha S and Cai D: Neuroinflammatory
basis of metabolic syndrome. Mol Metab. 2:356–363. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cani PD, Amar J, Iglesias MA, Poggi M,
Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et
al: Metabolic endotoxemia initiates obesity and insulin resistance.
Diabetes. 56:1761–1772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang D, Yang W, Tian Z, van Velkinburgh
JC, Song J, Wu Y and Ni B: Innate lymphoid cells as novel
regulators of obesity and its-associated metabolic dysfunction.
Obes Rev. 17:485–498. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi H, Kokoeva MV, Inouye K, Tzameli I,
Yin H and Flier JS: TLR4 links innate immunity and fatty
acid-induced insulin resistance. J Clin Invest. 116:3015–3025.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ley RE, Bäckhed F, Turnbaugh P, Lozupone
CA, Knight RD and Gordon JI: Obesity alters gut microbial ecology.
Proc Natl Acad Sci USA. 102:11070–11075. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Venkatesh M, Mukherjee S, Wang H, Li H,
Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, et al:
Symbiotic bacterial metabolites regulate gastrointestinal barrier
function via the xenobiotic sensor PXR and Toll-like receptor 4.
Immunity. 41:296–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang C, Zhang M, Wang S, Han R, Cao Y,
Hua W, Mao Y, Zhang X, Pang X, Wei C, et al: Interactions between
gut microbiota, host genetics and diet relevant to development of
metabolic syndromes in mice. ISME J. 4:232–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sekirov I, Russell SL, Antunes LC and
Finlay BB: Gut microbiota in health and disease. Physiol Rev.
90:859–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Turnbaugh PJ, Hamady M, Yatsunenko T,
Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA,
Affourtit JP, et al: A core gut microbiome in obese and lean twins.
Nature. 457:480–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trompette A, Gollwitzer ES, Yadava K,
Sichelstiel AK, Sprenger N, Ngom-Bru C, Blanchard C, Junt T, Nicod
LP, Harris NL and Marsland BJ: Gut microbiota metabolism of dietary
fiber influences allergic airway disease and hematopoiesis. Nat
Med. 20:159–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ley RE, Turnbaugh PJ, Klein S and Gordon
JI: Microbial ecology: Human gut microbes associated with obesity.
Nature. 444:1022–1023. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schwiertz A, Taras D, Schäfer K, Beijer S,
Bos NA, Donus C and Hardt PD: Microbiota and SCFA in lean and
overweight healthy subjects. Obesity (Silver Spring). 18:190–195.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Duncan SH, Lobley GE, Holtrop G, Ince J,
Johnstone AM, Louis P and Flint HJ: Human colonic microbiota
associated with diet, obesity and weight loss. Int J Obes (Lond).
32:1720–1724. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Montiel-Castro AJ, González-Cervantes RM,
Bravo-Ruiseco G and Pacheco-López G: The microbiota-gut-brain axis:
Neurobehavioral correlates, health and sociality. Front Integr
Neurosci. 7:702013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harris K, Kassis A, Major G and Chou CJ:
Is the gut microbiota a new factor contributing to obesity and its
metabolic disorders? J Obes. 2012:8791512012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Amar J, Chabo C, Waget A, Klopp P, Vachoux
C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S,
et al: Intestinal mucosal adherence and translocation of commensal
bacteria at the early onset of type 2 diabetes: Molecular
mechanisms and probiotic treatment. EMBO Mol Med. 3:559–572. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cox LM and Blaser MJ: Pathways in
microbe-induced obesity. Cell Metab. 17:883–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Burcelin R, Garidou L and Pomié C:
Immuno-microbiota cross and talk: The new paradigm of metabolic
diseases. Semin Immunol. 24:67–74. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mosser DM and Edwards JP: Exploring the
full spectrum of macrophage activation. Nat Rev Immunol. 8:958–969.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Eagle Red A and Chawla A: In obesity and
weight loss, all roads lead to the mighty macrophage. J Clin
Invest. 120:3437–3440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kratz M, Coats BR, Hisert KB, Hagman D,
Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS,
et al: Metabolic dysfunction drives a mechanistically distinct
proinflammatory phenotype in adipose tissue macrophages. Cell
Metab. 20:614–625. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu X, Grijalva A, Skowronski A, van Eijk
M, Serlie MJ and Ferrante AW Jr: Obesity activates a program of
lysosomal-dependent lipid metabolism in adipose tissue macrophages
independently of classic activation. Cell Metab. 18:816–830. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feuerer M, Herrero L, Cipolletta D, Naaz
A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S and
Mathis D: Lean, but not obese, fat is enriched for a unique
population of regulatory T cells that affect metabolic parameters.
Nat Med. 15:930–939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Strissel KJ, DeFuria J, Shaul ME, Bennett
G, Greenberg AS and Obin MS: T-cell recruitment and Th1
polarization in adipose tissue during diet-induced obesity in
C57BL/6 mice. Obesity (Silver Spring). 18:1918–1925. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Spits H, Artis D, Colonna M, Diefenbach A,
Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius
RE, et al: Innate lymphoid cells – a proposal for uniform
nomenclature. Nat Rev Immunol. 13:145–149. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Spits H and Cupedo T: Innate lymphoid
cells: Emerging insights in development, lineage relationships and
function. Annu Rev Immunol. 30:647–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Artis D and Spits H: The biology of innate
lymphoid cells. Nature. 517:293–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Villanova F, Flutter B, Tosi I, Grys K,
Sreeneebus H, Perera GK, Chapman A, Smith CH, Di Meglio P and
Nestle FO: Characterization of innate lymphoid cells in human skin
and blood demonstrates increase of NKp44+ ILC3 in
psoriasis. J Invest Dermatol. 134:984–991. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fuchs A and Colonna M: Innate lymphoid
cells in homeostasis, infection, chronic inflammation and tumors of
the gastrointestinal tract. Curr Opin Gastroenterol. 29:581–587.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cortez VS, Robinette ML and Colonna M:
Innate lymphoid cells: New insights into function and development.
Curr Opin Immunol. 32:71–77. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cortez VS and Colonna M: Diversity and
function of group 1 innate lymphoid cells. Immunol Lett. 179:19–24.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sun JC and Lanier LL: NK cell development,
homeostasis and function: Parallels with CD8+ T cells.
Nat Rev Immunol. 11:645–657. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Robinette ML, Fuchs A, Cortez VS, Lee JS,
Wang Y, Durum SK, Gilfillan S and Colonna M: Immunological Genome
Consortium: Transcriptional programs define molecular
characteristics of innate lymphoid cell classes and subsets. Nat
Immunol. 16:306–317. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Fuchs A, Vermi W, Lee JS, Lonardi S,
Gilfillan S, Newberry RD, Cella M and Colonna M: Intraepithelial
type 1 innate lymphoid cells are a unique subset of IL-12- and
IL-15-responsive IFN-γ-producing cells. Immunity. 38:769–781. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vivier E, Raulet DH, Moretta A, Caligiuri
MA, Zitvogel L, Lanier LL, Yokoyama WM and Ugolini S: Innate or
adaptive immunity? The example of natural killer cells. Science.
331:44–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cichicki F, Schlums H, Theorell J, Tesi B,
Miller JS, Ljunggren HG and Bryceson YT: Diversification and
functional specialization of human NK cell subsets. Curr Top
Microbiol Immunol. 395:63–94. 2016.PubMed/NCBI
|
|
45
|
Fuchs A: ILC1s in tissue inflammation and
infection. Front Immunol. 7:1042016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gasteiger G, Fan X, Dikiy S, Lee SY and
Rudensky AY: Tissue residency of innate lymphoid cells in lymphoid
and nonlymphoid organs. Science. 350:981–985. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Peng H, Jiang X, Chen Y, Sojka DK, Wei H,
Gao X, Sun R, Yokoyama WM and Tian Z: Liver-resident NK cells
confer adaptive immunity in skin-contact inflammation. J Clin
Invest. 123:1444–1456. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Klose CSN, Flach M, Möhle L, Rogell L,
Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira
D, et al: Differentiation of type 1 ILCs from a common progenitor
to all helper-like innate lymphoid cell lineages. Cell.
157:340–356. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Daussy C, Faure F, Mayol K, Viel S,
Gasteiger G, Charrier E, Bienvenu J, Henry T, Debien E, Hasan UA,
et al: T-bet and Eomes instruct the development of two distinct
natural killer cell lineages in the liver and in the bone marrow. J
Exp Med. 211:563–577. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Spits H, Bernink JH and Lanier L: NK cells
and type 1 innate lymphoid cells: Partners in host defense. Nat
Immunol. 17:758–764. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sonnenberg GF and Artis D: Innate lymphoid
cells in the initiation, regulation and resolution of inflammation.
Nat Med. 21:698–708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Vivier E, Tomasello E, Baratin M, Walzer T
and Ugolini S: Functions of natural killer cells. Nat Immunol.
9:503–510. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
53
|
Karta MR, Broide DH and Doherty TA:
Insights into group 2 innate lymphoid cells in human airway
disease. Curr Allergy Asthma Rep. 16:82016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Walker JA, Barlow JL and McKenzie AN:
Innate lymphoid cells – how did we miss them? Nat Rev Immunol.
13:75–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hoyler T, Klose CS, Souabni A,
Turqueti-Neves A, Pfeifer D, Rawlins EL, Voehringer D, Busslinger M
and Diefenbach A: The transcription factor GATA-3 controls cell
fate and maintenance of type 2 innate lymphoid cells. Immunity.
37:634–648. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wong SH, Walker JA, Jolin HE, Drynan LF,
Hams E, Camelo A, Barlow JL, Neill DR, Panova V, Koch U, et al:
Transcription factor RORα is critical for nuocyte development. Nat
Immunol. 13:229–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Moro K, Yamada T, Tanabe M, Takeuchi T,
Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H and Koyasu S:
Innate production of T(H)2 cytokines by adipose tissue-associated
c-Kit(+)Sca-1(+) lymphoid cells. Nature. 463:540–544. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Saenz SA, Siracusa MC, Perrigoue JG,
Spencer SP, Urban JF Jr, Tocker JE, Budelsky AL, Kleinschek MA,
Kastelein RA, Kambayashi T, et al: IL25 elicits a multipotent
progenitor cell population that promotes T(H)2 cytokine responses.
Nature. 464:1362–1366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Gentek R, Munneke JM, Helbig C, Blom B,
Hazenberg MD, Spits H and Amsen D: Modulation of signal strength
switches notch from an inducer of T cells to an inducer of ILC2.
Front Immunol. 4:3342013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Price AE, Liang HE, Sullivan BM, Reinhardt
RL, Eisley CJ, Erle DJ and Locksley RM: Systemically dispersed
innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad
Sci USA. 107:11489–11494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Tait Wojno ED and Artis D: Innate lymphoid
cells: Balancing immunity, inflammation and tissue repair in the
intestine. Cell Host Microbe. 12:445–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Neill DR, Wong SH, Bellosi A, Flynn RJ,
Daly M, Langford TK, Bucks C, Kane CM, Fallon PG, Pannell R, et al:
Nuocytes represent a new innate effector leukocyte that mediates
type-2 immunity. Nature. 464:1367–1370. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wilhelm C, Turner JE, Van Snick J and
Stockinger B: The many lives of IL-9: A question of survival? Nat
Immunol. 13:637–641. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Doherty TA and Broide DH: Group 2 innate
lymphoid cells: New players in human allergic diseases. J Investig
Allergol Clin Immunol. 25:1–11; quiz 2p following 11.
2015.PubMed/NCBI
|
|
65
|
Oliphant CJ, Hwang YY, Walker JA, Salimi
M, Wong SH, Brewer JM, Englezakis A, Barlow JL, Hams E, Scanlon ST,
et al: MHCII-mediated dialog between group 2 innate lymphoid cells
and CD4(+) T cells potentiates type 2 immunity and promotes
parasitic helminth expulsion. Immunity. 41:283–295. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Drake LY, Iijima K and Kita H: Group 2
innate lymphoid cells and CD4+ T cells cooperate to
mediate type 2 immune response in mice. Allergy. 69:1300–1307.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fajt ML, Gelhaus SL, Freeman B, Uvalle CE,
Trudeau JB, Holguin F and Wenzel SE: Prostaglandin D2
pathway upregulation: Relation to asthma severity, control and TH2
inflammation. J Allergy Clin Immunol. 131:1504–1512. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yu X, Pappu R, Ramirez-Carrozzi V, Ota N,
Caplazi P, Zhang J, Yan D, Xu M, Lee WP and Grogan JL: TNF
superfamily member TL1A elicits type 2 innate lymphoid cells at
mucosal barriers. Mucosal Immunol. 7:730–740. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Motomura Y, Morita H, Moro K, Nakae S,
Artis D, Endo TA, Kuroki Y, Ohara O, Koyasu S and Kubo M:
Basophil-derived interleukin-4 controls the function of natural
helper cells, a member of ILC2s, in lung inflammation. Immunity.
40:758–771. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Maazi H, Patel N, Sankaranarayanan I,
Suzuki Y, Rigas D, Soroosh P, Freeman GJ, Sharpe AH and Akbari O:
ICOS: ICOS-ligand interaction is required for type 2 innate
lymphoid cell function, homeostasis and induction of airway
hyperreactivity. Immunity. 42:538–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Salimi M, Barlow JL, Saunders SP, Xue L,
Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST,
McKenzie AN, et al: A role for IL-25 and IL-33-driven type-2 innate
lymphoid cells in atopic dermatitis. J Exp Med. 210:2939–2950.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Molofsky AB, Van Gool F, Liang HE, Van
Dyken SJ, Nussbaum JC, Lee J, Bluestone JA and Locksley RM:
Interleukin-33 and interferon-γ counter-regulate group 2 innate
lymphoid cell activation during immune perturbation. Immunity.
43:161–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Walford HH and Doherty TA: Diagnosis and
management of eosinophilic asthma: A US perspective. J Asthma
Allergy. 7:53–65. 2014.PubMed/NCBI
|
|
74
|
Oboki K, Ohno T, Kajiwara N, Arae K,
Morita H, Ishii A, Nambu A, Abe T, Kiyonari H, Matsumoto K, et al:
IL-33 is a crucial amplifier of innate rather than acquired
immunity. Proc Natl Acad Sci USA. 107:18581–18586. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Chang YJ, Kim HY, Albacker LA, Baumgarth
N, McKenzie AN, Smith DE, Dekruyff RH and Umetsu DT: Innate
lymphoid cells mediate influenza-induced airway hyper-reactivity
independently of adaptive immunity. Nat Immunol. 12:631–638. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Brestoff JR, Kim BS, Saenz SA, Stine RR,
Monticelli LA, Sonnenberg GF, Thome JJ, Farber DL, Lutfy K, Seale P
and Artis D: Group 2 innate lymphoid cells promote beiging of white
adipose tissue and limit obesity. Nature. 519:242–246. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Molofsky AB, Nussbaum JC, Liang HE, Van
Dyken SJ, Cheng LE, Mohapatra A, Chawla A and Locksley RM: Innate
lymphoid type 2 cells sustain visceral adipose tissue eosinophils
and alternatively activated macrophages. J Exp Med. 210:535–549.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hams E, Locksley RM, McKenzie AN and
Fallon PG: Cutting edge: IL-25 elicits innate lymphoid type 2 and
type II NKT cells that regulate obesity in mice. J Immunol.
191:5349–5353. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Spencer SP, Wilhelm C, Yang Q, Hall JA,
Bouladoux N, Boyd A, Nutman TB, Urban JF Jr, Wang J, Ramalingam TR,
et al: Adaptation of innate lymphoid cells to a micronutrient
deficiency promotes type 2 barrier immunity. Science. 343:432–437.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Satoh-Takayama N, Vosshenrich CA,
Lesjean-Pottier S, Sawa S, Lochner M, Rattis F, Mention JJ, Thiam
K, Cerf-Bensussan N, Mandelboim O, et al: Microbial flora drives
interleukin 22 production in intestinal NKp46+ cells
that provide innate mucosal immune defense. Immunity. 29:958–970.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Cella M, Fuchs A, Vermi W, Facchetti F,
Otero K, Lennerz JK, Doherty JM, Mills JC and Colonna M: A human
natural killer cell subset provides an innate source of IL-22 for
mucosal immunity. Nature. 457:722–725. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Satoh-Takayama N: Heterogeneity and
diversity of group 3 innate lymphoid cells: New cells on the block.
Int Immunol. 28:29–34. 2016.PubMed/NCBI
|
|
83
|
van de Pavert SA and Vivier E:
Differentiation and function of group 3 innate lymphoid cells, from
embryo to adult. Int Immunol. 28:35–42. 2016.PubMed/NCBI
|
|
84
|
Klose CS, Kiss EA, Schwierzeck V, Ebert K,
Hoyler T, d'Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver
Y and Diefenbach A: A T-bet gradient controls the fate and function
of CCR6-RORγt+ innate lymphoid cells. Nature.
494:261–265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hepworth MR, Fung TC, Masur SH, Kelsen JR,
McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W,
et al: Immune tolerance. Group 3 innate lymphoid cells mediate
intestinal selection of commensal bacteria-specific CD4+
T cells. Science. 348:1031–1035. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sawa S, Cherrier M, Lochner M,
Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP and Eberl G:
Lineage relationship analysis of RORγt+ innate lymphoid
cells. Science. 330:665–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Rankin LC, Groom JR, Chopin M, Herold MJ,
Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL and Belz GT:
The transcription factor T-bet is essential for the development of
NKp46+ innate lymphocytes via the Notch pathway. Nat
Immunol. 14:389–395. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Takatori H, Kanno Y, Watford WT, Tato CM,
Weiss G, Ivanov II, Littman DR and O'Shea JJ: Lymphoid tissue
inducer-like cells are an innate source of IL-17 and IL-22. J Exp
Med. 206:35–41. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hanash AM, Dudakov JA, Hua G, O'Connor MH,
Young LF, Singer NV, West ML, Jenq RR, Holland AM, Kappel LW, et
al: Interleukin-22 protects intestinal stem cells from
immune-mediated tissue damage and regulates sensitivity to graft
versus host disease. Immunity. 37:339–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Pickard JM, Maurice CF, Kinnebrew MA, Abt
MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG,
Turnbaugh PJ and Chervonsky AV: Rapid fucosylation of intestinal
epithelium sustains host-commensal symbiosis in sickness. Nature.
514:638–641. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Goto Y, Obata T, Kunisawa J, Sato S,
Ivanov II, Lamichhane A, Takeyama N, Kamioka M, Sakamoto M, Matsuki
T, et al: Innate lymphoid cells regulate intestinal epithelial cell
glycosylation. Science. 345:12540092014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gladiator A, Wangler N, Trautwein-Weidner
K and LeibundGut-Landmann S: Cutting edge: IL-17-secreting innate
lymphoid cells are essential for host defense against fungal
infection. J Immunol. 190:521–525. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Van Maele L, Carnoy C, Cayet D, Ivanov S,
Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG,
et al: Activation of type 3 innate lymphoid cells and interleukin
22 secretion in the lungs during Streptococcus pneumoniae
infection. J Infect Dis. 210:493–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kirchberger S, Royston DJ, Boulard O,
Thornton E, Franchini F, Szabady RL, Harrison O and Powrie F:
Innate lymphoid cells sustain colon cancer through production of
interleukin-22 in a mouse model. J Exp Med. 210:917–931. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Sawa S, Lochner M, Satoh-Takayama N,
Dulauroy S, Bérard M, Kleinschek M, Cua D, Di Santo JP and Eberl G:
RORγt+ innate lymphoid cells regulate intestinal
homeostasis by integrating negative signals from the symbiotic
microbiota. Nat Immunol. 12:320–326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Taube C, Tertilt C, Gyülveszi G, Dehzad N,
Kreymborg K, Schneeweiss K, Michel E, Reuter S, Renauld JC,
Arnold-Schild D, et al: IL-22 is produced by innate lymphoid cells
and limits inflammation in allergic airway disease. PLoS One.
6:e217992011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Osborn O and Olefsky JM: The cellular and
signaling networks linking the immune system and metabolism in
disease. Nat Med. 18:363–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jin C, Henao-Mejia J and Flavell RA:
Innate immune receptors: Key regulators of metabolic disease
progression. Cell Metab. 17:873–882. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Winer DA, Luck H, Tsai S and Winer S: The
intestinal immune system in obesity and insulin resistance. Cell
Metab. 23:413–426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Bostick JW and Zhou L: Innate lymphoid
cells in intestinal immunity and inflammation. Cell Mol Life Sci.
73:237–252. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hashiguchi M, Kashiwakura Y, Kojima H,
Kobayashi A, Kanno Y and Kobata T: IL-33 activates eosinophils of
visceral adipose tissue both directly and via innate lymphoid
cells. Eur J Immunol. 45:876–885. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Lee MW, Odegaard JI, Mukundan L, Qiu Y,
Molofsky AB, Nussbaum JC, Yun K, Locksley RM and Chawla A:
Activated type 2 innate lymphoid cells regulate beige fat
biogenesis. Cell. 160:74–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wensveen FM, Jelenčić V, Valentić S,
Šestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Štimac D,
Wunderlich FT, et al: NK cells link obesity-induced adipose stress
to inflammation and insulin resistance. Nat Immunol. 16:376–385.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lee BC, Kim MS, Pae M, Yamamoto Y, Eberlé
D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, et al: Adipose
natural killer cells regulate adipose tissue macrophages to promote
insulin resistance in obesity. Cell Metab. 23:685–698. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
O'Sullivan TE, Rapp M, Fan X, Weizman OE,
Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ and Sun JC:
Adipose-resident group 1 innate lymphoid cells promote
obesity-associated insulin resistance. Immunity. 45:428–441. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kim HY, Lee HJ, Chang YJ, Pichavant M,
Shore SA, Fitzgerald KA, Iwakura Y, Israel E, Bolger K, Faul J, et
al: IL-17 producing innate lymphoid cells and the NLRP3
inflammasome facilitate obesity-associated airway hyperreactivity.
Nat Med. 20:54–61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wang X, Ota N, Manzanillo P, Kates L,
Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J,
et al: Interleukin-22 alleviates metabolic disorders and restores
mucosal immunity in diabetes. Nature. 514:237–241. 2014.PubMed/NCBI
|
|
108
|
Hasnain SZ, Borg DJ, Harcourt BE, Tong H,
Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, et al:
Glycemic control in diabetes is restored by therapeutic
manipulation of cytokines that regulate beta cell stress. Nat Med.
20:1417–1426. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Luck H, Tsai S, Chung J, Clemente-Casares
X, Ghazarian M, Revelo XS, Lei H, Luk CT, Shi SY, Surendra A, et
al: Regulation of obesity-related insulin resistance with gut
anti-inflammatory agents. Cell Metab. 21:527–542. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Garidou L, Pomié C, Klopp P, Waget A,
Charpentier J, Aloulou M, Giry A, Serino M, Stenman L, Lahtinen S,
et al: The gut microbiota regulates intestinal CD4 T cells
expressing RORγt and controls metabolic disease. Cell Metab.
22:100–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ding S, Chi MM, Scull BP, Rigby R,
Schwerbrock NM, Magness S, Jobin C and Lund PK: High-fat diet:
Bacteria interactions promote intestinal inflammation which
precedes and correlates with obesity and insulin resistance in
mouse. PLoS One. 5:e121912010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Hashiguchi M, Kashiwakura Y, Kojima H,
Kobayashi A, Kanno Y and Kobata T: Peyer's patch innate lymphoid
cells regulate commensal bacteria expansion. Immunol Lett. 165:1–9.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Veilleux A, Mayeur S, Bérubé JC, Beaulieu
JF, Tremblay E, Hould FS, Bossé Y, Richard D and Levy E: Altered
intestinal functions and increased local inflammation in
insulin-resistant obese subjects: A gene-expression profile
analysis. BMC Gastroenterol. 15:1192015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Monteiro-Sepulveda M, Touch S, Mendes-Sá
C, André S, Poitou C, Allatif O, Cotillard A, Fohrer-Ting H, Hubert
EL, Remark R, et al: Jejunal T cell inflammation in human obesity
correlates with decreased enterocyte insulin signaling. Cell Metab.
22:113–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Johnson AM, Costanzo A, Gareau MG, Armando
AM, Quehenberger O, Jameson JM and Olefsky JM: High fat diet causes
depletion of intestinal eosinophils associated with intestinal
permeability. PLoS One. 10:e01221952015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Cox LM, Yamanishi S, Sohn J, Alekseyenko
AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D, et al: Altering
the intestinal microbiota during a critical developmental window
has lasting metabolic consequences. Cell. 158:705–721. 2014.
View Article : Google Scholar : PubMed/NCBI
|















