Introduction
Spontaneous subarachnoid hemorrhage (SAH) is a
common cerebral vascular condition that affects 9 out of 100,000
people in the Western world (1,2).
Although it accounts for ~5% of all strokes, it is a devastating
neurological condition with poor outcomes and high mortality and
morbidity (1–3). Early brain injury (EBI), acute
hydrocephalus, delayed cerebral vasospasm (CVS) and cerebral
infarction are important factors that may contribute to poor
outcome post-SAH. However, data from a previous study indicated
that treatment of CVS, using targeted medication in patients with
SAH, may not lead to improved neurological outcomes, and suggested
that CVS may not be the only cause of the delayed ischemic
complications (4). Additional
studies reported that the functional outcome did not improved with
reversal of angiographic vasospasm (5), and that treatment with an endothelin
receptor antagonist may significantly improve CVS post-SAH
(6), but it did not improve the
poor outcome.
Cerebral vessel autoregulation is the intrinsic
ability of the cerebral vasculature to maintain stable blood flow
even during changes in blood pressure, such as cerebral perfusion
pressure (7). A recent review of
the clinical effects of disturbed cerebral vessel autoregulation
following aneurysmal SAH aimed to determine whether patients with
SAH may benefit from therapeutic targeting of cerebral vessel
autoregulation (4). However,
large-scale analyses are required to determine clinical benefits,
and it was suggested that statins may serve an important role in
cerebral vessel autoregulation.
Statins, which are inhibitors of the
3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, are
widely used in cardiovascular therapy as cholesterol-lowering drugs
that may have pleiotropic effects, including anti-inflammatory
(8), antioxidative stress
(9), anti-CVS (10,11)
and inhibition of platelet aggregation (12,13).
Atorvastatin is a potent statin that inhibits HMG-CoA reductase;
however, the specific pathophysiological mechanisms of statins on
cerebral vessel autoregulation and EBI following SAH remain
unknown. Therefore, the aim of the present study was to evaluate
the effects of atorvastatin on SAH-induced EBI, CVS and function of
vascular endothelial cells. Results revealed that atorvastatin was
an effective and well-tolerated treatment for SAH in various
clinical settings and may protect the autoregulation of cerebral
vessels. The present study investigated whether clinical benefits
could be achieved by influencing the state of cerebral vessels
autoregulation.
Materials and methods
Animals and drugs
Animal's care and experimental protocols were
approved by the Animal Care and Use Committee of Anhui Medical
University (Hefei, China) and conformed to the Guide for the Care
and Use of Laboratory Animals as outlined by the National
Institutes of Health. New Zealand white rabbits used in the present
study were purchased from Animal Central of TaiHu Hospital (cat.
no. SYXK2012-0033; Wuxi, China). Rabbits were raised in a
comfortable room with normal atmospheric moisture and fed with a
standard diet every 3–4 h at the Animal Center of Taihu Hospital
for 10 days prior to the start of the experiments. The temperature
of the feeding and operation rooms was maintained at 22°C.
Atorvastatin (20 mg/kg/d; Pfizer, Inc., Wuxi, China) was
administered by gastric gavage once daily for 3 days prior to SAH
operation and 22 h post-SAH to maintain drug levels (14). Neurological deficits were assessed
24 h following SAH operation and rabbits were euthanized
immediately following evaluation. The data of blood pressure,
injected arterial blood gas data, body weight and blood pressure
were used for monitoring only, and were not shown in the results of
the present study.
Experimental design
A total of 48 adult male New Zealand white rabbits
(weight, 2.5–3.2 kg) were randomly assigned to 3 groups
(n=16/group): i) Sham group, which received an injection of saline
into the cisterna magna (2 ml); ii) SAH group; and iii) SAH +
atorvastatin group. As 3 rabbits died in the SAH group and 4
rabbits died in the SAH + atorvastatin group, 3 additional rabbits
were added for the SAH group and 4 additional rabbits were added
for the SAH + atorvastatin group. Atorvastatin (20
mg/kg/d) was administered when the SAH model was
established and continued every 24 for 72 h, as previously
described (10). A total of 8
rabbits from each group were sacrificed on day 3 with the
fixation-perfusion method in 10% formaldehyde. The hippocampus and
basilar artery (BA) were removed for terminal
deoxynucleotidyl-transferase-mediated dUTP nick-end labeling
(TUNEL), hematoxylin and eosin (H&E) and for
immunohistochemical staining analyses. The remaining eight rabbits
were exsanguinated and decollated, and the artery was removed and
frozen in deep cryogenic freezer for further biochemical studies.
Prior to sacrifice, serum samples were collected from each rabbit
to evaluate the protein expression levels of endothelin-1 (ET-1) by
ELISA.
SAH model rabbit establishment
SAH was induced according to a previously described
two-hemorrhage rabbit model (10,14).
Briefly, rabbits were anesthetized with an auricular marginal vein
injection of 10% chloral hydrate (2.5 ml/kg). Vital signs were kept
stable and a 23-gauge butterfly needle was inserted into the
cisterna magna. When cerebrospinal fluid was observed from the
butterfly needle, then 2 ml non-heparinized fresh autologous
auricular arteries blood was injected into the cisterna magna from
the butterfly needle with strict aseptic technique and the
injection lasted 1 min. To maintain the blood flow from cisterna
magna to the basilar cistern, all rabbits were held at a 30-degree
head-down position for 30 min. Rabbits were returned to the feeding
room following recovery from anesthesia. The second injection was
administered after 48 h in a similar manner.
Neurological scoring
Neurological scores were recorded by the same
independent observer who was blinded to the study. A previously
modified scoring table was used to evaluate the neurological
function every day (14,15). The Neurological study consisted of
the following three tests: i) Appetite. Scores indicate the
following: 2, Scarcely ate; 1, left meal unfinished; 0, finished
meal. ii) Activity. Scores indicate the following: 2, Almost always
lying down; 1, lying down, will stand and walk with some
stimulation; 0, active, alert or standing. iii) Deficits. Scores
indicate the following: 2, Impossible to walk and stand due to
ataxia and paresis; 1, unable walk due to ataxia or paresis; 0, no
deficits.
Fixation-perfusion
A total of 8 rabbits from each group were
anesthetized with an auricular marginal vein injection of 10%
chloral hydrate (4 ml/kg). The chest was quickly cut open for
cannula intubation in the left ventricle and the right atrium was
cut open. Perfusion started with 1,500 ml of physiological
phosphate buffered saline (PBS; pH 7.3) at 37°C, followed by 1,000
ml buffered formaldehyde (10%) under 120 cm H2O
perfusion pressure. Following fixation-perfusion, whole brain
tissues were removed and stored in 10% formalin at 22°C.
H&E staining
This tissue was then post-fixed in 4%
paraformaldehyde at 4°C overnight, dehydrated and embedded in
paraffin, and cut into sections (4 µm). To avoid the arterial
branches, the BAs were transected at the same middle position each
time (2 mm). Every fourth section along the coronal plane was
stained with H&E and observed under a microscope. The diameter,
perimeter and cross-sectional area of the BA were measured by an
independent investigator, blinded to the study, using an Olympus
light microscope (Olympus Corporation, Tokyo, Japan) and Image-Pro
Plus 6.0 Software (Media Cybernetics, Inc., Rockville, MD, USA).
The diameter, perimeter and cross-sectional area of BA were
calculated as previously described (16). T-score indicates the BA
perimeter/BA thickness.
Immunohistochemical staining
Brain tissues (brainstem) were isolated from brain
tissues before storing in formalin, and were routinely fixed,
embedded and cut into sections (4 µm) for immunohistochemistry.
Endogenous peroxidase was quenched with 3% hydrogen peroxide for 10
min at room temperature. Sections were incubated with anti-von
Willebrand factor (vWF; cat no. ab778; dilution 1:30; Abcam,
Cambridge, UK) and anti-thrombomodulin (TM; cat no. ab6980;
1:1,000; Abcam) primary antibodies overnight at 4°C, followed by
incubation with HRP-conjugated secondary antibodies (goat serum;
cat no. ab138478; Abcam) for 1 h at room temperature. Following
washing, sections were developed in 3,3′-diaminobenzidine (DAB)
solution and counterstained with hematoxylin for 5 min at room
temperature. Negative controls were included by omitting the
primary antibody. A light Olympus microscope (Olympus Corporation)
and Image-Pro Plus 6.0 software (Media Cybernetics, Inc.) were used
for analysis.
TUNEL staining and cell counting
The TUNEL Staining kit (Roche Diagnostics, Basel,
Switzerland) was used for hippocampus staining, and the
TUNEL-positive cells were analyzed by fluorescein-dUTP with dNTP or
POD with DAB (manufacturer's protocol for in situ apoptosis
detection kit (Roche Diagnostics) according to the methods
described previously (17). A
negative control was used by eliminating the TUNEL reaction
mixture. Cells exhibiting nuclear condensation/fragmentation and
apoptotic bodies in the absence of cytoplasmic TUNEL reactivity,
brown staining of nuclei were considered as apoptotic cells.
Apoptotic cells were confirmed with the help of a pathologist
blinded to the grouping. The number of TUNEL-positive cells in each
region (number/mm2) were counted in a high-powered field
(magnification, ×400) by an investigator who was blinded to the
study. A total of 8 rabbits from each group were used. A total of 5
fields were analyzed, and the experiment was repeated three
times.
Western blot analysis
Western blot analysis was performed as described
previously for evaluating the levels of Caspase-3 proteins
(18). The samples (20 µg
protein), as determined by using a bicinchoninic acid assay
(Abcam), were separated by 10% SDS-PAGE and transferred to a
nitrocellulose membrane. Membranes were probed with the following
primary antibodies: Rabbit anti-Caspase-3 (cat no. ab4051; 1:500;
Abcam) antibody. GAPDH (cat no. G5262; 1:6,000; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) was used as a loading control.
Following incubation with the primary antibodies for 1 h at room
temperature, membranes were washed with TBS + 5% Tween-20 (TBST)
and incubated with appropriate horseradish peroxidase-labeled
secondary antibodies (cat no. sc2357; 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 1 h at room temperature
in 1% nonfat milk in TBST for 1 h at room temperature. Following
two rinses and four washes with PBST, membranes were incubated with
Enhanced Chemiluminescence Western Blotting Detection Reagent (GE
Healthcare Life Sciences, Shanghai, China) for 60 sec and exposed
to autoradiography film for visualization of the bands. Results
were quantified by Quantity One version 4.5 software (Bio-Rad
Laboratories, Hercules, CA, USA). A total of 8 rabbits from each
group were used.
ELISA
At day 3 following surgical intervention, blood
samples were collected from anesthetized animals (n=8/group) and
analyzed for ET-1 expression levels using a rabbit ET-1 ELISA kit
(cat no. F2003; Westang Bio-Tech Co., Ltd., Shanghai, China)
specific for rabbits. Plasma was separated from the blood by
centrifugation at 3,000 × g for 15 min, and the supernatant was
assayed for the protein concentrations of ET-1, according to the
manufacturer's protocol. ET-1 concentrations (pg/ml) were
determined based on a standard curve, prepared using a known set of
serial dilutions of standard proteins. The experiment was repeated
three times.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from hippocampus brain
samples (n=8/group) using TRIzol Reagent (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), following the manufacture's
protocol. β-actin was used as an internal control. First-strand
cDNA was synthesized from the total RNA as previously described by
using a kit from Abcam (cat no. 185916) (10,19).
vWF and TM mRNA levels in each sample were determined by qPCR using
SYBR Green Master Mix (Toyobo Co., Ltd., Osaka, Japan). The qPCR
thermocycling conditions were as follows: 45°C (2 min) and 95°C (10
min), followed by 40 cycles of denaturation 95°C (15 sec);
annealing 60°C (1 min); extension 72°C (1 min). All samples were
analyzed in triplicate. The primers were as follows: TM, forward
CTC TAG CAC CTA CAA TAC CCC ATT, reverse CCC GAG TCC AGT GTC CCT CT
(146); vWF, forward TTT TCT TAT GTT CTC CAC GAA GGG, reverse CAT
TGA TGA GGC AGG GGT TCT (151); β-actin, forward CCC ATC TAT GAG GGT
TAC GC, reverse TTT AAT GTC ACG CAC GAT TTC (150) (20).
Statistical analysis
All data were presented as the mean ± standard
deviation. SPSS 14.0 statistical software (SPSS, Inc., Chicago, IL,
USA) was used for statistical analysis. Differences between the two
groups were analyzed using a two-tailed unpaired Student's t-test.
The differences among multiple groups were assessed using a one-way
analysis of variance followed by Tukey's post hoc test. Ranked data
between the two groups were evaluated using rank sum test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
General observations
No significant differences were identified in blood
pressure, injected arterial blood gas data, body weight and blood
pressure (data not shown). The mortality of SAH group was 18.75% (3
of 16), 25% (4 of 16) in SAH + atorvastatin group and 0% in the
Sham group. No significant difference was identified for the
mortality of SAH + atorvastatin treated group compared with the SAH
group (P>0.05). During the process of model establishment, the
dead and not eligible rabbits were excluded from the present study
and were replaced with new rabbits to maintain the number of
animals in each group.
Neurological scoring
The neurological scores of rabbits in SAH +
atorvastatin group were significantly lower than the scores in the
SAH group at 72 h following SAH induction (P<0.05; Fig. 1; Table
I). In addition, treatment with atorvastatin was demonstrated
to improve neurological functional post-SAH in experimental
rabbits.
 | Table I.Neurological scores among the three
experimental groups. |
Table I.
Neurological scores among the three
experimental groups.
| Group | n | Score |
|---|
| Sham | 16 | 0.38+0.19 |
| SAH | 16 | 2.25+0.39 |
| SAH +
atorvastatin | 16 | 1.75+0.28 |
H&E staining for morphometric
vasospasm
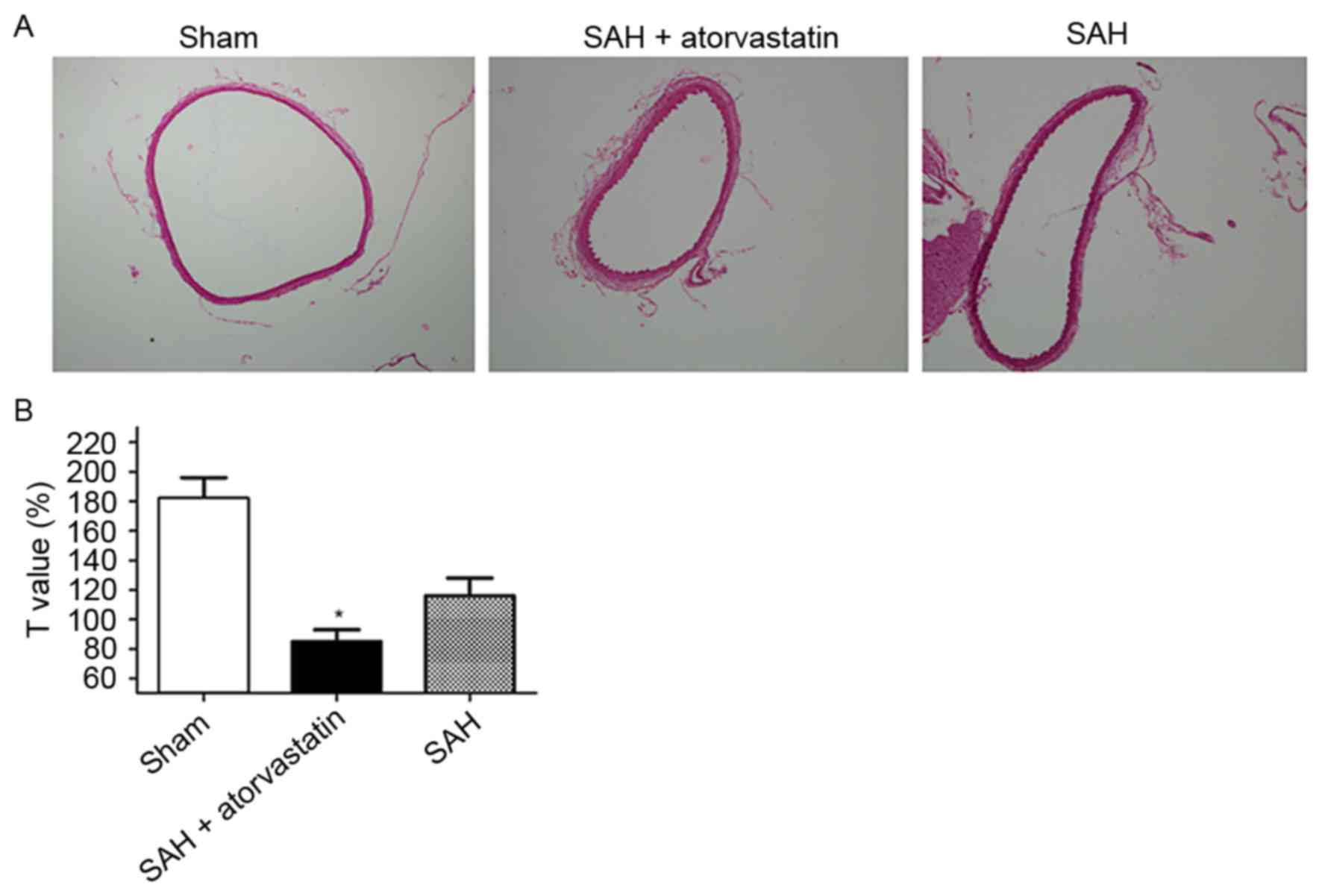
Notable differences were observed in BA morphology
among the three groups on day 3 post-SAH (Fig. 2A): The inner perimeter, diameter
and cross-sectional area of BA in the SAH group was smaller, and
the BA wall was thicker compared with the Sham group. To avoid the
individual differences, the relatively fixed values of T (T=inner
perimeter/wall thickness) were calculated (Fig. 2B). Compared with the SAH group, the
T value of the BA in the SAH + atorvastatin treatment group
exhibited a significant increase (115.4±11.0 vs. 89.6±9.11;
P<0.01).
Immunohistochemical staining for vWF
and TM
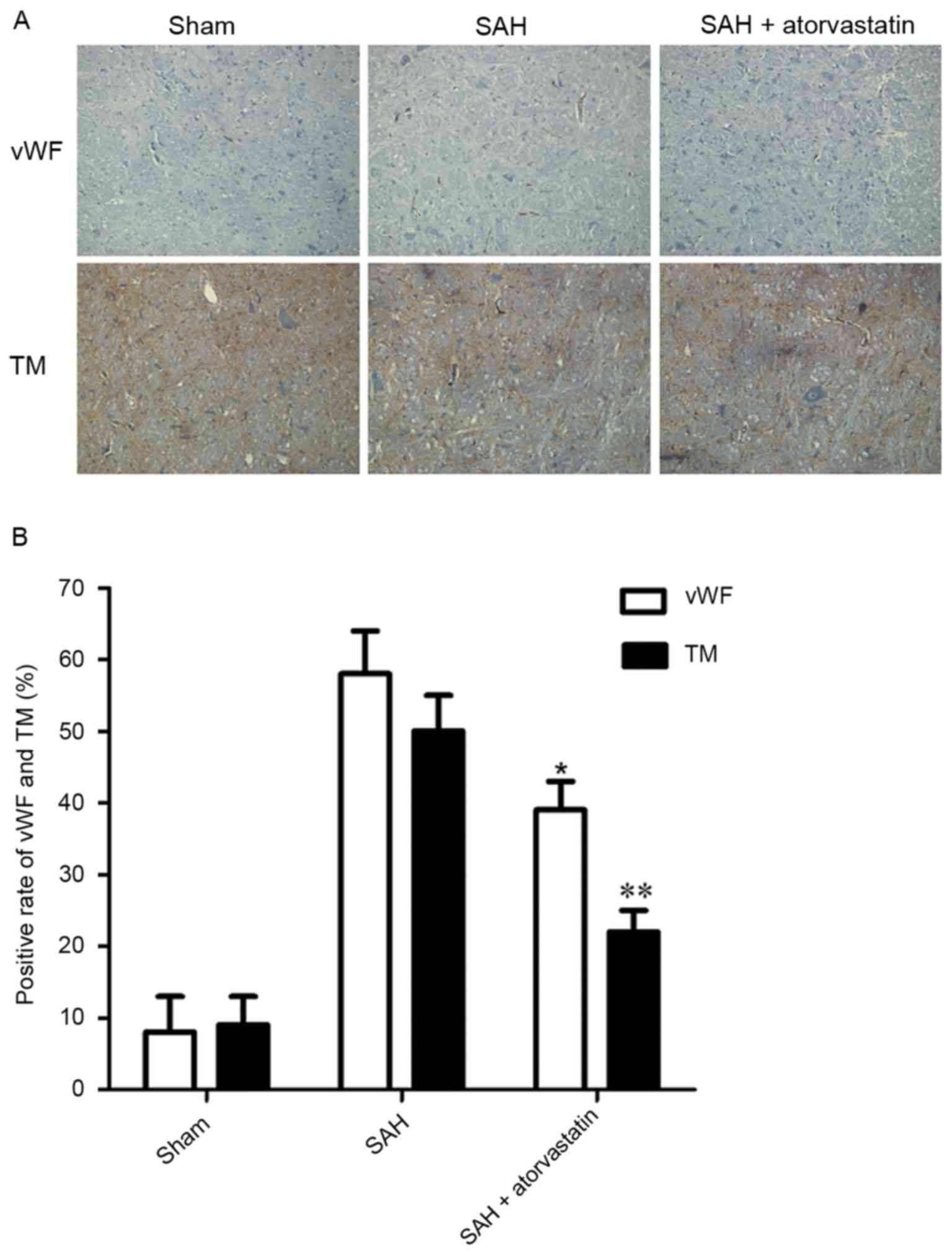
Immunohistochemical analysis revealed only a few
vWF- and TM-positive regions located in the vessels in the Sham
group (Fig. 3A). However, vWF and
TM expression levels were upregulated in the SAH and SAH +
atorvastatin treatment groups. The SAH group and the SAH +
atorvastatin treatment groups exhibited a significant increase in
the number of vWF- and TM-positive immunostained regions compared
with the Sham group (P<0.01; Fig
3B). Immunohistochemical analysis also indicated a significant
decrease in the expression vWF and TM in the SAH + atorvastatin
treatment group compared with the SAH group (P<0.01).
TUNEL staining and cell counting for
apoptosis
Few TUNEL-positive cells were detected in the
hippocampus of rabbits in the Sham group. However, TUNEL-positive
cells were significantly increased in SAH-induced rabbits at 72 h,
and TUNEL-positive cells were significantly decreased in the SAH +
atorvastatin treatment group compared with untreated SAH rabbits
(P<0.05; Fig. 4).
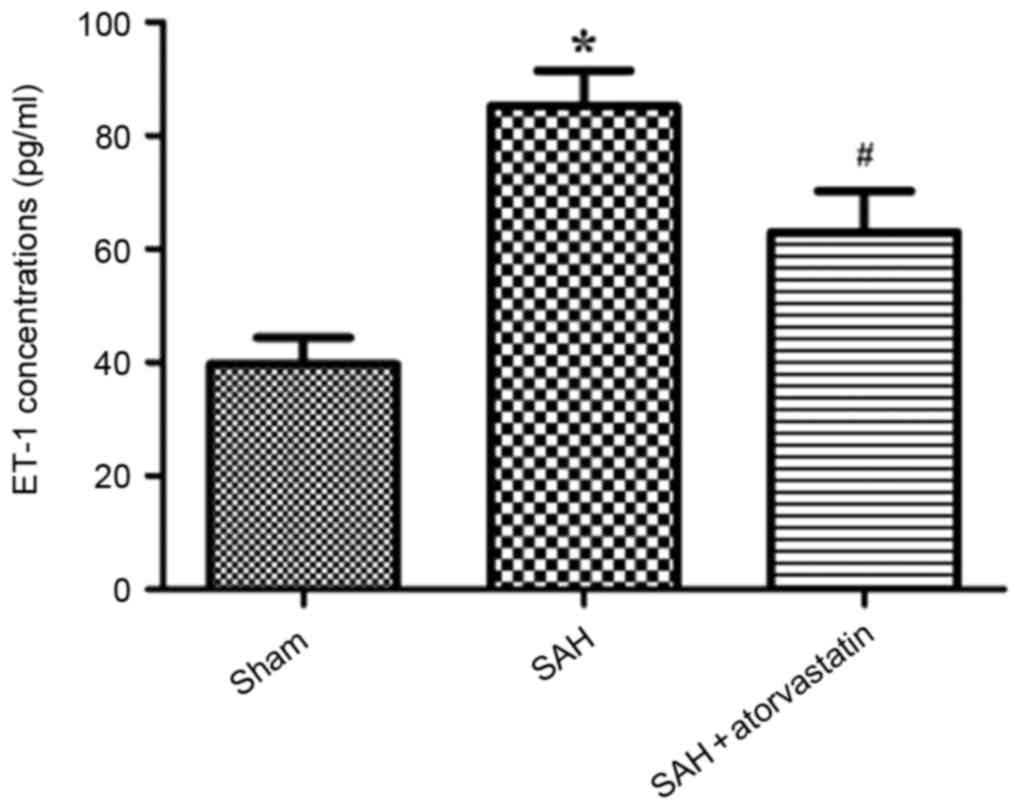
ELISA for ET-1 protein expression
ELISA was performed to examine the alterations in
protein concentration of the vasoconstrictor ET-1 to determine the
effects of atorvastatin on cerebral edema post-SAH. Compared with
the Sham group, ET-1 expression was significantly upregulated in
the SAH rabbits (85.24+6.25 vs. 39.72+4.67 pg/ml; P<0.01;
Fig. 5). Following atorvastatin
treatment, the elevation of plasma ET-1 concentration was
significantly lower compared with the SAH group (62.92+7.27 vs.
85.24+6.25 pg/ml; P<0.01; Fig.
5).
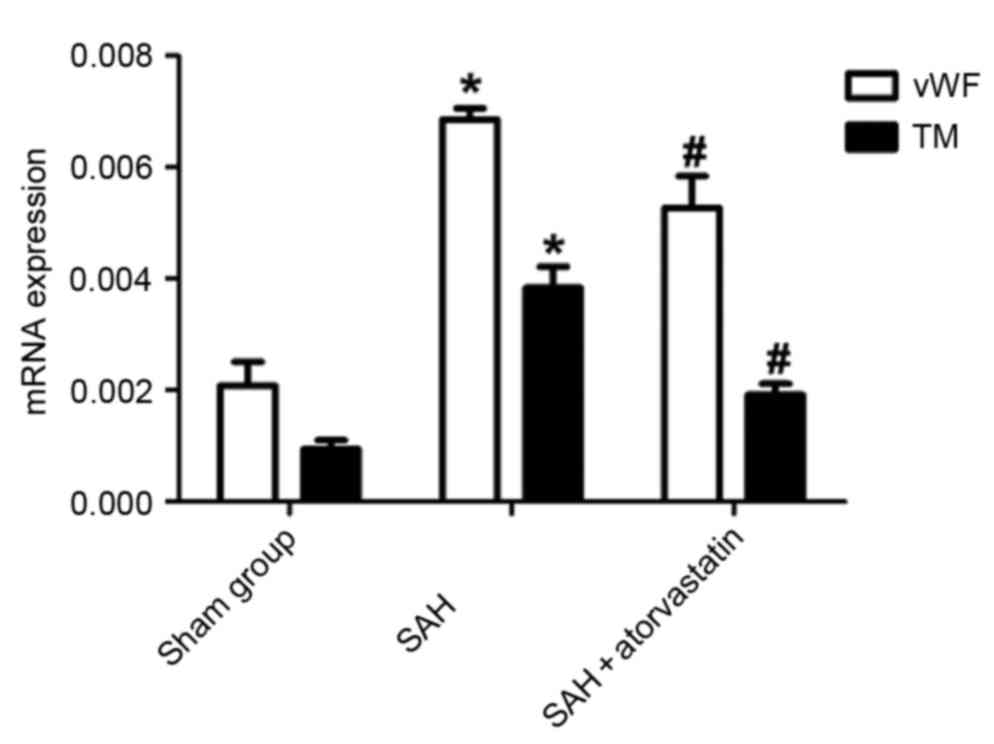
RT-qPCR for mRNA of vWF and TM
vWF and TM mRNA expression levels of were detected
by RT-qPCR: vWF and TM mRNAs were expressed at low levels in the
brain tissue in the Sham group, whereas the levels of vWF and TM
mRNA expression were significantly increased in the SAH and SAH +
atorvastatin treatment groups. Compared with the SAH group, the vWF
and TM mRNA expression levels were reduced in the SAH +
atorvastatin treatment group. (P<0.01; Fig. 6).
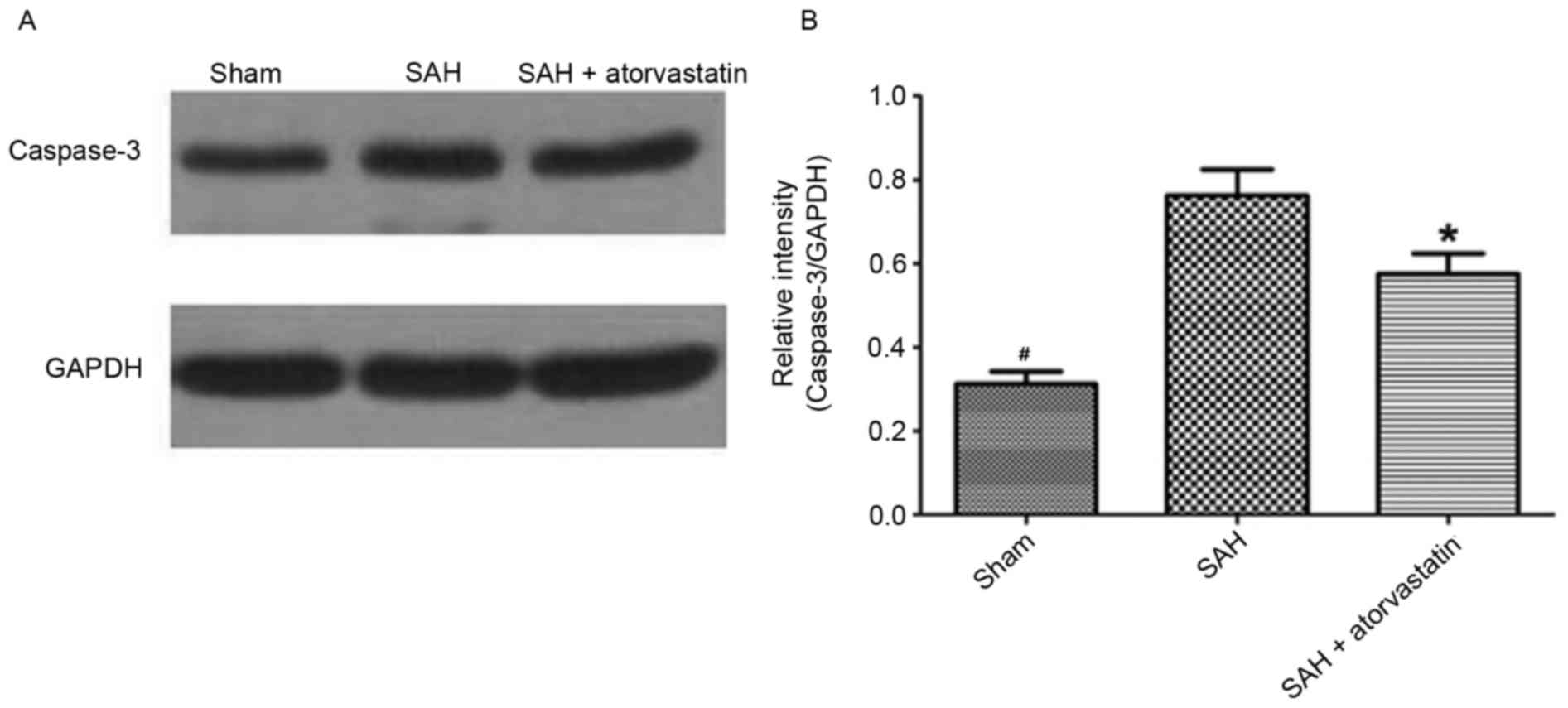
Caspase-3 protein expression
Caspase-3 protein expression in the hippocampus was
detected by western blot to observe neuronal apoptosis at 72 h
following SAH. SAH induced an evident increase of Caspase-3
expression levels in the hippocampus compared with the Sham group,
whereas the level of Caspase-3 was markedly decreased in the SAH +
atorvastatin group (P<0.05 vs. SAH; Fig. 7).
Discussion
To the best of our knowledge, the present study is
the first to demonstrate the therapeutic effects of atorvastatin on
early brain injury, CVS and cerebral vessels autoregulation
following experimental SAH in rabbits. CVS was evaluated by
examination and quantification of BA morphology, cerebral vessel
autoregulation by evaluating the expression levels of vWF and TM,
and early brain injury was examined by TUNEL staining.
Several previous studies have demonstrated that
statins may significantly inhibit the activity of matrix
metalloproteinase (MMP)-2 and MMP-9 to maintain the stability of
the blood/brain barrier (BBB) (17,21).
Another study reported that pitavastatin exerted its
neuroprotective effects by inhibiting the activation of c-Jun
N-terminal kinase p46/p55 and reducing the expression levels of
cleaved Caspase-9a and MMP-9 (22). Simvastatin has also been
demonstrated to protect the cerebrum from neuronal excitotoxicity
and cytotoxic edema by reducing the expression of phosphorylated
Calcium/calmodulin-dependent protein kinase type II and AQP4 in
experimental animals with ischemic stroke (18). Tseng et al (11) were the first to demonstrate by a
phase II randomized placebo-controlled trial that acute treatment
with pravastatin following SAH was safe and was able to improve CVS
and cerebral autoregulation, and reduce vasospasm-related delayed
ischemic deficits, and the poor prognostic rate was reduced. One
other study reported that simvastatin was safe for the prevention
of delayed cerebral ischemia following SAH in a randomized
placebo-controlled trial with 39 patients (23). The present study demonstrated that
atorvastatin may ameliorate early CVS and may protect cerebral
vascular endothelial cells in SAH model rabbits, suggesting its
potential as a treatment strategy to reverse CVS and EBI in
patients suffering from SAH.
However, previous studies (24,25)
have reported contradictory results in which statins were not
indicated to have a significant impact on brain edema, delayed
cerebral ischemia and brain injury post-SAH. A multicenter,
randomized, controlled, double-blinded clinical trial demonstrated
that high- or low-dose simvastatin treatment exerted no long-term
effects in the incidence of delayed ischemic deficits or in the
rate of favorable outcomes following SAH (24). Another study reported a similar
conclusion that no benefit was detected in the use of simvastatin
for long-term or short-term outcome in patients with SAH in a
famous Simvastatin in Aneurysmal SAH trial (25). The present study, however,
demonstrated that atorvastatin may have a vasodilatory effect on
the intracranial vessels of BA in a rabbit SAH model, indicating
its potential as a treatment strategy to alleviate CVS in patients
suffering from SAH.
Atorvastatin is an inhibitor of the HMG-CoA
reductase, and has been widely used in cardiovascular medicine as a
cholesterol-lowering drug. A previous study reported that
atorvastatin was a neuroprotective drug that was able to preserve
BBB permeability, decrease brain edema, increase neurological
scores and ameliorate CVS, and that atorvastatin may function by
inhibiting the Caspase-dependent proapoptotic pathway (10). Atorvastatin was previously
demonstrated to reduce the level of ET-1 expression, which
corresponded to its antivasospastic effects in chronic vasospasm
following SAH-induced vasospasm (26).
A mechanism underlying the anti-CVS or
neuroprotective functions of atorvastatin, whether or not a
critical reduction in cerebral blood flow has occurred, may be
explained by the subsequent constriction of intracerebral vessels,
good condition of vascular endothelial cell function and
improvement of cerebral blood flow. However, a specific molecular
mechanism remains unclear.
To explore the molecular mechanisms underlying
atorvastatin function, the expression of three major factors need
to be studied as follows: the function of vascular endothelial
cell, cerebral vessels autoregulation and contribution to CVS
post-SAH. All of them were potent vasoactive peptides synthesized
and released by the vascular endothelium and were markers of
endothelial function. vWF is a macromolecule glycoprotein with
adhesive functions that is mainly stored in platelet
activation-dependent granules and Weibel-Palade bodies of the
vascular endothelial cells. It is involved with platelet adhesion
to the extracellular matrix of vascular endothelial cells and
promotes platelet aggregation (27). TM is a single-strand glycoprotein
that is also synthesized and stored in vascular endothelial cells;
it has anticoagulation effects by combining with thrombin and
converts Protein C into activated protein C (27). ET-1 has been demonstrated to bind
to specific receptors on smooth muscle cells and leads to the
constriction of blood vessels and the proliferation of endothelial
cells (28), and has deleterious
effects on water homeostasis, cerebral edema and BBB integrity. In
normal physiological conditions, the expression of these cytokines
was minimal. When organism was stimulated severely or the function
of cerebral endothelial cells autoregulation suffered damage, the
expression of vWF, TM and ET-1 will increase. Therefore, vWF, TM
and ET-1 were considered as the ‘gold standard’ to evaluate the
function of cerebral endothelial cells and the cerebral vessel
autoregulation (27,29). In the previous study, endothelins
was recognized as potent vasoconstrictors, promitogens and
inflammatory mediators in the pathogenesis of vasospasm after SAH,
it may be critical in the pathogenesis of CVS (26,30,31).
Tang et al (32) also found
that the expression of vWF was significantly increased at CVS group
than the no-CVS group and control group by clinical research. Xu
et al (33) demonstrated
that TM had protective effects in preserving microvascular
integrity after SAH through preserving endothelial junction
proteins and quenching apoptosis/inflammation in endothelial cells,
and the underlying mechanism may be via blocking of the
p38MAPK-p53/NF-κB(p65) pathway. Su et al (34) also found TM analog solution
promotes reperfusion and reduces infarct volume in a thrombotic
model of stroke.
The present study demonstrated that atorvastatin
treatment significantly decreased the expression of vWF, TM and
ET-1 following SAH induction. These results suggested that
atorvastatin may alleviate CVS by regulating the expression of vWF,
TM and ET-1. The results indicated that the function of vascular
endothelial cell was destroyed and it also led to dysfunction of
cerebral autoregulation, the other side. Therefore, this finding
suggested that atorvastatin may alleviate CVS by regulating the
plasma concentrations of ET-1, vWF and TM in the clinic. A precise
mechanism of action for atorvastatin remains to be elucidated.
Neuronal apoptosis serves an important role of
mechanism about delayed neurologic deficits after SAH. Previous
studies have demonstrated that p53 also serves an important role in
apoptotic cell death following experimental SAH induction (10,35).
The present study performed TUNEL analysis to identify and quantify
the number of apoptotic cells in the hippocampus of SAH model
rabbits treated with and without atorvastatin. Caspases are a
family of cysteine proteases that serve essential roles in
apoptosis (10,14,36).
Caspases-3 is a member of the caspase family and serves an
important role in the execution-phase of apoptosis. The expression
of the Caspase-3 was reported to be increased in brain neurons
following SAH and may promote neuron apoptosis (3,10).
The present study demonstrated that the number of TUNEL-positive
cells was significantly increased in SAH rabbits, but was
significantly decreased in the SAH + atorvastatin group. These
results suggested that atorvastatin treatment may preclude CVS and
improve circulation of the brain, and may have neuroprotective
effects.
In conclusion, results from the present study
suggested that atorvastatin may be used for the treatment of CVS,
thus protecting vascular endothelial cell function and maintaining
cerebral vessel autoregulation. It is a relatively convenient and
affordable method for SAH and may be used in widespread clinical
practice. The increased expression of ET-1, vWF and TM may be
partly responsible for the therapeutic effects of atorvastatin;
atorvastatin treatment may reduce neuronal apoptosis and may be
another method for the treatment of SAH and improved outcome.
However, the precise mechanism of action of atorvastatin remains to
be investigated.
References
|
1
|
Komotar RJ, Schmidt JM, Starke RM,
Claassen J, Wartenberg KE, Lee K, Badjatia N, Connolly ES Jr and
Mayer SA: Resuscitation and critical care of poor-grade
subarachnoid hemorrhage. Neurosurgery. 64:397–411. 2009. View Article : Google Scholar
|
|
2
|
Rosengart AJ, Schultheiss KE, Tolentino J
and Macdonald RL: Prognostic factors for outcome in patients with
aneurysmal subarachnoid hemorrhage. Stroke. 38:2315–2321. 2007.
View Article : Google Scholar
|
|
3
|
Steiner T, Juvela S, Unterberg A, Jung C,
Forsting M and Rinkel G: European Stroke Organization: European
Stroke Organization guidelines for the management of intracranial
aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 35:93–112.
2013. View Article : Google Scholar
|
|
4
|
Budohoski KP, Czosnyka M, Kirkpatrick PJ,
Smirlewski P, Steiner LA and Pickard JD: Clinical relevance of
cerebral autoregulation following subarachnoid haemorrhage. Nat Rev
Neurol. 9:152–163. 2013. View Article : Google Scholar
|
|
5
|
Laskowitz DT and Kolls BJ: Neuroprotection
in subarachnoid hemorrhage. Stroke. 41 10 Suppl:S79–S84. 2010.
View Article : Google Scholar :
|
|
6
|
Macdonald RL, Higashida RT, Keller E,
Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wangke I, Bach D, Frey
A, et al: Clazosentan, an endothelin receptor antagonist, in
patients with aneurysmal subarachnoid haemorrhage undergoing
surgical clipping: A randomised, double-blind, placebo-controlled
phase 3 trail (CONSCIOUS-2). Lancet Neurol. 10:618–625. 2011.
View Article : Google Scholar
|
|
7
|
Kontos HA, Wei EP, Navari RM, Levasseur
JE, Rosenblum WI and Patterson JL Jr: Responses of cerebral
arteries and arterioles to acute hypotension and hypertension. Am J
Physiol. 234:H371–H383. 1978.
|
|
8
|
Sehba FA, Pluta RM and Zhang JH:
Metamorphosis of subarachnoid hemorrhage research: From delayed
vasospasm to early brain injury. Mol Neurobiol. 43:27–40. 2011.
View Article : Google Scholar
|
|
9
|
Tousoulis D, Antoniades C, Katsi V,
Bosinakou E, Kotsopoulou M, Tsioufis C and Stefanadis C: The impact
of early administration of low-dose atorvastatin treatment on
inflammatory process, in patients with unstable angina and low
cholesterol level. Int J Cardiol. 109:48–52. 2006. View Article : Google Scholar
|
|
10
|
Cheng G, Wei L, Zhi-Dan S, Shi-Guang Z and
Xiang-Zhen L: Atorvastatin ameliorates cerebral vasospasm and early
brain injury after subarachnoid hemorrhage and inhibits
caspase-dependent apoptosis pathway. BMC Neurosci. 10:72009.
View Article : Google Scholar :
|
|
11
|
Tseng MY, Czonsnyka M, Richards H, Pickard
JD and Kirkpatrick PJ: Effects of acute treatment with pravastatin
on cerebral vasospasm, autoregulation and delayed ischemic deficits
after aneurismal subarachnoid hemorrhage: A phase 2 randomized
placebo-controlled trial. Stroke. 36:1627–1632. 2005. View Article : Google Scholar
|
|
12
|
Lee YM, Chen WF, Chou DS, Jayakumar T, Hou
SY, Lee JJ, Hsiao G and Sheu JR: Cyclic nucleotides and
mitogen-activated protein kinase: Regulation of simvastatin in
platelet. J Biomed Sci. 17:452010. View Article : Google Scholar :
|
|
13
|
Luzak B, Boncler M, Rywaniak J, Wilk R,
Stanczyk L, Czyz M, Rysz J and Watala C: The effect of a platelet
cholesterol modulation on the acetylsalicylic acid-mediated blood
platelet inhibition in hypercholesterolemic patients. Eur J
Pharmacol. 658:91–97. 2011. View Article : Google Scholar
|
|
14
|
Chen JH, Yang LK, Chen L, Wang YH, Wu Y,
Jiang BJ, Zhu J and Li PP: Atorvastatin ameliorates early brain
injury after subarachnoid hemorrhage via inhibition of AQP4
expression in rabbits. Int J Mol Med. 37:1059–1066. 2016.
View Article : Google Scholar
|
|
15
|
Zhou C, Yamaguchi M, Kusaka G, Schonholz
C, Nanda A and Zhang JH: Caspase inhibitors prevent endothelial
apoptosis and cerebral vasospasm in dog model of experimental
subarachnoid hemorrhage. J Cerebral Blood Flow Metab. 24:419–431.
2004. View Article : Google Scholar
|
|
16
|
Hu N, Wu Y, Chen BZ, Han JF and Zhou MT:
Protective effect of stellate ganglion block on delayed cerebral
vasospasm in an experimental rat model of subarachnoid hemorrhage.
Brain Res. 1585:63–71. 2014. View Article : Google Scholar
|
|
17
|
Seo JH, Guo S, Lok J, Navaratna D, Whalen
MJ, Kim KW and Lo EH: Neurovascular matrix metalloproteinases and
the blood-brain barrier. Curr Pharm Des. 18:3645–3648. 2012.
View Article : Google Scholar :
|
|
18
|
Zhu MX, Lu C, Xia CM, Qiao ZW and Zhu DN:
Simvastatin pretreatment protects cerebrum from neuronal injury by
decreasing the expressions of phosphor-CaMK II and AQP4 in ischemic
stroke rats. J Mol Neurosci. 54:591–601. 2014. View Article : Google Scholar
|
|
19
|
Koyama Y, Maebara Y, Hayashi M, Nagae R,
Tokuyama S and Michinaga S: Endothelins reciprocally regulate
VEGF-A and angiopoietin-1 production in cultured rat astrocytes:
Implications on astrocytic proliferation. Glia. 60:1954–1963. 2012.
View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Rosenberg GA and Navratil M:
Metalloproteinase inhibition blocks edema in intracerebral
hemorrhage in the rat. Neurolgy. 48:921–926. 1997. View Article : Google Scholar
|
|
22
|
Chang CZ, Wu SC, Kwan AL and Lin CL:
Preconditioning with pitavastatin, an HMG-CoA reductase inhibitor,
attenuates C-Jun N-terminal kinase activation in experimental
subarachnoid hemorrhage-induced apoptosis. Acta Neurochir (Wien).
157:1031–1041. 2015. View Article : Google Scholar
|
|
23
|
Chou SH, Smith EE, Badjatia N, Nogueira
RG, Sims JR II, Ogilvy CS, Rordorf GA and Ayata C: A randomized,
double-blind, placebo-controlled pilot study of simvastatin in
aneurysmal subarachnoid hemorrhage. Stroke. 39:2891–2893. 2008.
View Article : Google Scholar
|
|
24
|
Wong GK, Chan DY, Siu DY, Zee BC, Poon WS,
Chan MT, Gin T and Leung M: HDS-SAH Investigators: High-dose
simvastatin for aneurysmal subarachnoid hemorrhage: Multicenter
randomized controlled double-blinded clinical trial. Stroke.
46:382–388. 2015. View Article : Google Scholar
|
|
25
|
Kirkpatrick PJ, Turner CL, Smith C,
Hutchinson PJ and Murray GD: STASH Collaborators: Simvastatin in
aneurysmal subarachnoid haemorrhage (STASH): A multicentre
randomised phase 3 trial. Lancet Neurol. 13:666–675. 2014.
View Article : Google Scholar
|
|
26
|
Chang CZ, Wu SC, Lin CL, Hwang SL, Howng
SL and Kwan AL: Atorvastatin preconditioning attenuates the
production of endothelin-1 and prevents experimental vasospasm in
rats. Acta Neurochir (Wien). 152:1399–1406. 2010. View Article : Google Scholar
|
|
27
|
Blann AD and Tabemer DA: A reliable marker
of endothelial cell dysfunction: Does it exist. Br J Haemataol.
90:224–228. 1995.
|
|
28
|
Chow M, Dumont AS and Kasselletal NF:
Endothelin receptor antagonists and cerebral vasospasm: An update.
Neurosurgery. 51:1333–1342. 2002. View Article : Google Scholar
|
|
29
|
Califano F, Giovanniello T, Pantone P,
Campana E, Parlapiano C, Alegiani F, Vincentelli GM and Turchetti
P: Clinical importance of thrombomodulin serum levels. Eur RevMed
Pharmacol Sci. 4:59–66. 2000.
|
|
30
|
Kästner S, Oertel MF, Scharbrodt W, Krause
M, Böker DK and Deinsberger W: Endothelin-1 in plasma, cisternal
CSF and microdialysate following aneurysmal SAH. Acta Neurochir
(Wien). 147:1271–1279. 2005. View Article : Google Scholar
|
|
31
|
Juvela S: Plasma endothelin concentrations
after aneurysmal subarachnoid hemorrhage. J Neurosurg. 92:390–400.
2000. View Article : Google Scholar
|
|
32
|
Tang QF, Lu SQ, Zhao YM and Qian JX: The
changes of von willebrand factor/a disintegrin-like and
metalloprotease with thrombospondin type I repeats-13 balance in
aneurysmal subarachnoid hemorrhage. Int J Clin Exp Med.
8:1342–1348. 2015.
|
|
33
|
Xu T, Zhang WG, Sun J, Zhang Y, Lu JF, Han
HB, Zhou CM and Yan JH: Protective effects of thrombomodulin on
microvascular permeability after subarachnoid hemorrhage in mouse
model. Neuroscience. 299:18–27. 2015. View Article : Google Scholar
|
|
34
|
Su EJ, Geyer M, Wahl M, Mann K, Ginsburg
D, Brohmann H, Petersen KU and Lawrence DA: The thrombomodulin
analog Solulin promotes reperfusion and reduces infarct volume in a
thrombotic stroke model. J Thromb Haemost. 9:1174–1182. 2011.
View Article : Google Scholar :
|
|
35
|
Cahill J, Calvert JW, Marcantonio S and
Zhang JH: p53 may play an orchestrating role in apoptotic cell
death after experimental subarachnoid hemorrhage. Neurosurgery.
60:531–545. 2007. View Article : Google Scholar
|
|
36
|
Alnemri ES, Livingston DJ, Nicholson DW,
Salvesen G, Thomberry NA, Wong WW and Yuan J: Human ICE/CED-3
protease nomenclature. Cell. 87:1711996. View Article : Google Scholar
|