Introduction
Factor VII (FVII) is a vitamin K dependent clotting
factor that participates in the early phases of blood coagulation.
It is synthesized by the liver as a single chain 50 kDa zymogen
with 406 amino acids and is secreted into blood at a concentration
of 500 ng/ml (1). FVII initiates
the extrinsic pathway in an activated two-chain form called FVIIa,
which is generated from the proteolysis of FVII at Arg152-Ile153.
It has a light chain containing a γ-carboxy glutamic acid (Gla)
domain and two epidermal growth factor-like domains and a heavy
chain comprised of a catalytic domain. The two chains are held
together by a disulfide bond formed between cysteines 135 and 262
(2). Subsequent to vascular injury
and in the presence of calcium ions, plasma FVIIa binds to tissue
factor (TF) exposed on extravascular cells and produces a TF-FVIIa
complex. This complex is involved in proteolytic activation of
coagulation factors IX and X and thrombin production (3).
The F7 gene is located on chromosome 13
(13q34), 2.8 kb upstream of the gene encoding factor X and
comprises 9 exons spanning ~12.5 kb (4). Hereditary FVII deficiency is commonly
inherited as an autosomal recessive disorder and has an incidence
of 1 per 500,000 in the general population (5). Clinical manifestations of FVII
deficiency range from mild bruising to life threatening hemorrhages
(6). The hemorrhagic diathesis in
affected patients is highly variable and does not necessarily
associate with plasma FVII procoagulant activity (FVII:C) levels
(7).
A large number of different F7 gene variants,
including missense, nonsense, promoter and splice site mutations,
have been reported. In addition, the functional impact of certain
variants has been investigated in order to provide evidence for the
underlying molecular mechanisms of FVII deficiency (8,9).
The present study investigated the functional
characteristics of H348R and S282R mutations within the serine
protease domain of FVII, which was detected in compound
heterozygous status in an Iranian patient with FVII deficiency.
Ser282 and His348 residues are located on exon 8 of the F7
gene and are highly conserved across different species. These
mutations were previously reported (10,11);
however, this patient appears to be the first case harboring the
two mutations simultaneously. In the present study, the mutated
F7 gene was expressed in mammalian cells to determine
different functional aspects of FVII biosynthesis including RNA
expression, protein secretion, coagulation activity and
intracellular localization of the protein. The confirmation of
pathogenicity of these mutations paves the way for the management
of the disease and genetic counseling for prenatal diagnosis in
affected families.
Materials and methods
Cell culture and construction of
expression vectors
HepG2 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS), 100
U/ml penicillin, 100 µg/ml streptomycin and 5.5 mM D-glucose
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). These
cells are known to express high levels of human FVII; however, the
HepG2 cell line is misidentified, according to http://web.expasy.org/cellosaurus/CVCL_0027. Total RNA
was extracted using TRIzol reagent (Thermo Fisher Scientific, Inc.)
and used for cDNA synthesis (1 µg), using a reverse and first
strand cDNA synthesis kit (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Specific primers with
XhoI and EcoRI recognition sites were used to isolate the F7 coding
region. The resulting polymerase chain reaction (PCR) product had
an XhoI recognition site on one end and an EcoRI site on the other
end. The sequence of oligonucleotides used to amplify F7 cDNA were
as follows: Forward, 5′-AGAATTCTTCATCATGGTCTCCCAGG-3′ and reverse,
5′-TCTCGAGGCTAGGGAAATGGGGCTCG-3′. The purified PCR product and
pcDNA3.1/neo plasmid were doubly digested with XhoI and EcoRI
enzymes (Thermo Fisher Scientific, Inc.) and the digestion products
were purified using a Genjet PCR purification kit (Thermo Fisher
Scientific, Inc.). Using a fast DNA ligation kit (Bio Basic, Inc.,
Markham, ON, Canada), purified F7 cDNA was inserted into the
pcDNA3.1/neo vector (Invitrogen; Thermo Fisher Scientific, Inc.).
The resulting recombinant vector [pcDNA/wild-type (WT)] was
amplified in competent DH5α bacterial cells. Amplified plasmids
were isolated and sequenced to verify the cloning process.
The desired mutations were introduced into pcDNA/WT
by the insertion of mutation-harboring fragments using suitable
restriction enzymes. In order to insert the S282R substitution, a
PCR reaction was performed on a patient's genomic DNA to amplify a
fragment containing the S282R mutation. The fragment had two
Van91I recognition sites on both sides of the S282R
substitution. The PCR product was digested by the Van91I
restriction enzyme (Thermo Fisher Scientific, Inc.). The linearized
pcDNA/WT vector was also prepared by Van91I digestion. Then,
a mutation containing fragment was inserted into the vector and the
recombinant vector (pcDNA/S282R) was amplified in competent DH5α
cells and sequenced.
In the same way, the gene fragment containing the
H348R substitution on a patient's DNA was amplified by PCR. The
reverse primer comprised an XhoI site at its 5′-end. First,
the PCR product was partially digested by Van91I and then
completely digested by XhoI. The digestion product was
inserted into the linearized pcDNA/WT and doubly digested by
XhoI and Van91I. The resulting recombinant vector
(pcDNA/H348) was amplified and verified by sequencing.
CHO-K1 cell culture and
transfection
CHO-K1 cells were grown in Glutamax DMEM-F12 (Gibco,
KBC, Iran) medium supplemented with 10% FBS and 1% penicillin and
streptomycin. The adherent cells were suspended using trypsin and
4×104 cells were transferred to 60-mm dishes for each
transfection reaction. According to the manufacturer's
instructions, turbofect (Thermo Fisher Scientific, Inc.) was used
to transiently transfect the cells with pcDNA/WT, pcDNA/S282R and
pcDNA/H348R vectors. To estimate transfection efficiency, a vector
containing green fluorescence protein (GFP) reporter gene
(pcDNA/GFP) and the empty vector (pcDNA) were used as transfection
controls. Three microscopic fields (×40) of each cell culture
dishes were randomly observed 24 h following transfection and GFP
positive as well as GFP negative cells were counted. The
transfection efficiency was calculated using the formula:
Transfection efficiency=number of GFP
positive cellstotal number of counted cells
Transfected cells were incubated for 48 and 72 h at
37°C in 5% CO2 prior to harvesting. Conditioned medium
of each dish was collected for FVII protein measurement. Total RNA
content of the cells was extracted using TRIzol reagent
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and cell lysates
were prepared using freeze-thaw method (3 cycles of 70°C/37°C). All
samples were stored at −70°C until further analysis.
Reverse transcription-PCR (RT-PCR) and
protein expression assays
To confirm F7 RNA expression by the transfected
cells, RT-PCR analysis was performed on the RNA extracted from
transfected CHO-K1 cells using the QIAzol reagent (cat. no. 79306;
Qiagen GmbH, Hilden, Germany) as recommended by the manufacturer's
instructions. The concentration and quality of the extracted RNA
was determined using a NanoDrop spectrophotometer (NanoDrop
Technologies; Thermo Fisher Scientific, Inc.). The extracted RNAs
were used directly for cDNA synthesis by PrimeScript™ RT kit (cat.
no. RR014A; Takara Biotechnology Co., Ltd., Dalian, China) using
the following temperature program; RNA denaturation: 65°C for 5
min, reverse transcription: 42°C for 15 min followed by 85°C for 5
min to inactivate the reverse transcriptase enzyme. The primers for
F7 CD2 and β-actin were as follows, respectively; F7 CD2 forward,
TGTGTGAACGAGAACGGCG and reverse, ACCTTCCGTGACTGCTGC; and β-actin
forward, AGAGCTACGAGCTGCCTGAC and reverse, AGCACTGTGTTGGCGTACAG,
which were used to amplify the F7 CD2 and β-actin transcript cDNA
with PCR master mix (cat. no. K0171; Thermo Fisher Scientific,
Inc.) under the following conditions; denaturation at 95°C for 45
sec, annealing for 40 sec at 56°C, elongation at 72°C for 50 sec,
for a total of 30 cycles. The PCR products on electrophoresis
agarose gel (2%) were stained with SYBR Green I dye (1:10,000;
Thermo Fisher Scientific, Inc.). Relative mRNA ratio was calculated
using ImageJ software 2.0 (National Institutes of Health, Bethesda,
MD, USA) with β-actin as an internal control (12).
FVII procoagulant activity (FVII:C) and antigen
levels (FVII:Ag) were measured in conditioned media and cell lysate
samples, 48 and 72 h following transfection. FVII:C was determined
by the one-stage prothrombin time (PT)-based method (1) using FVII-deficient plasma as a
substrate and commercial human thromboplastin preparation. This
procedure was performed by an automated Sysmex CA-1500 coagulation
analyzer system. The concentration of FVII protein in conditioned
media and cell lysates was analyzed using a commercial ELISA assay
(cat. no. ab108829, human factor VII ELISA kit; Abcam, Cambridge,
UK).
Immunocytochemistry
In order to study intracellular localization of WT
and mutated FVII protein, immunocytochemistry was performed on CHO
cells. Transfected cells were seeded onto glass coverslips and
fixed with 3% paraformaldehyde for 1 h at room temperature.
Permeabilization was performed with 0.1% Triton X-100
(Sigma-Aldrich; Merck KGaA) for 10 min at room temperature and the
cover slips were blocked with 1% bovine serum albumin in PBS
(Sigma-Aldrich; Merck KGaA). A rabbit anti-human FVII antibody
(1:150, cat. no. ab97614; Abcam) was added to each coverslip and
incubated for 1 h at 37°C. Following incubation and PBS washing
steps, the cells were incubated with Dylight 488 goat anti-rabbit
IgG (1:250, cat. no. ab96899; Abcam) for 30 min at 37°C and 10
fields of each slide were analyzed under a fluorescent microscope
(Olympus Corporation, Tokyo, Japan).
Statistical analysis
The comparison between mean levels of protein
expression in different study groups was performed using a one-way
analysis of variance followed by a Tukey's multiple comparison test
using GraphPad Prism V.7.03 (GraphPad software, Inc., La Jolla, CA,
USA). Data are expressed as the mean ± standard deviation.
P<0.05 was determined to indicate a statistically significant
difference.
Results
Mutagenesis and transfection of CHO
cells
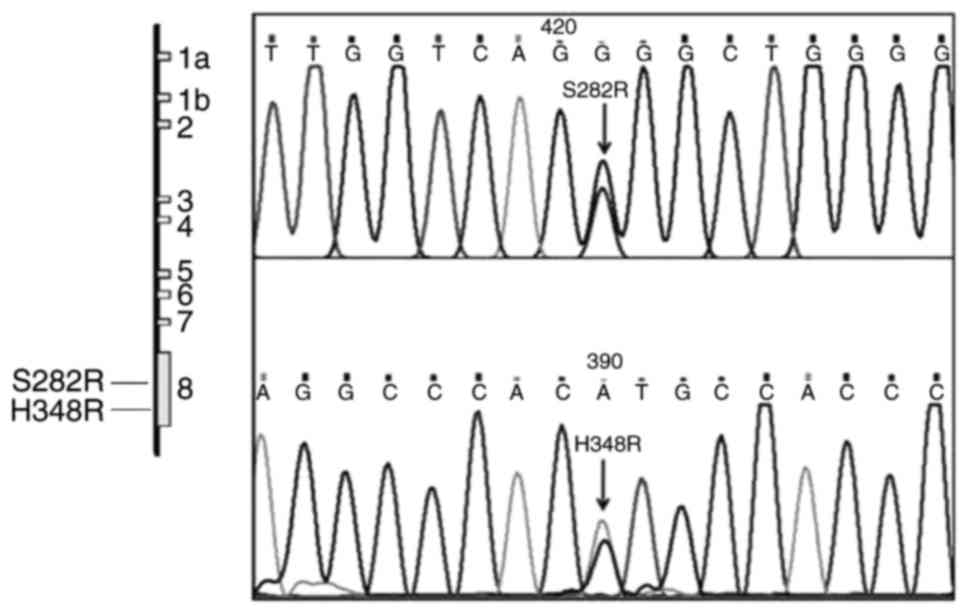
The sequencing of pcDNA/WT, pcDNA/S282R and
pcDNA/H348R vectors indicated that the 2 mutations were
successfully created at the desired positions on F7 cDNA (Fig. 1). The control CHO cells transfected
with pcDNA/GFP in parallel with the mutated F7 expression vectors
indicated appropriate transfection efficiency with 63±4.1%
GFP-positive cells (Fig. 2).
Expression of the mutated transcripts
and proteins
RT-qPCR demonstrated that transfected cells
expressed the WT, S282R and H348R transcripts, indicating that the
mutations had no effect on F7 mRNA expression (data not shown).
Ser282 and His348 residues on exon 8 are highly conserved amino
acids across different species, which have remained unaltered
through evolution (Table I). To
investigate the effects of mutations on procoagulant activity of
FVII (FVII:C), conditioned media was analyzed using a one-stage
PT-based method. No FVII procoagulant activity was detected in
conditioned media of the cells transfected with the mutated
constructs compared with cells expressing the WT construct
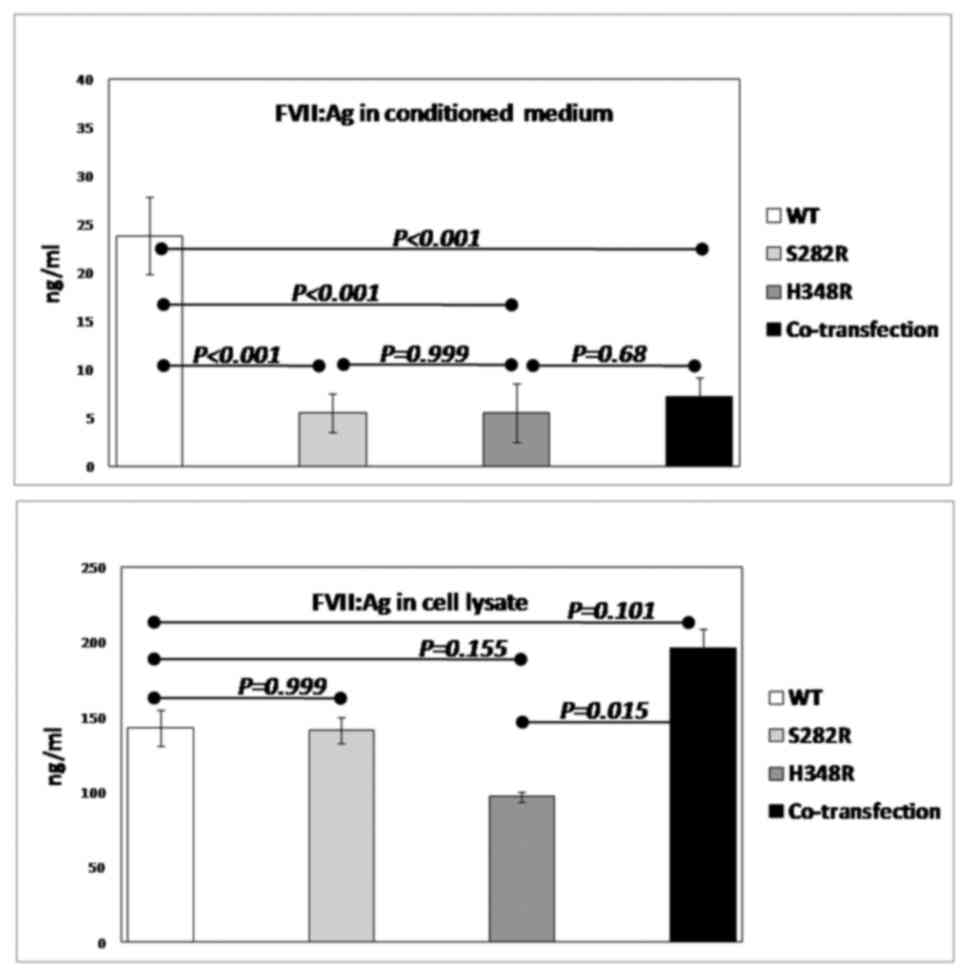
(Table II). Also, an ELISA assay
was used to determine the concentration of FVII antigen (FVII:Ag)
in the conditioned media as well as in the lysate of transfected
cells (Fig. 3). The mean
concentration of the FVII antigen in conditioned media of the cells
harboring WT vector was 23.84 ng/ml, whereas in the pcDNA/S282R or
pcDNA/H348R groups, it was 4.3-fold (5.5 ng/ml; Table II and Fig. 3). In addition, conditioned media of
the cells that were co-transfected with pcDNA/S282R and pcDNA/H348R
had reduced extracellular FVII levels (Fig. 3).
 | Table I.Conservation of S282 and H348 residues
across different species. |
Table I.
Conservation of S282 and H348 residues
across different species.
| Protein ID
species | Protein sequence
(S282R) | Protein sequence
(H348R) |
|---|
|
sp|P22457|FA7_BOVIN |
AFVRFSAVSGWGQLLERGVT |
SKDACKGDSGGPHATRFRGTWFL |
|
sp|P08709|FA7_HUMAN |
AFVRFSLVSGWGQLLDRGAT |
SKDSCKGDSGGPHATHYRGTWYL |
|
sp|Q2F9P2|FA7_PANTR |
AFVRFSLVSGWGQLLDRGAT |
SKDSCKGDSGGPHATHYRGTWYL |
|
sp|Q2F9P4|FA7_PANPA |
AFVRFSLVSGWGQLLDRGAT |
SKDSCKGDSGGPHATHYRGTWYL |
|
sp|Q8K3U6|FA7_RAT |
ASIRFSRVSGWGQLLDRGAT |
FKDACKGDSGGPHATHYHGTWYL |
|
sp|P70375|FA7_MOUSE |
ARIRFSRVSGWGQLLDRGAT |
TKDACKGDSGGPHATHYHGTWYL |
| Table II.Transient expression of recombinant
FVII variants by transfected CHO-K1 cells. |
Table II.
Transient expression of recombinant
FVII variants by transfected CHO-K1 cells.
| FVII variants | WT | S282R | H348R | Co-transfection |
|---|
| FVII:Ag (conditioned
medium, ng/ml) | 23.84 | 5.5 | 5.5 | 7.2 |
| FVII:Ag (cell lysate,
ng/ml) | 142.65 | 141.22 | 96.85 | 195.8 |
| FVII:C (conditioned
medium, %) | 100 | ND | ND | ND |
In the lysate of the cells expressing the H348R
allele, FVII antigen level was reduced by~32% (96.85 ng/ml)
compared with the cells expressing the WT allele (142.65 ng/ml).
FVII antigen in the lysate of the cells expressing S282R allele was
comparable to that of the WT allele. However, in co-transfected
cells, the protein expression levels were enhanced (195.8 ng/ml)
compared with the WT group (Table
II).
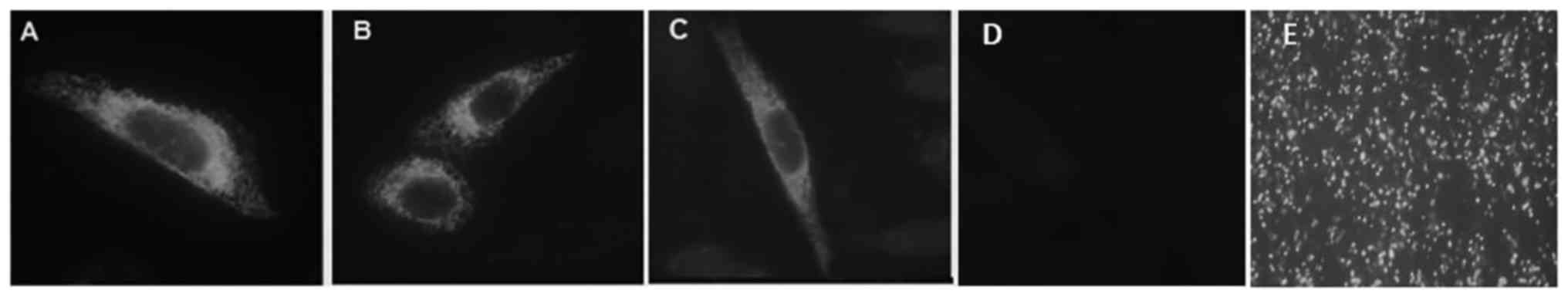
Immunocytochemistry
Intracellular localization of WT, S282R and H348R
FVII proteins was determined by fluorescent microscopy. Perinuclear
weak staining was observed in the cytoplasm of the cells expressing
WT protein (Fig. 2A). The same
observation of perinuclear staining was observed in the cells
expressing the mutated FVII (Fig. 2B
and C). The mutated proteins exhibited similar
immunofluorescence intensity to that of the WT protein, suggesting
that the mutations had no marked effect on the intracellular
localization of FVII.
Discussion
In the present study, H348R and S282R mutations
within serine protease domain of FVII were investigated, as these
mutations were detected in compound heterozygous status in an
Iranian patient with an FVII deficiency. Ser282 and His348 residues
on exon 8 are highly conserved amino acids across different
species, which have remained unaltered through evolution. Exon 8 is
the largest exon of the gene and accommodates the majority of
missense mutations scattered across the F7 gene. This exon
encodes for the catalytic domain of FVII protein, which has serine
protease activity and mediates factor X activation by proteolytic
cleavage (13). Mutations in this
domain may severely affect the coagulation activity and the
secretion of the FVII protein. However, considerable evidence now
supports the concept that there is no clear genotype-phenotype
association in FVII deficiency (14). Preceded by several publications
based on various mutations, it has been demonstrated that the
disease manifestations, including epistaxis, hemarthrosis and
menorrhagia, do not always associate with FVII:C (1,15).
Recently, Quintavalle et al (14) demonstrated the variations in
clinical and molecular aspects of the FVII deficiency disease. They
reported an association between the type of F7 variant and
FVII:C levels, but not for bleeding tendency and FVII:C.
Using immunofluorescent staining, the present study
demonstrated that perinuclear localization of both S282R and H348R
protein variants was similar to that of the WT protein (Fig. 2). Normally, the FVII protein is
predominantly localized to the perinucleus with cytoplasmic
expression. This suggests that FVII accumulates in the endoplasmic
reticulum (ER) and golgi apparatus in order to achieve correct
folding and modifications prior to secretion (3). Certain mutations may alter this
localization due to impaired secretion pathway or degradation of
FVII in various cellular compartments (16).
Generally, there is a certain degree of difference
between intra- and extra-cellular levels of various secreted
proteins. This difference may be due to the accumulation of the
protein in the ER or golgi apparatus prior to secretion. In the
case of FVII, the protein accumulates inside the cell until it
gains the correct post-transcriptional modifications and
conformational properties. In order to reveal the effect of the
mutations on FVII secretion, FVII:Ag was measured in conditioned
media of transiently transfected cells. The present study indicated
that S282R and H348R substitutions reduced the secretion of the
protein variants into the conditioned media. This result is
consistent with the results of previous studies on other F7
gene mutations (17,18). In the ELISA assay, which was
performed on the lysates of the transfected cells, there was a
discrepancy regarding the impact of the two mutations on the
intra-cellular FVII levels. The quantitative measurement of
intracellular FVII in the cells transfected with the H348R variant
exhibited reduced levels of the protein compared with the WT
protein, whereas the intracellular levels of the mutated S282R FVII
was equal to the WT control. The difference in the expression and
stability of FVII protein variants has been previously reported
indicating the distinct effect of each mutation on the overall
intracellular FVII protein content in transfected cells (5,19).
The enzymatic activity (FVII:C) of the recombinant
FVII variants in conditioned media was investigated using a
PT-based assay, which indicated no detectable coagulation activity
in S282R and H348R media. However, the concentration of the
secreted proteins (5.5 ng/ml) was very low in the media, which may
lead to undetectable FVII:C activity. However, the present study
did not study the function of the proteins in the cell lysate
samples. Therefore, further functional analysis of the mutations is
required to determine the activity of the two protein variants and
to explain the discrepancy in their intra-cellular levels.
In conclusion, the results of the present study
indicated that S282R and H348R substitutions within the FVII serine
protease domain affected the secretion of the enzyme. Although the
pattern of intracellular localization of the mutated proteins
remained unaltered, there were differences between S282R and H348R
intra-cellular levels. The validation of pathogenic effects of
these mutations may be implemented for genetic counseling and
prenatal diagnosis of FVII deficiency in families with affected
children in the future.
Acknowledgements
The present study was funded in part by a grant from
Iran National Science Foundation (INSF; grant no. 90004948) to SS
and in the other part by research deputy of Tarbiat Modares
University to AM.
References
|
1
|
Hellstern P, Beeck H, Fellhauer A, Fischer
A and Faller-Stöckl B: Measurement of factor VII and of activated
factor VII in healthy individuals and in prothrombin complex
concentrates. Thromb Res. 86:493–504. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Millar DS, Kemball-Cook G, McVey JH,
Tuddenham EG, Mumford AD, Attock GB, Reverter JC, Lanir N, Parapia
LA, Reynaud J, et al: Molecular analysis of the genotype-phenotype
relationship in factor VII deficiency. Hum Genet. 107:327–342.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tanaka R, Nakashima D, Suzuki A, Miyawaki
Y, Fujimori Y, Yamada T, Takagi A, Murate T, Yamamoto K, Katsumi A,
et al: Impaired secretion of carboxyl-terminal truncated factor VII
due to an F7 nonsense mutation associated with FVII deficiency.
Thromb Res. 125:262–266. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
O'Hara PJ, Grant FJ, Haldeman BA, Gray CL,
Insley MY, Hagen FS and Murray MJ: Nucleotide sequence of the gene
coding for human factor VII, a vitamin K-dependent protein
participating in blood coagulation. Proc Natl Acad Sci USA. 84:pp.
5158–5162. 1987; View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagaizumi K, Inaba H, Suzuki T, Hatta Y,
Hagiwara T, Amano K, Arai M and Fukutake K: Two double heterozygous
mutations in the F7 gene show different manifestations. Br J
Haematol. 119:1052–1058. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shahbazi S: Nonsense-mediated mRNA decay
among coagulation factor genes. Iran J Basic Med Sci. 19:344–349.
2016.PubMed/NCBI
|
|
7
|
Mariani G and Bernardi F: Factor VII
deficiency. Semin Thromb Hemos. 35:400–406. 2009. View Article : Google Scholar
|
|
8
|
Mariani G, Herrmann FH, Dolce A, Batorova
A, Etro D, Peyvandi F, Wulff K, Schved JF, Auerswald G, Ingerslev
J, et al: Clinical phenotypes and factor VII genotype in congenital
factor VII deficiency. Thromb Haemost. 93:481–487. 2005.PubMed/NCBI
|
|
9
|
Liu N, Aldea S, Francois D, Cherqui-Michel
M, Giansily-Blaizot M and Fischler M: Recombinant activated factor
VII for a patient with factor VII deficiency undergoing urgent
intracerebral haematoma evacuation with underlying cavernous
angioma. Br J Anaesth. 103:858–860. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahmed RP, Biswas A, Kannan M, Bhattacharya
M, Geisen C, Seifried E, Oldenburg J and Saxena R: First report of
a FVII-deficient Indian patient carrying double heterozygous
mutations in the FVII gene. Thromb Res. 115:535–536. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Peyvandi F, Jenkins PV, Mannucci PM,
Billio A, Zeinali S, Perkins SJ and Perry DJ: Molecular
characterisation and three-dimensional structural analysis of
mutations in 21 unrelated families with inherited factor VII
deficiency. Thromb Haemost. 84:250–257. 2000.PubMed/NCBI
|
|
12
|
Ma W, Shi X, Lu S, Wu L and Wang Y:
Hypoxia-induced overexpression of DEC1 is regulated by HIF-1α in
hepatocellular carcinoma. Oncol Rep. 30:2957–2962. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang P, Xue D, Zhang Y, Ye L, Liu Y,
Makale M, Kesari S, Edgington TS and Liu C: The extrinsic
coagulation cascade and tissue factor pathway inhibitor in
macrophages: A potential therapeutic opportunity for
atherosclerotic thrombosis. Thromb Res. 133:657–666. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Quintavalle G, Riccardi F, Rivolta GF,
Martorana D, Di Perna C, Percesepe A and Tagliaferri A; Ad-Hoc
Study Group, : F7 gene variants modulate protein levels in a large
cohort of patients with factor VII deficiency. Results from a
genotype-phenotype study. Thromb Haemost. 117:1455–1464. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Girolami A, Cosi E, Ferrari S, Girolami B
and Lombardi AM: Bleeding manifestations in heterozygotes with
congenital FVII deficiency: A comparison with unaffected family
members during a long observation period. Hematology. 22:375–379.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hunault M, Arbini AA, Carew JA, Peyvandi F
and Bauer KA: Characterization of two naturally occurring mutations
in the second epidermal growth factor-like domain of factor VII.
Blood. 93:1237–1244. 1999.PubMed/NCBI
|
|
17
|
Branchini A, Ferrarese M, Lombardi S, Mari
R, Bernardi F and Pinotti M: Differential functional readthrough
over homozygous nonsense mutations contributes to the bleeding
phenotype in coagulation factor VII deficiency. J Thromb Haemost.
14:1994–2000. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hao X, Cheng X, Ye J, Wang Y, Yang L, Wang
M and Jin Y: Severe coagulation factor VII deficiency caused by a
novel homozygous mutation (p. Trp284Gly) in loop 140s. Blood Coagul
Fibrinolysis. 27:461–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
D'Andrea G, Bossone A, Lupone MR, Peyvandi
F, Maisto G, Perricone F, Grandone E and Margaglione M: Molecular
characterization of a factor VII deficient patient supports the
importance of the second epidermal growth factor-like domain.
Haematologica. 89:979–984. 2004.PubMed/NCBI
|